
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of TMAO and urea on the structure of water studied by inelastic X-ray scattering
Christoph J.
Sahle
*a,
Martin A.
Schroer
bc,
Iina
Juurinen
d and
Johannes
Niskanen
d
aEuropean Synchrotron Radiation Facility, 71 Avenue des Martyrs, 38000 Grenoble, France. E-mail: christoph.sahle@esrf.fr
bDeutsches Elektronen-Synchrotron DESY, Notkestrasse 85, 22607 Hamburg, Germany
cThe Hamburg Centre for Ultrafast Imaging (CUI), Luruper Chaussee 149, 22761 Hamburg, Germany
dDepartment of Physics, University of Helsinki, Gustaf Hällströmin katu 2a, P. O. Box 64, FI-00014, Finland
First published on 25th May 2016
Abstract
We present a study on the influence of the naturally occurring organic osmolytes tri-methylamine N-oxide (TMAO) and urea on the bulk structure of water using X-ray Raman scattering spectroscopy. Addition of TMAO is known to stabilize proteins in otherwise destabilizing aqueous urea solutions. The experimental X-ray Raman scattering spectra change systematically with increasing solute concentration revealing different effects on the structure of water due to the presence of the two osmolytes. Although these effects are distinct for both molecular species, they have mutually compensating influences on the spectra of the ternary water–TMAO–urea mixtures. This compensation effect seen in the spectra vanishes only at the highest studied ternary concentration of 4 M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 M (TMAO:urea). Our experiment shows that the hydrogen-bonding structure of water remains rather intact in the presence of the aforementioned osmolytes if both of them are present.
:4 M (TMAO:urea). Our experiment shows that the hydrogen-bonding structure of water remains rather intact in the presence of the aforementioned osmolytes if both of them are present.
1 Introduction
The two organic osmolytes tri-methylamine N-oxide (TMAO) and urea are commonly occuring substances in the metabolisms of animals.1,2 TMAO has protein stabilizing effects,3–5 that deep-sea organisms use to counteract the high pressure perturbation in their natural habitat. Urea is found as a waste product in mammalian kidneys and is a strong denaturant at high concentrations. Interestingly, TMAO is known to counteract the protein-destabilizing influence of urea3,5–9 (for an exhaustive review, see ref. 10). While these properties of the two osmolytes are well characterized, the underlying mechanisms are not entirely understood.10One proposed destabilization mechanism of urea is the direct interaction with the protein backbone11–13 or with the amino acid side chains.14–17 The second hypothesis is an indirect mechanism where urea alters the water structure,18,19 which is not a likely explanation.10 In fact, recent experiments20–23 and simulations24,25 show that the contrary is the case: urea fits well into the water network by substituting for a water dimer. Some studies suggest that this happens at the cost of substantial disruptions of the water network21 and some do not.25,26 Finally, Yoshida et al. suggest that urea strengthens the water structure slightly rather than weakening it.27
TMAO is known to be excluded from the surface of proteins.28–30 Among other possible explanations, its effects on the structure of water have been proposed as a mechanism to stabilize proteins.31,32 Strong interactions between TMAO and water have been reported frequently.32–35 Several experiments support immobile water in TMAO–water complexes,32,35 in accordance with simulations.36 Strongly immobilized OH groups have been found in the vicinity of TMAO,33 but despite the slow dynamics of the molecules in the hydration shell, a water-like disordered structure was still concluded. Simulations of Laage and coworkers explain the slow reorientation without the need for iceberg formation.37
The counter-acting effects, i.e. the protein stabilizing influence of TMAO in otherwise denaturating urea solutions, have been proposed to originate from direct interactions38–40 and from water-mediated interactions. Some studies show that TMAO and urea hydrogen bond strongly and thus TMAO is able to refrain urea from the proteins' backbone/surface.38,39 A combined MD simulation and neutron scattering study on TMAO:urea solutions by Meersman et al.40 found evidence for the direct interaction between urea and TMAO and explained the counter-acting effect by the formation of TMAO:urea complexes, but the result was later revised.41 On the other hand, molecular dynamics (MD) simulations42 and dielectric relaxation spectroscopy in combination with viscosity measurements43 found no evidence for a strong direct interaction between urea and TMAO. In their work, Hunger et al.43 propose a water-mediated interaction between TMAO and urea involving hydrated, long lived TMAO·3H2O complexes where the hydration waters of TMAO provide one or several protecting layers between TMAO, and the donor sites of urea. The idea of these long-lived complexes is in agreement with the observation of immobile OH groups of Rezus and co-workers.33
In this work, we present a study of the influence of TMAO and urea on the structure of water using X-ray Raman scattering (XRS) spectroscopy at the oxygen K-edge of aqueous solutions of urea, TMAO, and mixtures of the two. XRS has been shown to be sensitive to the structure of water and its hydrogen bonding topology across the extensive phase diagram,44–50 as well as in aqueous solutions.51–54 The use of XRS, thus, allows us to gain element specific spectroscopic information about the systems under relevant thermodynamic conditions without, for example, isotopic substitutions. The information of the local electronic structure is entangled with the local atomic structure and bonding topology around the scattering atom and thus leads to unexplored aspects of the local structure and bonding in these relevant systems. Using this energy loss spectroscopy technique, we find a large influence of TMAO on the shape of the oxygen K-edge, and only a slightly smaller influence of urea and mixtures of urea and TMAO on the shape of K-edge spectra. As all molecular species have one intramolecular oxygen and thus yield an oxygen K-edge signal, we address the problem of decomposing the according spectra by using two different approaches: first, multi-component fitting, and second non-negative matrix factorization (NNMF).55 From the observations and data analysis we conclude that the spectra are characterized by the solvation effects of the two molecules. In the binary solutions, both molecules influence the structure of water whereas in the ternary solution, the solvation of the two solute molecules compensates their distinct effects on the XRS spectra. This compensation ceases at the highest concentration investigated.
