
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO adsorption on the GaPd(![[1 with combining macron]](https://www.rsc.org/images/entities/h2_char_0031_0304.gif) ) surface: a comparative DFT study using different functionals
) surface: a comparative DFT study using different functionals
S.
Alarcón Villaseca
ab,
S. V.
Levchenko
b and
M.
Armbrüster
*ac
aMax-Planck-Institut für Chemische Physik fester Stoffe, Nöthnitzer Str. 40, 01187 Dresden, Germany. E-mail: marc.armbruester@chemie.tu-chemnitz.de
bFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4–6, D-14195 Berlin, Germany
cFaculty of Natural Sciences, Institute of Chemistry, Materials for Innovative Energy Concepts, Technische Universität Chemnitz, Straße der Nationen 62, D-09111 Chemnitz, Germany
First published on 5th May 2016
Abstract
CO adsorption on the polar (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) surface of the intermetallic compound GaPd is examined within ab initio methods using an all-electron full-potential electronic structure approach. Comparison between the PW-LDA, GGA-PBE, GGA-RPBE, GGA-revPBE, and hybrid HSE06 functionals is considered through bulk, clean surface and CO adsorption calculations. The choice of the functional is found to have a strong influence in the description of single CO adsorption on the surface model proposed in literature. As expected from the so called “CO adsorption puzzle”, differences in the obtained results demonstrate that classic LDA and PBE functionals can only partially describe the complex CO adsorption bonding scenario on a surface containing transition metal elements (in this case Pd atoms), where the energies of the substrate–adsorbate electronic states are shifted, yielding important differences in the absolute values of the adsorption energies, vibrational frequencies and surface–adsorbate interaction. So far the hybrid functional HSE06 correctly retrieves all the tendencies observed experimentally as confirmed by comparing our first-principles results to experimental findings.
) surface of the intermetallic compound GaPd is examined within ab initio methods using an all-electron full-potential electronic structure approach. Comparison between the PW-LDA, GGA-PBE, GGA-RPBE, GGA-revPBE, and hybrid HSE06 functionals is considered through bulk, clean surface and CO adsorption calculations. The choice of the functional is found to have a strong influence in the description of single CO adsorption on the surface model proposed in literature. As expected from the so called “CO adsorption puzzle”, differences in the obtained results demonstrate that classic LDA and PBE functionals can only partially describe the complex CO adsorption bonding scenario on a surface containing transition metal elements (in this case Pd atoms), where the energies of the substrate–adsorbate electronic states are shifted, yielding important differences in the absolute values of the adsorption energies, vibrational frequencies and surface–adsorbate interaction. So far the hybrid functional HSE06 correctly retrieves all the tendencies observed experimentally as confirmed by comparing our first-principles results to experimental findings.
I. Introduction
The intermetallic compound GaPd is experiencing great interest due to its catalytic properties in the semi-hydrogenation of acetylene, an essential cleaning step for the polyethylene production.1–3 Recently, different studies about the structure determination and chemical reactivity of the polar surface GaPd() have been published.4–9 The work of Rosenthal et al. is an experimental study that joins a variety of surface science techniques.4 In particular, results of thermal desorption spectroscopy (TDS) of adsorbed CO molecules indicated that different surface terminations are obtained depending on the preparation temperature (670 K or 870 K). Scanning tunneling microscopy (STM) images revealed flat terraces separated by a unique step height. Low-energy electron diffraction (LEED) together with X-ray photoelectron diffraction (XPD) showed no signs of surface reconstruction or segregation. It was proposed that the surface prepared at 870 K can be explained with an atomistic model derived from a bulk truncation containing in the topmost atomic layer a single protruding Pd atom per surface unit cell. For the surface prepared at 670 K, a denser surface model that could contain either Pd-rich or Ga-rich terminations was proposed. Another study, a joint application of experimental LEED, STM, and plane-wave quantum chemical calculations using the PBE-GGA functional,10 was performed by Prinz et al.5 Calculated relative surface energies and qualitative comparison between experimental and simulated STM images in addition to LEED results, lead the authors to the conclusion that the GaPd() surface can be explained with a model containing only one protruding Pd atom per surface unit cell, located in the center of 3 Ga atoms, in agreement with the previous findings.4 However, a different surface model was proposed in the work of Krajčí et al.6 In this study, the determination of the structure of the GaPd() surface was performed exclusively by plane-wave quantum chemical calculations using the PW91-GGA functional. As the previous publication by Prinz et al. it includes relative surface energy calculations and qualitative comparison of the electronic structure with STM images and photoemission spectroscopy. However, their conclusion points towards a Ga-terminated surface model and not to a Pd-terminated model as in the previous works. The publication of Krajčí et al. includes a study of the CO adsorption in different surface models,6 but as in previous ref. 7–9 calculations were performed at the GGA level of theory, thus not addressing the “CO adsorption puzzle”.11–15
For understanding processes occurring at solid surfaces, the atomic structure determination must be complemented by the analysis of the electronic structure, with both depending on the interplay between the chemical species present at the surface at their corresponding coverages. Thus, to comprehend and predict the behavior of solid surfaces, an atomistic approach is vital. The density functional theory (DFT), offering good accuracy at low computational cost, has become the main tool in the last decades for understanding properties of molecules, aggregates, and materials on the atomic scale.16,17 DFT is in principle exact.18 However, since the exact form of the exchange–correlation density functional is not known, approximations must be introduced.19 The most widely used approximations are the local density approximation (LDA) and the generalized gradient approximation (GGA). In complex and more realistic systems, the choice of the right functional describing the exchange and correlation interactions has a strong impact on the accuracy of the obtained results, sometimes leading to considerable errors in the predicted energies, misleading final interpretations.11–15,20–26 Particularly, DFT within the Perdew–Wang parametrization for the LDA (PW-LDA),27 the Perdew–Burke–Ernzerhof (PBE) functional for the GGA,10 and the modified PBE functional, namely the RPBE,28 fail to predict the correct adsorption site for the CO molecule on most of the surfaces of transition- and noble-metal elements.11–15 The underlying problem, known as the “CO-adsorption puzzle”, is due to the inherent lack of accuracy of both LDA and GGA to describe at the same time the electronic structure of small molecules, such as CO, the electronic surface structure of metals containing d (and/or f) states, as well as the interactions of the surface and the molecule. This leads to a wrong positioning of the orbitals of the CO molecule upon surface adsorption, causing an overestimation of the adsorption energies.11,14,15 Additionally, the error also depends on the atomic structure of the adsorption site, which leads to wrong site-preference, i.e. wrong energetic ordering of the different adsorption sites.12,14,15,29,30 Thus, the quest for an appropriate DFT method that includes a treatable exchange–correlation term to solve the problems linked to the “CO adsorption puzzle” for intermetallic compounds containing transition-metals at their surfaces must be faced before any comparison with available experimental data. In the present work, a comparative DFT study of the CO adsorption on the Pd1-terminated model4,5 of the () surface of the intermetallic compound GaPd is conducted using different exchange–correlation density-functional approximations. This case study constitutes an excellent opportunity to unveil the effects coming from both surface and sub-surface transition-metal elements on the adsorption of the small, yet very important CO molecule.
II. Computational details
Calculations are conducted using the all-electron full-potential code FHI-aims.31 Effects of different approximations in exchange–correlation functional are explored by comparing results obtained with (i) the local density approximation (LDA), (ii) the generalized gradient approximation (GGA) and (iii) the hybrid Hartree–Fock/GGA approximation. Atomic relaxations, electronic structure, and energetics are calculated using the Perdew–Wang parametrization for the LDA (PW-LDA),27 the GGA Perdew–Burke–Ernzerhof (PBE) functional,10 the modified PBE functional RPBE,28 the revised PBE functional revPBE,32 and the hybrid Hartree–Fock/GGA-PBE Heyd–Scuseria–Ernzerhof functional (HSE06).33 In the latter, the fraction of the Hartree–Fock exchange α and the screening parameter ω are set to 0.25 and 0.11 bohr−1, respectively, as recommended.33 FHI-aims employs numeric atomic orbital (NAO) basis sets. Final results are obtained using “tight” numerical settings, basis set level up to tier 1 for Ga and Pd, and up to tier 2 for C and O species.31,34,35 Relativistic effects are considered for both core and valence electrons employing the scaled zero-order regular approximation (ZORA).31,36,37 Spin polarization is considered in both bulk and surface calculations, where a fixed quantization (collinear) axis is used to define the spin density. Atomic relaxations are conducted by calculating first energy-derivatives (forces) with respect to the nuclear coordinates, and their minimization using the trust radius method of the Broyden–Fletcher–Goldfarb–Shanno BFGS algorithm.38,39 For the bulk material, also unit cell size/shape relaxation is considered. In all our calculations, forces are converged to values better than 10−4 eV Å−1, and total energies and charge densities are converged to values better than 10−6 eV and 10−5 e Å−3, respectively. A well-converged Γ-centred mesh of 20 × 20 × 20 k-points is used to sample the Brillouin zone in bulk calculations. For the slab calculations an 8 × 8 × 1 k-point mesh was used. Tests in bulk calculations – for all here tested functionals – using an 8 × 8 × 8 mesh and a 20 × 20 × 20 mesh result in energy differences smaller than 10 meV, while no significant differences are found in the calculated bulk and surface atomic and electronic structures.PBE and HSE06 calculations including both the Tkatchenko–Scheffler (TS) correction40 for van der Waals interactions and the modified Tkatchenko–Scheffler (TSsurf) correction that includes short-range screening effects, particularly developed for the study of surface–adsorbate systems,41–43 have been considered. The results obtained with the HSE06 (PBE) functional show that the dispersion