2 Methods
Non-resonant inelastic X-ray scattering from core-level electrons, also known as X-ray Raman scattering (XRS), allows for the investigation of shallow absorption edges using hard X-rays as a probe. The use of hard X-rays makes XRS a highly bulk sensitive method without constraints on the sample environment. Thus, XRS allows for the study of liquids under ambient conditions, which, using soft X-rays or electrons, is almost prohibitively complicated by the need for a vacuum.A typical XRS experiment measures the double differential scattering cross section
 | (1) |
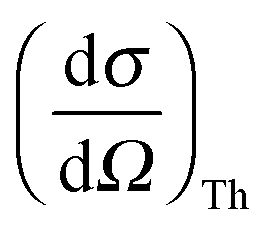 is Thomson's scattering cross section and S(q,ω) is the dynamic structure factor that holds all information about the sample obtainable by XRS. In the inelastic scattering process, the energy ω = ω1 − ω2 and the momentum q = k1 − k2 are transferred to the sample.
is Thomson's scattering cross section and S(q,ω) is the dynamic structure factor that holds all information about the sample obtainable by XRS. In the inelastic scattering process, the energy ω = ω1 − ω2 and the momentum q = k1 − k2 are transferred to the sample.
Even though individual XRS scattering events occur locally, the experiment probes the ensemble average of the dynamic structure factor in eqn (1). In the current case, the obtained constants N, P and T yield S(q,ω) = 〈S(q,ω,R)〉NPT, where
 | (2) |
Here, |i〉| and f〉 are the initial and final N-electron states, which depend on the set of nuclear coordinates R, over which the ensemble average is taken. In other words, XRS is sensitive to the ensemble average of instantaneous local structures around the scattering sites.
We studied the aqueous solutions of TMAO and urea as a function of the osmolyte concentration, namely 1 M, 2 M, 4 M, 6 M aqueous solutions of TMAO, 2 M, 4 M, 6 M, and 8 M aqueous solutions of urea, and 1 M:1 M, 2 M:1 M, 2 M:2 M, 4 M:2 M, and 4 M:4 M solutions of urea:TMAO in water. In addition, we measured the oxygen K-edge XRS from pure water as well as TMAO and urea powder samples as references. The powder samples were purchased from Sigma Aldrich (TMAO: C3H9NO·2H2O, >99.0% purity and urea: CH4N2O anion traces: chloride (Cl−): ≤5 ppm) and were used without further processing. For the preparation of the aqueous liquids, we used milli-q water (resistance > 18 MΩ). A stick-and-ball representation of an urea molecule (part (a)) and a TMAO molecule (part (b)) and a respective typical local environment of the molecules in aqueous solution (part (c) and (d)) are shown in Fig. 1.
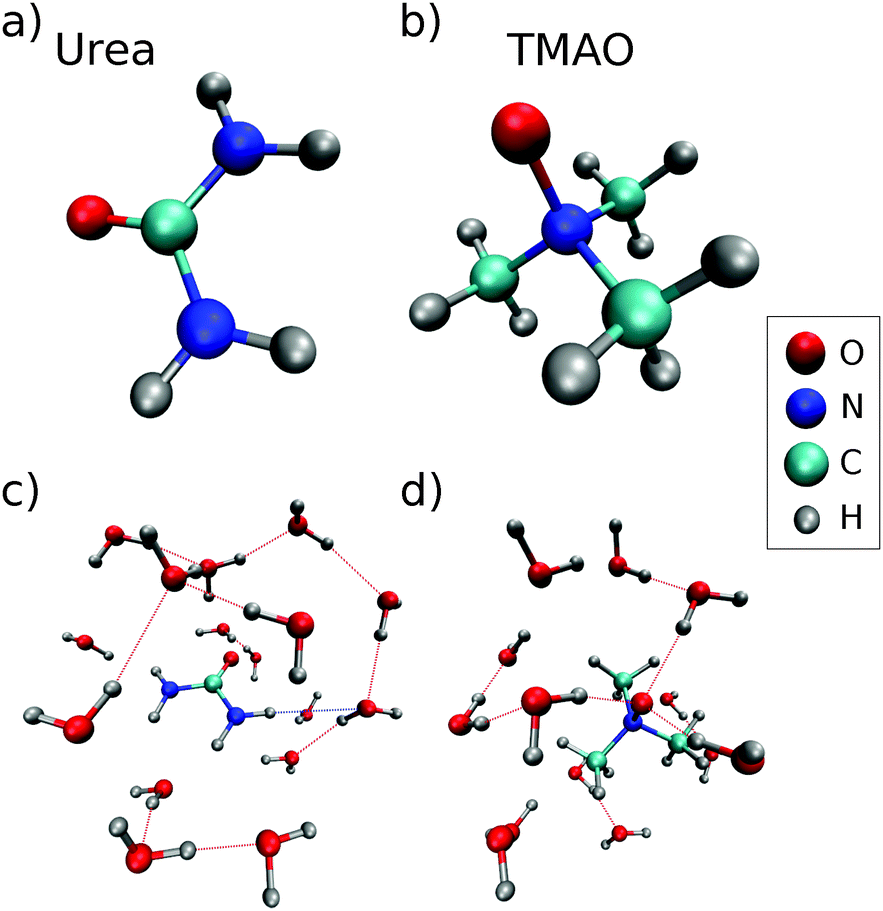 | ||
| Fig. 1 Representations of (a) urea, (b) TMAO, (c) nearest neighbors of urea from a random MD snapshot, and (d) a typical solvation shell of TMAO from a random MD snapshot. | ||
All measurements were performed at the inelastic scattering beamline ID20 of the ESRF, Grenoble, France. This beamline is equipped with four U26 undulators to generate a pink beam. Using a Si(111) high-heat-load monochromator and a consecutive Si(311) channel cut monochromator the incident X-ray beam bandwidth was cut down to 0.3 eV. We used the multi-analyzer-crystal spectrometer employing 12 spherically bent Si(660) analyzer crystals at a mean scattering angle of 85°. This way, we collected data of q = 6.8 ± 0.2 Å−1 momentum transfer at an overall energy resolution of 0.6 eV. The liquid samples were filled into a prototype version of a miniature liquid flow cell (described elsewhere56) sealed with Kapton. The liquids were pumped at approx. 100 ml min−1 through the closed miniature pump system to prevent radiation damage. Typical measuring times were 2–3 h per sample/concentration. For the measurement of the powder reference samples, we prepared disc shaped powder pellets of 13 mm diameter and ca. 2 mm thickness. We measured these powder samples in reflection geometry employing the same momentum transfer as for the liquid samples. In all cases, we measured several spectra, checked them for consistency and averaged them consecutively. The data are normalized to the integrated intensity between 530.0 and 550.0 eV energy loss. A detailed account of the data analysis is presented in ref. 57.