corrections of both TS and TSsurf contribute with ≈0.1 eV (≈0.2 eV) to the final adsorption energies of the studied adsorbate–substrate system. Furthermore, their contribution to the CO adsorption energy differences between the most favoured adsorption sites T1 and H1 is <10 meV (<50 meV), for the results obtained with the HSE06 (PBE) functional. Differences between TS- and TSsurf-corrected adsorption energies for the studied adsorbate–substrate system are <10 meV (<20 meV) for the results obtained with the HSE06 (PBE) functional. Since for all the here tested functionals, the CO adsorption energy difference between the sites T1 and H1 is ≈1 eV (see Table 4), van der Waals dispersion corrections are found to play only a minor role in the CO adsorption on the GaPd() surface and are not further considered in the present study. These results are in line with the fact that CO is a very small and stable closed-shell molecule and that Pd is the element that presents the smallest correction for the many-body collective response (screening) of the C6 coefficients of different metallic surfaces.41–43
The C–O stretching vibrational frequency of the free molecule (gas phase) together with the vibrational frequencies for CO adsorbed at different surface sites are calculated in the harmonic approximation by diagonalizing the Hessian matrix obtained by finite differences of analytic forces, using “tight” numerical settings and basis set level up to tier 1 for all atomic species. The motion of the C and O atoms, as well as the two topmost atomic layers Pd1 and Ga3 is considered, with displacement steps of 0.002 Å for each atom along each Cartesian coordinate. Lower-lying atomic layers of the slab are kept frozen at their corresponding relaxed positions.
Chemical bonding analysis in real space is carried out using the concept of electron localizability indicator (ELI).44 The ELI in the ELI-D representation45,46 is computed by the help of an interface to the FHI-aims code. For both the electron density (ED) and the ELI-D, the attractor locations, basins and the number of electrons contained inside the basins are determined by the DGrid package.47
III. GaPd crystal structure and bulk calculations
The intermetallic compound GaPd crystallizes in the cubic FeSi type of structure, space group P213 (No. 198, Pearson symbol cP8), containing 8 atoms per unit cell.48 At 295 K and atmospheric pressure, the lattice parameter of GaPd is a = 4.89695(6) Å, with a volume V = 117.4 Å3.48 The first coordination shell of the Pd (Ga) atoms consists entirely of Ga (Pd) atoms. The shortest inter-atomic distance corresponds to a Pd–Ga distance equal to 2.5399(2) Å, located along the threefold axis, and the closest Pd–Pd distance is 3.0084(1) Å.48 Because of the absence of inversion symmetry, two possible enantiomorphic forms of the GaPd compound exist, namely form A and form B.4,48 The calculations of the atomic and electronic structure of the GaPd compound are initially based on lattice parameters and atomic coordinates from its enantiomorphic form B, as described in ref. 4 and 48 (but accidentally labelled form A in ref. 48). Fig. 1 shows the structure model of form B. Each enantiomorph possesses a characteristic but inverse stacking sequence of four non-equivalent atomic layers along the polar [111] direction. Following the notation developed by Rosenthal et al.,4 in the case of form B, the sequence of atomic planes is Pd1/Ga3/Pd3/Ga1 as shown in Fig. 1. | ||
| Fig. 1 Projected crystal structure of the enantiomorph B of the intermetallic compound GaPd.48 The red box depicts the unit cell, gray (green) spheres represent Pd (Ga) atoms. | ||
The structural stability of crystals typically correlates with the lattice parameters.49 The structural stability of solids is characterized by the enthalpy of formation ΔHf, which is linked to the cohesive energy and the elasticity of a given compound.50 For the intermetallic compound GaPd, a rough approximation of ΔHf at 0 K, no pressure, and without considering vibrational contributions – here called “formation energy” ΔEf – can be obtained by calculating the difference between the total energy of bulk GaPd unit cell, and four times the respective bulk energies of elemental Ga and Pd. Table 1 presents the lattice parameter a and the formation energy ΔEf of GaPd, obtained with different functionals. The relative errors (in %) of the calculated values with respect to the experimentally obtained ones (at room temperature) are given in parenthesis, where a positive (negative) sign indicates an over(under)estimation. To compute ΔEf, the stable α-modification of Ga and the Cu type of structure of Pd, were considered.51–53 As expected,49 the LDA calculation underestimates the lattice parameter of GaPd with respect to the experimental value, while GGA calculations overestimate it. Bulk calculations using the hybrid functional HSE06 were also conducted. The corresponding optimized lattice parameter is the closest (+0.6%) to the experimental value. The choice of the functional has also an important effect on the obtained values of ΔEf. Although the latter is negative in all cases, which is in agreement with the high melting point of GaPd (1338 K),54 both LDA and GGA calculations overestimate ΔEf compared to the experimental value of ΔHf = −5.96 eV.55 The RPBE functional gives the highest deviation from the experimental value (+16.38%). In general, all GGA-based calculations give similar results, with the LDA value being this time closer to the experimental ΔHf (+5.89%) than the GGA values. Although the nearest value to the experimental ΔHf is the one obtained with the hybrid HSE06 functional (−5.87%), this is not much better than the LDA result.