We study the changes of the O K-edge spectra as a function of concentration, and the deviation from a linear combination of individual molecular spectra to pinpoint the effects of the interaction. In our analysis, we use two approaches: (i) fingerprint fitting using the powder spectra and the spectrum of pure water as references and (ii) NNMF,55–59 in which all spectra in the raw data series are assumed to be linear combinations of fewer spectra, only the weights of which vary along the raw data set. In the procedure, the data of different concentrations are presented as column vectors of a non-negative matrix Dn×m. Each of the recorded spectra is described as a linear combination of k < m component spectra presented by the column vectors of a non-negative matrix Fn×k. The coefficients are then presented by a non-negative matrix Ck×m, one row vector for each component spectrum along the concentration series. Optimization of the elements in F and C is then performed so that D ≈ FC is best fulfilled. For meaningful results in terms of C, normalization of the spectra in D and F is required, and when fulfilled, the NNMF coefficients transfer directly to molecular fractions in the system, because there is one O atom in each of the studied molecules.
In the binary solutions, we used three components in the NNMF procedure. One for the signal of the solute (constrained to the spectrum of the urea and TMAO powder, respectively), one for the signal of the water solvent (constrained to the spectrum of the pure water reference) and one free component spectrum (FCS). This free spectrum is allowed to take any functional form for these spectra and their weights to best fulfill the matrix factorization. It can be seen as a representative of the normalized average K-edge signal changed due to the molecular interaction, which cannot be described by the pure (constrained) component spectra. We assign these changes to the solvation effects, i.e. the interaction between the solute and the solvent and the concentration dependent interaction between the solvent and the solvent.
In the ternary urea:TMAO:water solutions, we allowed for four components in the NNMF procedure. Three of them were constrained to the urea powder, TMAO powder, and pure water spectrum, respectively, and one FCS was again varied freely to best fulfill the matrix factorization.
We performed the constrained optimization described above by minimizing the quadratic cost function
 | (3) |
3 Results
Fig. 2(a and b) show the oxygen K-edge spectra of the binary solutions of TMAO–water and urea–water, respectively. The spectra of the ternary TMAO–urea–water solutions are shown in Fig. 2(c). XRS at the oxygen K-edge probes all oxygen atoms, in the solvent and in the solute alike. Therefore, we also measured the O K-edge spectra from polycrystalline powder samples of TMAO and urea. The resulting spectra are shown in Fig. 2(d) together with the spectrum of pure water. The differences in the spectra of solutions from that of pure water are shown directly below the respective figures of the original spectra. | ||
| Fig. 2 Oxygen K-edge spectra and spectral differences with respect to pure water. (a) TMAO–water, (b) urea–water, (c) urea–TMAO–water, and (d) the powder spectra and the spectrum of pure water. The statistical error bars for these spectra are of the order of the line width. The spectral differences between the aqueous solutions and pure water are shown directly below the respective figures. | ||
Although the spectra of the powder references largely overlap with the oxygen K-edge of pure water (Fig. 2d), each of the three spectra has distinct shapes and exhibits distinct features at various energy positions. Most prominently, the spectra from the TMAO and urea powder samples show extensive spectral weight below the onset of the K-edge in water, where the urea powder spectrum also shows a pronounced peak at 533.5 eV energy loss.
In all series of spectra, the additional feature around 533.5 eV energy loss evolves with increasing concentration. Clearly, this feature originates from contributions of the oxygen atom of the TMAO and urea molecules in the solution. This contribution is relatively sharp in the urea–water solution as the urea powder shows a sharp peak at this energy loss position. In the TMAO–water solutions this low energy loss feature is smaller.
In the pre-edge region, which is around 535.0 eV energy loss in pure water, we observe subtle changes as a function of osmolyte concentration in all three solutions. The main-edge region, from approximately 536.6 to 539.5 eV energy loss displays the biggest spectral changes in comparison with the pure water spectrum for all osmolyte solutions. In the TMAO–water spectra (Fig. 2a) we find a stark loss of spectral weight in this region as a function of the TMAO concentration, whereas the post-edge region remains unchanged with respect to the pure water spectrum. The spectra of the urea–water concentration series (Fig. 2b) show a smaller intensity drop in both the main- and post-edge region as a function of concentration and a non-linear increase of spectral weight around 545 eV energy loss between 4 M and 6 M. In the ternary solutions (Fig. 2c), we again observe a loss of intensity in the main-edge region as a function of concentration but smaller changes in the post edge region when compared to the changes observed in this energy loss region in the binary urea–water solutions.
For a more detailed view of the osmolytes' influence on the water structure, we used linear combination fitting of the spectra using the powder spectra and the spectrum of pure water as references. The results of these fits are presented in Fig. 3. Deviations from a linear combination of the pure water spectrum and the powder spectra indicate solvent–solute interactions in the solution. In the case of the TMAO–water solution, shown in Fig. 3(a), this binary fit grossly overestimates the main-edge (537.0–539.0 eV) and underestimates the post-edge feature (ca. 540.0–542.0 eV). We find a better agreement for the binary urea–water solutions in Fig. 3(b). For the spectra of ternary urea–TMAO–water solutions in Fig. 3(c), also the main-edge is overestimated and the post-edge underestimated like in the binary TMAO–water solution, but to a lesser degree than in the binary TMAO–water cases. For the binary systems the fit-coefficients of the reference spectra show an almost linear trend, whereas for the ternary system the behavior is more complex, as seen in Fig. 3(d and e). We assign the deviation from the fits and the nonlinear behavior of the coefficients of the ternary fit to molecular interactions in the solutions. Deviations from the fits are biggest for the pure TMAO solution as a sign for the strong interaction of TMAO with water and smallest for the urea solutions.
 | ||
| Fig. 3 The oxygen K-edge spectra fitted by a linear combination of the spectrum of pure water and the respective powder samples. (a) TMAO–water, (b) urea–water and (c) urea–TMAO–water. Below the edges, we plot the residuals between the spectra and their corresponding best fits. (d) The relative weight of TMAO and urea spectra in the binary fits, (e) the relative weights of the TMAO spectrum and the urea spectrum in the ternary solution. | ||
The component spectra and their weights from the NNMF procedure are presented in Fig. 4. In the binary systems, three components were used: the powder (constrained), pure water (constrained), and a free component spectrum (FCS). For the ternary system, both powder spectra and the water spectrum were fixed, again, with a FCS. The NNMF procedure therefore finds the single FCS and all coefficients so that the component spectra and their coefficients best represent the variation in the data set along the series of concentrations. With integral-normalized spectra, and since all molecules contain one oxygen atom, the weights directly relate to the molecular fractions.