Another important property that can be correlated to the structural stability of a solid is its electronic structure. Fig. 2 presents the bulk density of states (DOS) of GaPd calculated with different functionals, with the lattice parameter relaxed for each functional (see Table 1). Table 2 summarizes the main features of the obtained DOS (defined in Fig. 2), including the opening of the DOS between ≈−5 and ≈−7 eV (position of its center OC and length OL), the peak Pd of the Pd 4d band, the Pd 4d bandwidth Wd, and the DOS at the Fermi level n(EF). In line with previous results, in the valence region the most important partial contribution to the total DOS of GaPd is given by the Pd 4d states, that are shifted below the Fermi level in comparison to pure Pd.1,56–59 Although the GGA functionals perform similarly, e.g., |ΔPd| ≤ 0.11 eV, |ΔWd| ≤ 0.18 eV, the differences between the DOS calculated with LDA and GGA functionals are significant, e.g., ΔWLDA-RPBEd = 0.75 eV, ΔPLDA-RPBEd = −0.50 eV. However, a PBE calculation at the geometry optimized with the PW-LDA functional (denoted below PBE*) removes almost completely the observed differences, e.g.,  . Thus, the differences in the electronic structure obtained with LDA and GGA functionals can partly be tracked down to the differences in their respective optimized geometries. On the other hand, the DOS obtained with LDA and GGA functionals present important differences in comparison to the results obtained with the hybrid HSE06 functional. With respect to the latter, all GGA (LDA) calculations predict both a narrower opening and a smaller Pd 4d bandwidth, i.e., ΔOHSE06-GGAL = 0.18 eV (ΔOHSE06-LDAL = 0.23 eV) and ΔWHSE06-GGAd ≥ 1.03 eV (ΔWHSE06-LDAd = 0.46 eV). Furthermore, the DOS is shifted to higher energies in the GGA (LDA) calculations in comparison to the HSE06 results, e.g., |ΔPHSE06-GGAd| ≥ 0.95 eV (|ΔPHSE06-LDAd| = 0.56 eV). Here, the obtained differences cannot be explained as mainly a geometric effect. The observed differences are due to the self-interaction error present in the LDA and GGA functionals, which is partly removed in hybrid functionals including the HSE06.14,33,60–65 As a result, the latter provide a better description of the electronic structure of molecular systems, and have been demonstrated to yield better alignment of the metallic band in solids, but can overestimate its width.14,33,60–65 This can explain the difference between the HSE06 and LDA/GGA positions of the Pd 4d band in the bulk GaPd DOS. Since the chemical reactivity of a surface depends on the electronic structure of both the adsorbed molecular species and the surface, the obtained results suggest that the predicted chemical properties of the surfaces of GaPd will be sensitive to the approximations used in the employed exchange–correlation functional.
. Thus, the differences in the electronic structure obtained with LDA and GGA functionals can partly be tracked down to the differences in their respective optimized geometries. On the other hand, the DOS obtained with LDA and GGA functionals present important differences in comparison to the results obtained with the hybrid HSE06 functional. With respect to the latter, all GGA (LDA) calculations predict both a narrower opening and a smaller Pd 4d bandwidth, i.e., ΔOHSE06-GGAL = 0.18 eV (ΔOHSE06-LDAL = 0.23 eV) and ΔWHSE06-GGAd ≥ 1.03 eV (ΔWHSE06-LDAd = 0.46 eV). Furthermore, the DOS is shifted to higher energies in the GGA (LDA) calculations in comparison to the HSE06 results, e.g., |ΔPHSE06-GGAd| ≥ 0.95 eV (|ΔPHSE06-LDAd| = 0.56 eV). Here, the obtained differences cannot be explained as mainly a geometric effect. The observed differences are due to the self-interaction error present in the LDA and GGA functionals, which is partly removed in hybrid functionals including the HSE06.14,33,60–65 As a result, the latter provide a better description of the electronic structure of molecular systems, and have been demonstrated to yield better alignment of the metallic band in solids, but can overestimate its width.14,33,60–65 This can explain the difference between the HSE06 and LDA/GGA positions of the Pd 4d band in the bulk GaPd DOS. Since the chemical reactivity of a surface depends on the electronic structure of both the adsorbed molecular species and the surface, the obtained results suggest that the predicted chemical properties of the surfaces of GaPd will be sensitive to the approximations used in the employed exchange–correlation functional.
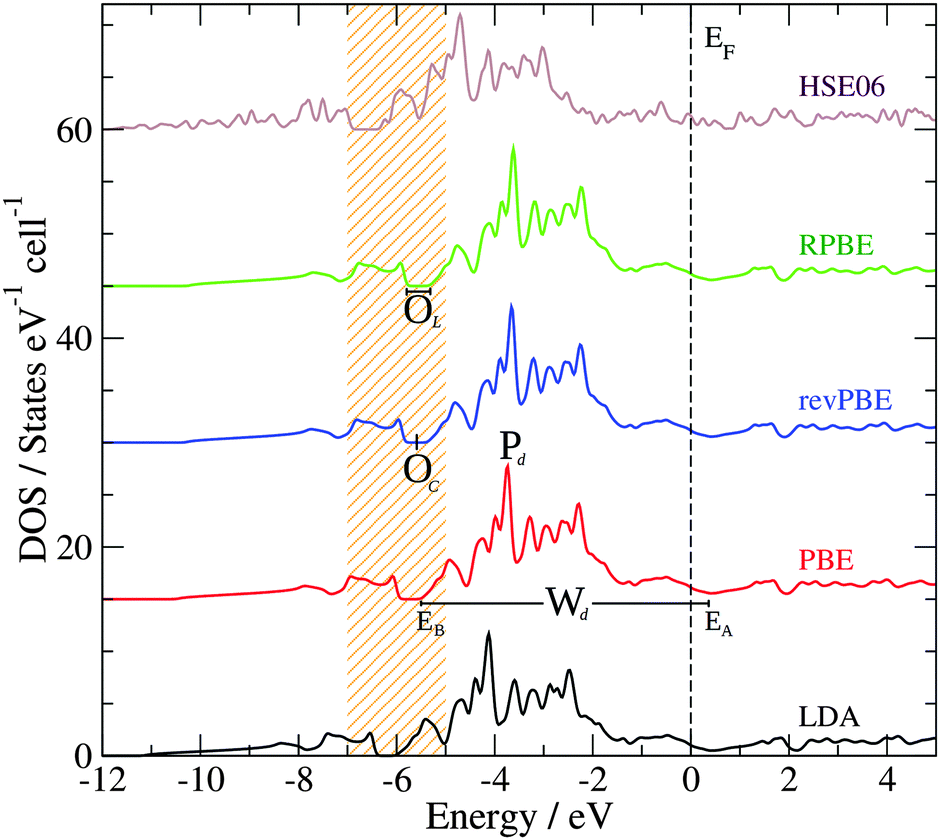 | ||
| Fig. 2 Bulk DOS of GaPd calculated with different functionals. The HSE06 DOS calculation was performed using an 8 × 8 × 8 k-point mesh. Wd = EA − EB corresponds to the Pd 4d bandwidth, while OL and OC depict the length and the center of the opening of the DOS around −6 eV. The zero of the energy scale is set to the Fermi level (EF, vertical dashed line). | ||
IV. Pd1-terminated GaPd(![[1 with combining macron]](https://www.rsc.org/images/entities/h3_char_0031_0304.gif) ) surface
) surface
Following the bulk stacking sequence of atomic planes Pd1/Ga3/Pd3/Ga1 along [], the construction of the Pd1-terminated model of the GaPd() surface is achieved by simple bulk truncation, resulting in a (√3 × √3)Rot30° surface unit cell.4,5Fig. 3 shows a top view of the Pd1-terminated model. The topmost atomic layer (Pd1) consists of only one Pd atom per surface unit cell. The second (third) layer Ga3 (Pd3) consists of three Ga (Pd) atoms per surface unit cell – connected with dotted lines in Fig. 3.