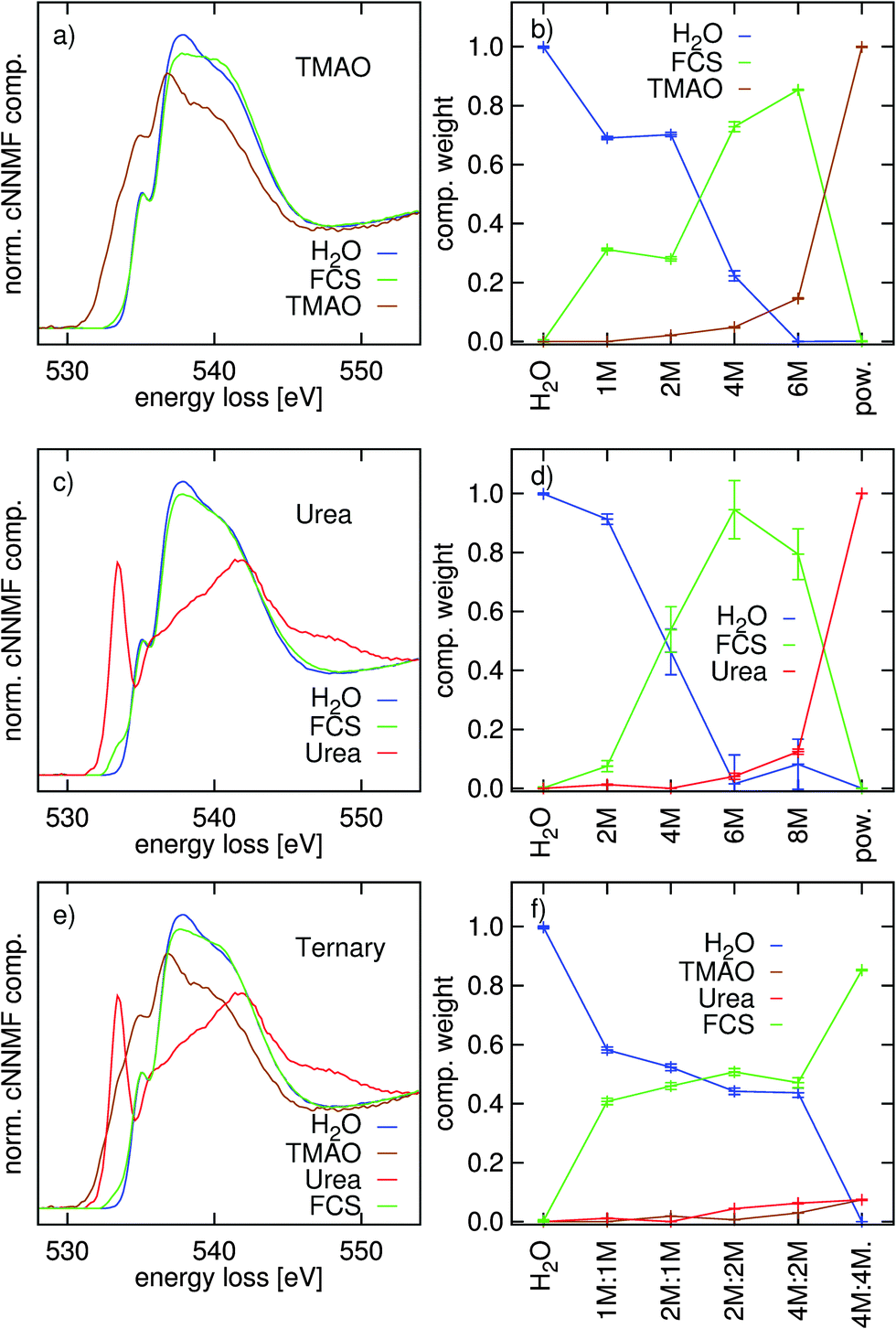 | ||
| Fig. 4 Constrained non-negative matrix factorization of (a and b): TMAO–water data, (c and d): urea–water data, and (e and f): TMAO–urea–water data. | ||
From Fig. 4 it is apparent that the independent component always looks similar to the O K-edge spectrum of water (with slight modifications). As the NNMF scheme finds the spectrum that best describes the variation along the series of differently concentrated spectra, the fact that the FCS resembles that of neat water much more than either of those of the osmolytes infers that changes in the oxygen K-edge of water dominate those of the solute oxygen K-edge in the data set as the solute concentration is increased. Thus, the change in the FCS reflects the effects of solvation on the spectra and we can observe a diminished main- and prominent post-edge in the FCS of the TMAO solution and only a slightly decreased main-edge in the FCS of urea with respect to the component representing neat or intact water. The CNNMF weights (Fig. 4(b and d)) are in agreement with this interpretation: for both binary systems, the fraction of “intact” water decreases rather monotonously and the FCS gains weight as long as free/un-associated water molecules that can hydrate the solute molecules remain. In contrast, the coefficients of the ternary system (Fig. 4(f)) show a plateau in the FCS and “intact” water spectra along the whole range of concentrations, except the highest 4 M:4 M concentration. Such a plateau is not observed in the spectra of the binary systems.
4 Discussion
The qualitative assessment of the oxygen K-edge spectra and the results of the linear combination fitting suggest that TMAO interacts strongest with the water molecules, whereas the interaction between urea and water is weaker. The behavior of the CNNMF coefficients and the shape of the FCS reveal that in 1:1 and 2:1 combinations the two osmolytes compensate their mutual impact on the XRS spectra, and therefore on the structure of the solution. Unless very high concentrations are used, the exact ratio of the osmolyte concentration has no apparent effect. The changes observed in the experimental XRS data are certainly related to changes in the structure and bonding in the respective solutions. However, this relation between the structure and spectra may not be one-to-one and needs careful interpretation on which we will elaborate in the following.
Over the past decade a common consensus about the interpretation of the oxygen K-edge in water and aqueous systems has been reached and the sensitivity of this edge to the details of the atomic structure and bonding topology of water is well established.47,48 Usually, the oxygen K-edge is divided into three regions, the pre-edge, the main-edge, and the post-edge region. Spectral intensity in the pre-edge region is commonly connected to weakening or breaking of hydrogen-bonds and a prominent pre-edge peak can be found e.g. in supercritical water.49 Similarly, researchers commonly connect a prominent main-edge to a heavily distorted and weakly hydrogen bonded network and e.g. Tse et al. report intensity shifts from the main- to the post-edge region when the hydrogen bond network becomes more ordered.45 Changes in the main-edge have also been attributed to a density increase in which the hydrogen bonding does not necessarily change, for example, in high density ices.46 The post-edge, in contrast, is prominent in the presence of tetrahedral order and a strong hydrogen-bond network. Thus, the oxygen K-edges of several ice phases exhibit increased spectral weight in the post-edge region.45,46
In the present systems of aqueous solutions, the interpretation of the oxygen K-edge data is more challenging since both urea and TMAO molecules have one intramolecular oxygen atom that contributes to the overall signal. Following the interpretation scheme usually applied to the oxygen K-edge of data from pure water, both in the TMAO–water solution and the ternary TMAO–urea–water solution, the hydrogen bond network is strengthened, evidenced from the decreased spectral weight in the main-edge region with respect to the pure water spectrum. Such a strengthening of the water structure and the water–water hydrogen bonds has been reported earlier31 and several studies, experimental and theoretical, found strong hydrogen bonding between the hydrophilic N–O group of TMAO and water.61 Dielectric and vibrational spectroscopy found a 2–3 times longer lifetimes and slower rotational dynamics for these stable TMAO–water complexes as compared to the bulk hydrogen bond lifetimes.32 Using femtosecond mid-infrared pump–probe spectroscopy Rezus et al.35 found a fraction of relatively immobile water hydroxyl groups that they associate to be involved in the solvation of TMAO and a relatively mobile group associated with the bulk water. However, in contrast to other amphiphilic solutes, this mobile bulk fraction in aqueous TMAO solutions is more mobile, which they explain with an increased number of hydrogen-bond defects with increasing TMAO concentration. The trend in the oxygen K-edge is quite clear showing a systematic decrease in main-edge intensity (Fig. 2a) and our two-component fit of the TMAO powder spectrum and bulk water spectrum systematically overestimates the main-edge and underestimates the post-edge (Fig. 3(a)). These strong deviations from a simple linear superposition of the reference spectra point to a contribution from water molecules that form strong hydrogen bonds in a stable network in line with the most recent experimental reports on long lived strongly H-bonded TMAO–water complexes and MD simulations.62,63 The results of the CNNMF procedure support this interpretation: the FCS found in the concentration series of aqueous TMAO solutions has a decreased main- and increased post-edge compared to neat water (Fig. 4(a)), despite the fact that the TMAO powder sample has a maximum in the vicinity of the main-edge and decreased intensity in the post-edge region.
The urea–water solutions, in contrast, show much smaller deviations from a simple powder-water-superposition fit than TMAO–water (Fig. 3(b)). This implies that there is not as strong an interaction between the urea molecules and water and a significant alteration of the water structure by urea is unlikely. This is in line with recent experimental21,22,26 and theoretical25,26 findings. The dynamics of the urea water system was probed by mid-infrared pump–probe spectroscopy and Rezus et al.22 found the relaxation times, even at high urea concentrations, to be largely unchanged as compared to bulk water values. However, they do find a small fraction of strongly immobilized water molecules and relate this finding to the formation of long lived urea–water complexes, where one water molecule forms two hydrogen bonds with a urea molecule, in accord with the results of THz absorption spectroscopy.23 Such doubly H-bonded water molecules were not found in a recent combined vibrational spectroscopy and MD simulation study.26 From our data and the performed two-component fit, this fraction of stable water molecules must be very small at most, which is in line with the finding of Rezus et al.22 that even high concentrations of urea do not alter the reorientation time of the majority of the water molecules. In accordance with this view, the FCS found in the CNNMF analysis of the concentration series of the urea solutions shows relatively small deviations from the pure water spectrum (Fig. 4(b)). The FCS is a representative of the changes in the measured K-edge with respect to the pure (constrained) component spectra, which implies relatively little influence of urea on the structure of water.
Naturally, the question arises how these two observations in the binary solutions add up when forming ternary TMAO–urea–water solutions. To answer this question we repeated the linear combination fitting of the ternary solutions using the corresponding spectra of the binary solutions as references (i.e. for the 1 M:1 M urea:TMAO mixture we used the spectrum of the 2 M urea- and the 2 M TMAO-solution as references, etc., except for the highest concentrated ternary solution, for which we used a superposition of the spectrum of the 8 M urea solution and that of the 6 M TMAO-solution). This direct comparison between the two sorts of fits is shown in Fig. 5, where we plot the series of fits using the powder reference samples in part (a) (the same as in Fig. 3) and the series of fits using the binary solutions as references in part (b). Whereas the ternary powder–water fits will again give an estimate of the influence of the solutes on the structure of water, the fits using the spectra of the binary solutions as references will readily provide an estimate of the additivity of the effects of the two solutes. The overall agreement of these fits is higher for the series using the binary solutions (part (b) in Fig. 3). As the hydration of the individual species is already fully accounted for in the binary-solution spectra, the better agreement of the fits indicates that the two solutes maintain their hydration structure and that the effects of the direct interaction are smaller in magnitude.
 | ||
| Fig. 5 (a) Ternary powder–water fits to the TMAO–urea–water solutions (the same as in Fig. 3), (b) two-component fit using spectra binary TMAO–water and urea–water systems that produce the molarity of the corresponding ternary solution. The residual is shown below the spectra. | ||
Both, binary and ternary TMAO/urea systems have been studied extensively using classical10,25,42,62 and first principles MD.26,63 The observed additive behavior has also been found in other experimental43 and computer simulation based work: Canchi et al. show that calculated osmotic pressures are additive when comparing those of binary and ternary mixtures, suggesting no significant direct interaction between TMAO and urea.30 Likewise, in their MD study, Kokubo et al. report roughly cumulative total van der Waals contributions to the free energy of a decaalanine peptide when comparing binary vs. ternary solutions.42
On the one hand, we find simple additive behavior, i.e. weak or no direct interaction between TMAO and urea, evidenced by our fingerprinting analysis. On the other hand we observe a plateau in CNNMF coefficients, i.e. a clear mutual influence of the two solutes in the ternary solution. Thus, we observe a strong TMAO–water interaction together with a clear indirect interaction between TMAO and urea. This is in line with aforementioned MD studies and in contrast to the suggestion of a strong direct interaction40,41 between urea and TMAO, which should lead to a visible deviation in our fit using the two binary solutions as reference.