 | ||
| Fig. 3 Top view of the Pd1-terminated model of the GaPd() clean surface. The red box depicts the surface unit cell. The topmost atomic layers Pd1, Ga3 and Pd3 are highlighted (arrows, dotted lines), while deeper layers appear faded. Color code as in Fig. 1. | ||
The simulation of the Pd1-terminated model is achieved through asymmetric slabs. In this configuration, the slab is composed of a certain number NF of atomic layers that are kept fixed at the optimized bulk atomic positions (lying at the bottom of the slab), while another number NR of atomic layers on top of them (including the surface of interest) can fully relax. The thickness of the vacuum region separating two consecutive slabs is set to 20 Å. The area of the surface unit cell calculated with the LDA, PBE, and HSE06 functionals is 40.6 Å2, 42.6 Å2, and 42.3 Å2, respectively. Calculations conducted with the PBE functional and an 11-layer-thick slab consisting of different NR and NF layers – keeping the lattice parameters of the supercell fixed – show that the total energy of the slab changes with the ratio NF/NR, decreasing with NR. The most noticeable differences are between the here tested extreme values NF/NR = 1/10 and NF/NR = 8/3 (ΔEslab = 104.5 meV). To determine the thickness of the slab sufficient to simulate the Pd1-terminated model, surface energy differences and layer-resolved surface density of states (sDOS) calculations were compared for an 11-, a 20-, and a 29-layer-thick slab using the same conditions as described above, but (i) keeping constant the number of fixed layers lying at the bottom (NF = 4) or (ii) keeping constant the number of atomic layers that can fully relax (NR = 7), all slabs having identical bottom termination. Thus, the here calculated surface energy differences contain only contributions from the surface of interest (top), since the surface energy of the “frozen” bottom termination is the same for all slabs and cancels out when calculating surface energy differences. The calculations show differences smaller than 1 meV Å−2 (4 meV Å−2) for the surface energy difference between the 20-(11-) and 29-(20-)layer-thick slabs. Furthermore, only negligible changes of the calculated sDOS with the slab thickness were found, all slabs reaching bulk-like behavior in the central layers. Therefore, the 11-layer-thick slab with NF = 4 at the bottom and NR = 7 at the top (including the surface of interest) is used in all surface calculations.
Fig. 4 shows the sDOS of the Pd1 topmost layer of the clean surface model calculated with different functionals. As for the bulk calculations, the differences between the sDOS calculated with the PBE, revPBE and RPBE functionals, at the geometry optimized with the PBE functional, are negligible (smaller than 0.04 eV for OC, OL, Pd and Wd). However, the PW-LDA calculation at the geometry optimized with the PBE functional presents a small shift (0.3 eV) towards lower energies of the opening of the sDOS and the Pd 4d band, in comparison to the PBE results. More significantly, the PBE sDOS shows a shift to higher energies (|ΔOC| ≈ |ΔPd| = 0.9 eV) and a reduction of the Pd 4d bandwidth (|ΔWd| ≈ 1.0 eV), when compared to the HSE06 sDOS, in line with the bulk calculations. The bottom panel in Fig. 4 presents the partial contributions to the total HSE06 sDOS (valence region) of the Pd1 topmost layer. As expected from a Pd-terminated surface, the analysis shows that its main structure – between −6 and −1.7 eV – is primarily dominated by the Pd 4d states. The total area below the sDOS in this energy window is 4.46 states per cell, where 95% of it corresponds to Pd 4d states. The area under the sDOS between −7 and −6.8 eV is 0.12 states per cell and is also dominated by Pd 4d states, but to a lesser extent (69% Pd d states, 12.5% Pd p states and 18.5% Pd s states). Between −11.6 and −8.4 eV, the number of states in the sDOS is negligible.