5 Conclusions
We presented an experimental X-ray Raman scattering spectroscopy study of the micro-solvation structure and bonding topology of water upon solvation of the naturally occurring osmolytes trimethylamine N-oxide (TMAO) and urea. Oxygen K-edge data from concentration series of the binary urea–water and TMAO–water mixtures suggest a strong influence of TMAO on the water network and a weaker interaction between urea and water. The strong interaction between TMAO molecules and the water solute leads to a more structured hydrogen-bond network in the context of the conventional qualitative interpretation of the oxygen K-edge shape. Two different approaches to disentangle the oxygen K-edge response from both, the solvent and solute, namely via linear superposition fitting and by the use of non-negative matrix factorization, support this interpretation.In addition to the binary systems, we studied ternary urea–TMAO–water mixtures for different equimolar solutions and 2:1 (urea:TMAO) molar concentration ratios. Both, our multi-component fits using powder samples as well as the binary solutions as references, and an analysis based on constrained non-negative matrix factorization show that the two solutes largely maintain their respective hydration structure and the direct interaction between the two molecules is small in magnitude. Mutual solvation of both osmolytes cancels the influence of each osmolyte on the shape of the oxygen K-edge over a wide range of concentrations and for both studied mixing ratios (1:1 and 2:1 urea:TMAO). Only at the highest (4 M:4 M) concentration we find the influence of TMAO to dominate that of urea, which again demonstrates the stronger TMAO–water interaction compared to the interaction of urea and water. The interpretation of these experimental X-ray Raman scattering data points to an indirect interaction mechanism in line with most recent MD simulation studies.
Acknowledgements
We thank the European Synchrotron Radiation Facility for providing synchrotron radiation and the technical and scientific staff of ESRF beamline ID20 for support and valuable discussions. MAS thanks the excellence cluster ‘The Hamburg Centre for Ultrafast Imaging – Structure, Dynamics and Control of Matter at the Atomic Scale’ of the DFG. We acknowledge F. Lehmkühler for fruitful discussions.References
- P. H. Yancey, Water stress, osmolytes and proteins, Am. Zool., 2001, 41(4), 699–709 CAS.
- P. H. Yancey, Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses, J. Exp. Biol., 2005, 208(15), 2819–2830 CrossRef CAS PubMed.
- P. H. Yancey, M. E. Clark, S. C. Hand, R. D. Bowlus and G. N. Somero, Living with water stress: evolution of osmolyte systems, Science, 1982, 217(4566), 1214–1222 CAS.
- T. Arakawa and S. N. Timasheff, The stabilization of proteins by osmolytes, Biophys. J., 1985, 47(3), 411 CrossRef CAS PubMed.
- B. J. Bennion and V. Daggett, Counteraction of urea-induced protein denaturation by trimethylamine N-oxide: a chemical chaperone at atomic resolution, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(17), 6433–6438 CrossRef CAS PubMed.
- A. Wang and D. W. Bolen, A naturally occurring protective system in urea-rich cells: mechanism of osmolyte protection of proteins against urea denaturation, Biochemistry, 1997, 36(30), 9101–9108 CrossRef CAS PubMed.
- I. Baskakov, A. Wang and D. W. Bolen, Trimethylamine-N-oxide counteracts urea effects on rabbit muscle lactate dehydrogenase function: a test of the counteraction hypothesis, Biophys. J., 1998, 74(5), 2666–2673 CrossRef CAS PubMed.
- C. Krywka, C. Sternemann, M. Paulus, M. Tolan, C. Royer and R. Winter, Effect of Osmolytes on Pressure Induced Unfolding of Proteins: A High Pressure SAXS Study, ChemPhysChem, 2008, 9(18), 2809–2815 CrossRef CAS PubMed.
- M. A. Schroer, Y. Zhai, D. C. F. Wieland, C. J. Sahle, J. Nase, M. Paulus, M. Tolan and R. Winter, Exploring the Piezophilic Behavior of Natural Cosolvent Mixtures, Angew. Chem., Int. Ed., 2011, 50(48), 11413–11416 CrossRef CAS PubMed.
- D. R. Canchi and A. E. Garcia, Cosolvent effects on protein stability, Annu. Rev. Phys. Chem., 2013, 64, 273–293 CrossRef CAS PubMed.
- M. Auton and D. W. Bolen, Predicting the energetics of osmolyte-induced protein folding/unfolding, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(42), 15065–15068 CrossRef CAS PubMed.
- M. Auton, L. Marcelo F. Holthauzen and D. W. Bolen, Anatomy of energetic changes accompanying urea-induced protein denaturation, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15317–15322 CrossRef CAS PubMed.
- D. W. Bolen and G. D. Rose, Structure and energetics of the hydrogen-bonded backbone in protein folding, Annu. Rev. Biochem., 2008, 77, 339–362 CrossRef CAS PubMed.
- M. C. Stumpe and H. Grubmüller, Interaction of urea with amino acids: implications for urea-induced protein denaturation, J. Am. Chem. Soc., 2007, 129(51), 16126–16131 CrossRef CAS PubMed.
- S. Lee, Y. L. Shek and T. V. Chalikian, Urea interactions with protein groups: a volumetric study, Biopolymers, 2010, 93(10), 866–879 CrossRef CAS PubMed.
- E. J. Guinn, L. M. Pegram, M. W. Capp, M. N. Pollock and M. T. Record, Quantifying why urea is a protein denaturant, whereas glycine betaine is a protein stabilizer, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(41), 16932–16937 CrossRef CAS PubMed.
- S. Micciulla, J. Michalowsky, M. A. Schroer, C. Holm, R. von Klitzing and J. Smiatek, Concentration dependent effects of urea binding to poly(N-isopropylacrylamide) brushes: a combined experimental and numerical study, Phys. Chem. Chem. Phys., 2016, 18(7), 5324–5335 RSC.
- H. S. Frank and F. Franks, Structural approach to the solvent power of water for hydrocarbons; urea as a structure breaker, J. Chem. Phys., 1968, 48(10), 4746–4757 CrossRef CAS.