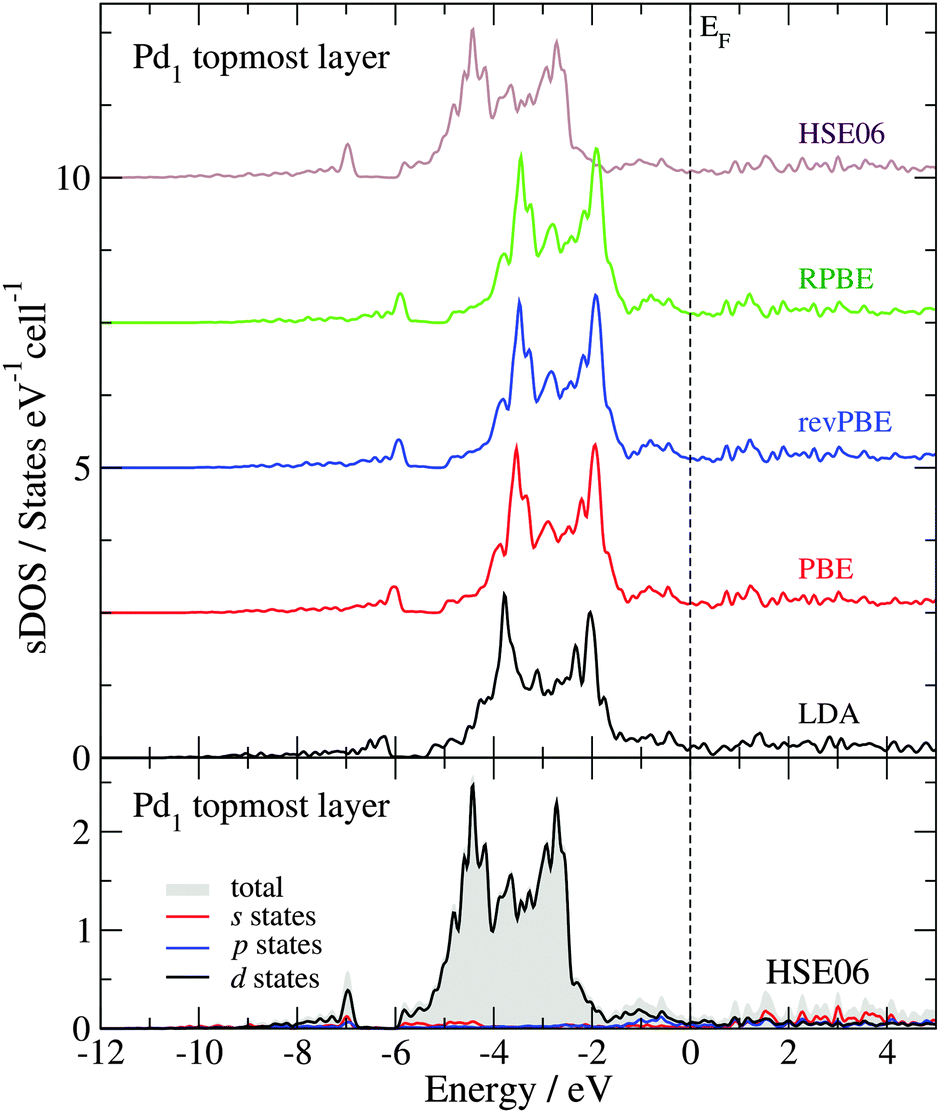 | ||
| Fig. 4 Surface density of states (sDOS) of the topmost atomic layer Pd1 of the Pd1-terminated surface model, obtained with different functionals (top panel). The partial contributions to the total sDOS calculated with the HSE06 functional are shown in the bottom panel. The zero of the energy scale is set to the Fermi level (EF). | ||
Fig. 5 shows the HSE06 sDOS convoluted with a Gaussian function66–68 (Δε = 0.25 eV) and compared to the He II ultraviolet photoelectron spectra (UPS) of the GaPd() clean surface recorded at room temperature of the sample prepared at 870 K.4 To estimate the contribution of the sub-surface atomic layers, the convoluted surface DOS is calculated considering (a) only the topmost atomic layer Pd1, (b) both surface and subsurface atomic layers Pd1 and Ga3, and (c) the three topmost atomic layers Pd1, Ga3 and Pd3 (emphasized by colored areas in Fig. 5). Moving towards lower energies, the structure of the convoluted sDOS presents two small peaks at 0.2 eV (P1) and −0.7 eV (P2), the two main peaks of the Pd 4d band at −3.0 eV (P3) and −4.3 eV (P4), and a shoulder-like peak at −5.6 eV (P5). After an opening at −6.3 eV, a peak at −7.0 eV (P6) appears together with two subsequent shoulder-like peaks. As can be seen in Fig. 5, there is an excellent agreement between the convoluted HSE06 sDOS and the experimental data.