- K. A. Dill, Dominant forces in protein folding, Biochemistry, 1990, 29(31), 7133–7155 CrossRef CAS PubMed.
- A. Shimizu, K. Fumino, K. Yukiyasu and Y. Taniguchi, NMR studies on dynamic behavior of water molecule in aqueous denaturant solutions at 25 C: effects of guanidine hydrochloride, urea and alkylated ureas, J. Mol. Liq., 2000, 85(3), 269–278 CrossRef CAS.
- A. K. Soper, E. W. Castner and A. Luzar, Impact of urea on water structure: a clue to its properties as a denaturant?, Biophys. Chem., 2003, 105(2), 649–666 CrossRef CAS PubMed.
- Y. L. A. Rezus and H. J. Bakker, Effect of urea on the structural dynamics of water, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(49), 18417–18420 CrossRef CAS PubMed.
- S. Funkner, M. Havenith and G. Schwaab, Urea, a Structure Breaker? Answers from THz Absorption Spectroscopy, J. Phys. Chem. B, 2012, 116(45), 13374–13380 CrossRef CAS PubMed.
- E. P. O'Brien, R. I. Dima, B. Brooks and D. Thirumalai, Interactions between hydrophobic and ionic solutes in aqueous guanidinium chloride and urea solutions: lessons for protein denaturation mechanism, J. Am. Chem. Soc., 2007, 129(23), 7346–7353 CrossRef PubMed.
- D. Bandyopadhyay, S. Mohan, S. K. Ghosh and N. Choudhury, Molecular dynamics simulation of aqueous urea solution: is urea a structure breaker?, J. Phys. Chem. B, 2014, 118(40), 11757–11768 CrossRef CAS PubMed.
- J. K. Carr, L. E. Buchanan, J. R. Schmidt, M. T. Zanni and J. L. Skinner, Structure and Dynamics of Urea/Water Mixtures Investigated by Vibrational Spectroscopy and Molecular Dynamics Simulation, J. Phys. Chem. B, 2013, 117(42), 13291–13300 CrossRef CAS PubMed.
- K. Yoshida, K. Ibuki and M. Ueno, Pressure and temperature effects on 2H spin-lattice relaxation times and 1H chemical shifts in tert-butyl alcohol-and urea-D2O solutions, J. Chem. Phys., 1998, 108, 1360–1367 CrossRef CAS.
- T.-Y. Lin and S. N. Timasheff, Why do some organisms use a urea-methylamine mixture as osmolyte? Thermodynamic compensation of urea and trimethylamine N-oxide interactions with protein, Biochemistry, 1994, 33(42), 12695–12701 CrossRef CAS PubMed.
- E. S. Courtenay, M. W. Capp, C. F. Anderson and M. T. Record, Vapor pressure osmometry studies of osmolyte–protein interactions: implications for the action of osmoprotectants in vivo and for the interpretation of osmotic stress experiments in vitro, Biochemistry, 2000, 39(15), 4455–4471 CrossRef CAS PubMed.
- D. R. Canchi, P. Jayasimha, D. C. Rau, G. I. Makhatadze and A. E. Garcia, Molecular mechanism for the preferential exclusion of TMAO from protein surfaces, J. Phys. Chem. B, 2012, 116(40), 12095–12104 CrossRef CAS PubMed.
- Q. Zou, B. J. Bennion, V. Daggett and K. P. Murphy, The molecular mechanism of stabilization of proteins by TMAO and its ability to counteract the effects of urea, J. Am. Chem. Soc., 2002, 124(7), 1192–1202 CrossRef CAS PubMed.
- J. Hunger, K.-J. Tielrooij, R. Buchner, M. Bonn and H. J. Bakker, Complex formation in aqueous trimethylamine-N-oxide (TMAO) solutions, J. Phys. Chem. B, 2012, 116(16), 4783–4795 CrossRef CAS PubMed.
- Y. L. A. Rezus and H. J. Bakker, Observation of immobilized water molecules around hydrophobic groups, Phys. Rev. Lett., 2007, 99(14), 148301 CrossRef CAS PubMed.
- A. Panuszko, P. Bruzdziak, J. Zielkiewicz, D. Wyrzykowski and J. Stangret, Effects of urea and trimethylamine-N-oxide on the properties of water and the secondary structure of hen egg white lysozyme, J. Phys. Chem. B, 2009, 113(44), 14797–14809 CrossRef CAS PubMed.
- Y. L. A. Rezus and H. J. Bakker, Destabilization of the Hydrogen-Bond Structure of Water by the Osmolyte Trimethylamine N-oxide, J. Phys. Chem. B, 2009, 113(13), 4038–4044 CrossRef CAS PubMed.
- L. Larini and J.-E. Shea, Double resolution model for studying TMAO/water effective interactions, J. Phys. Chem. B, 2013, 117(42), 13268–13277 CrossRef CAS PubMed.
- D. Laage, G. Stirnemann and J. T. Hynes, Why water reorientation slows without iceberg formation around hydrophobic solutes, J. Phys. Chem. B, 2009, 113(8), 2428–2435 CrossRef CAS PubMed.
- S. Paul and G. N. Patey, Structure and interaction in aqueous urea–trimethylamine-N-oxide solutions, J. Am. Chem. Soc., 2007, 129(14), 4476–4482 CrossRef CAS PubMed.
- J. Rösgen and R. Jackson-Atogi, Volume exclusion and H-bonding dominate the thermodynamics and solvation of trimethylamine-N-oxide in aqueous urea, J. Am. Chem. Soc., 2012, 134(7), 3590–3597 CrossRef PubMed.
- F. Meersman, D. Bowron, A. K. Soper and M. H. J. Koch, Counteraction of urea by trimethylamine N-oxide is due to direct interaction, Biophys. J., 2009, 97(9), 2559–2566 CrossRef CAS PubMed.
- F. Meersman, D. Bowron, A. K. Soper and M. H. J. Koch, An X-ray and neutron scattering study of the equilibrium between trimethylamine N-oxide and urea in aqueous solution, Phys. Chem. Chem. Phys., 2011, 13(30), 13765–13771 RSC.
- H. Kokubo, C. Y. Hu and B. M. Pettitt, Peptide conformational preferences in osmolyte solutions: transfer free energies of decaalanine, J. Am. Chem. Soc., 2011, 133(6), 1849–1858 CrossRef CAS PubMed.
- J. Hunger, N. Ottosson, K. Mazur, M. Bonn and H. J. Bakker, Water-mediated interactions between trimethylamine-N-oxide and urea, Phys. Chem. Chem. Phys., 2015, 17(1), 298–306 RSC.
- Ph. Wernet, D. Nordlund, U. Bergmann, M. Cavalleri, M. Odelius, H. Ogasawara and L. Å. Näslund, et al., The structure of the first coordination shell in liquid water, Science, 2004, 304(5673), 995–999 CrossRef CAS PubMed.
- J. S. Tse, D. M. Shaw, D. D. Klug, S. Patchkovskii, G. Vanko, G. Monaco and M. Krisch, X-ray Raman spectroscopic study of water in the condensed phases, Phys. Rev. Lett., 2008, 100(9), 095502 CrossRef PubMed.
- T. Pylkkänen, V. M. Giordano, J.-C. Chervin, A. Sakko, M. Hakala, J. A. Soininen, K. Hämäläinen, G. Monaco and S. Huotari, Role of non-hydrogen-bonded molecules in the oxygen K-edge spectrum of ice, J. Phys. Chem. B, 2010, 114(11), 3804–3808 CrossRef PubMed.
- T. Pylkkänen, A. Sakko, M. Hakala, K. Hämäläinen, G. Monaco and S. Huotari, Temperature dependence of the near-edge spectrum of water, J. Phys. Chem. B, 2011, 115(49), 14544–14550 CrossRef PubMed.
- A. Nilsson and L. G. M. Pettersson, Perspective on the structure of liquid water, Chem. Phys., 2011, 389(1), 1–34 CrossRef CAS.
- Ch. J. Sahle, C. Sternemann, C. Schmidt, S. Lehtola, S. Jahn, L. Simonelli and S. Huotari, et al., Microscopic structure of water at elevated pressures and temperatures, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(16), 6301–6306 CrossRef CAS PubMed.
- F. Lehmkühler, Y. Forov, T. Büning, Ch. J. Sahle, I. Steinke, K. Julius, T. Buslaps, M. Tolan, M. Hakala and C. Sternemann, Intramolecular structure and energetics in supercooled water down to 255 K, Phys. Chem. Chem. Phys., 2016, 18(9), 6925–6930 RSC.
- H. Conrad, F. Lehmkühler, C. Sternemann, A. Sakko, D. Paschek, L. Simonelli, S. Huotari, O. Feroughi, M. Tolan and K. Hämäläinen, Tetrahydrofuran clathrate hydrate formation, Phys. Rev. Lett., 2009, 103(21), 218301 CrossRef PubMed.
- I. Juurinen, T. Pylkkänen, K. O. Ruotsalainen, C. J. Sahle, G. Monaco, K. Hämäläinen, S. Huotari and M. Hakala, Saturation Behavior in X-ray Raman Scattering Spectra of Aqueous LiCl, J. Phys. Chem. B, 2013, 117(51), 16506–16511 CrossRef CAS PubMed.
- I. Juurinen, T. Pylkkänen, Ch. J. Sahle, L. Simonelli, K. Hämäläinen, S. Huotari and M. Hakala, Effect of the hydrophobic alcohol chain length on the hydrogen-bond network of water, J. Phys. Chem. B, 2014, 118(29), 8750–8755 CrossRef CAS PubMed.
- J. Niskanen, Ch. J. Sahle, I. Juurinen, J. Koskelo, S. Lehtola, R. Verbeni, H. Müller, M. Hakala and S. Huotari, Protonation Dynamics and Hydrogen Bonding in Aqueous Sulfuric Acid, J. Phys. Chem. B, 2015, 119(35), 11732–11739 CrossRef CAS PubMed.
- M. W. Berry, M. Browne, A. N. Langville, V. P. Pauca and R. J. Plemmons, Algorithms and applications for approximate nonnegative matrix factorization, Comput. Stat. Data Anal., 2007, 52(1), 155–173 CrossRef.
- Ch. J. Sahle, C. Henriquet, M. A. Schroer, I. Juurinen, J. Niskanen and M. Krisch, A miniature closed-circle flow cell for high photon flux X-ray scattering experiments, J. Synchrotron Radiat., 2015, 22, 1555–1558 CrossRef PubMed.
- Ch. J. Sahle, A. Mirone, J. Niskanen, J. Inkinen, M. Krisch and S. Huotari, Planning, performing and analyzing X-ray Raman scattering experiments, J. Synchrotron Radiat., 2015, 22(2), 400–409 CrossRef CAS PubMed.
- J. Inkinen, J. Niskanen, T. Talka, Ch. J. Sahle, H. Müller, L. Khriachtchev, J. Hashemi, A. Akbari, M. Hakala and S. Huotari, X-ray induced dimerization of cinnamic acid: time-resolved inelastic X-ray scattering study, Sci. Rep., 2015, 5, 15851 CrossRef CAS PubMed.
- J. Niskanen, Ch. J. Sahle, K. O. Ruotsalainen, H. Müller, M. Kavčič, M. Žitnik, K. Bučar, M. Petric, M. Hakala and S. Huotari, Sulphur Kβ emission spectra reveal protonation states of aqueous sulfuric acid, Sci. Rep., 2016, 6 DOI:10.1038/srep21012.
- MATLAB, The MathWorks Inc. 3 Apple Hill Drive Natick, Massachusetts 01760 USA Search PubMed.
- L. Knake, G. Schwaab, K. Kartaschew and M. Havenith, Solvation Dynamics of Trimethylamine N-oxide in Aqueous Solution Probed by Terahertz Spectroscopy, J. Phys. Chem. B, 2015, 119(43), 13842–13851 CrossRef CAS PubMed.
- P. Ganguly, T. Hajari, J.-E. Shea and N. F. A. van der Vegt, Mutual Exclusion of Urea and Trimethylamine N-oxide from Amino Acids in Mixed Solvent Environment, J. Phys. Chem. Lett., 2015, 6(4), 581–585 CrossRef CAS PubMed.
- S. Imoto, H. Forbert and D. Marx, Water structure and solvation of osmolytes at high hydrostatic pressure: pure water and TMAO solutions at 10 kbar versus 1 bar, Phys. Chem. Chem. Phys., 2015, 17(37), 24224–24237 RSC.
| This journal is © the Owner Societies 2016 |