 | ||
| Fig. 5 Convoluted sDOS obtained with the HSE06 functional (top panel) and experimental He II UPS spectra of the clean GaPd() surface (bottom panel).4 In the top panel, the blue curve (the envelope of the white area denoted “a”) is the convoluted DOS due to only the top-most layer of the slab. The envelope of the red patterned area (area b) is due to the top and the subsurface layers, while the envelope of the blue patterned area (area c) is due to the three top-most layers of the slab. The inset shows the same three sDOS in the region above the Fermi level (EF). | ||
V. CO adsorption on the Pd1-terminated GaPd() surface
Based on the results and discussion presented above, the CO adsorption on the Pd1-terminated model of the GaPd() surface was thoroughly studied applying different functionals. Several starting geometries of the molecule were considered taking into account the atomic structure of the three topmost layers of the Pd1-terminated surface model. The CO molecule was allowed to fully relax starting from different positions, 3 Å above the initial adsorption site. The initial C–O bond length was set to the calculated value for CO in gas phase (see Table 4). Fifteen crystallographically unique possible adsorption sites can be identified. Fig. 6 summarizes the initial adsorption sites considered in this study: top (T), bridge (B), and hollow (H) sites. In the Ti sites (i = 1 to 3), the CO molecule is initially adsorbed atop one Ga or one Pd atom of the substrate. In the case of Bj sites (j = 1 to 7), the CO molecule initially forms a bridge between two atoms of the substrate, while in the case of Hk sites (k = 1 to 5) the CO molecule is initially located between three substrate atoms.
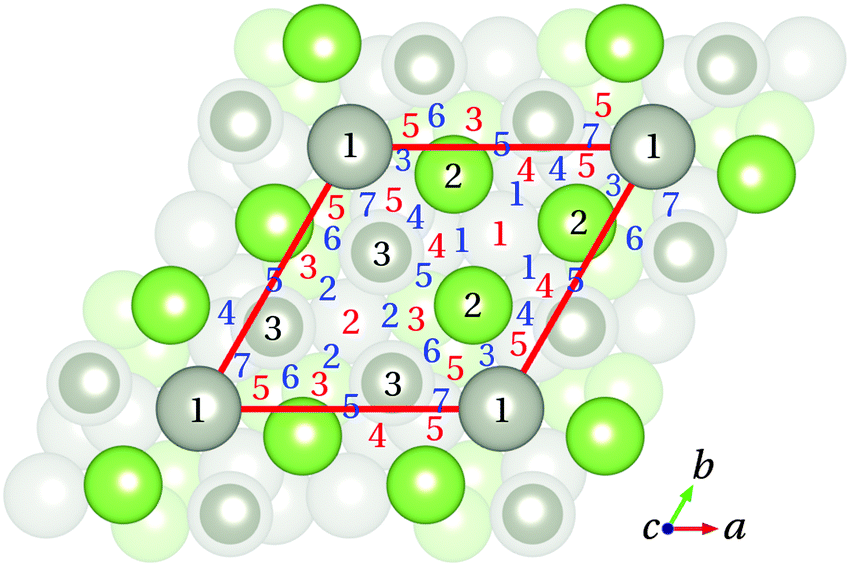 | ||
| Fig. 6 Top view of the Pd1-terminated model with the initial adsorption sites considered in this study. Top, bridge, and hollow sites are indicated by black (1 to 3), blue (1 to 7) and red (1 to 5) numbers, respectively. Color code is as in Fig. 1 and 3. | ||
Total adsorption energies Eads(αβ) for the different adsorption sites αβ were calculated using
| Eads(αβ) = Eslab + ECO − Eslab+CO(αβ), | (1) |
In calculations based on the HSE06 functional, the most favoured adsorption site is T1 with CO adsorption energy of 1.07 eV (103.24 kJ mol−1). In this site, the CO molecule relaxes perpendicular to the surface with the C atom pointing towards the topmost Pd atom of the substrate (d(C–Pd1) = 1.98 Å, see Table 4). When the CO molecule is initially above the sites B3, B6, B7, and H5 it also relaxes to a position above T1. In the case of the starting position above T2, the CO molecule desorbs upon relaxation. Similarly, the CO molecules initially above the hollow sites H2 and H3 desorb. On the other hand, the CO molecule initially above T3 moves towards the hollow site H1, relaxing perpendicular to the surface, with the C atom pointing towards the substrate (d(C–Ga3) = 2.88 Å, see Table 4). This is also the case for the CO molecule initially relaxed from above the bridge sites B1, B2, B4, B5, and hollow sites H1 and H4. The site H1 is composed of a triangle of Ga atoms with a central Pd atom at 2.04 Å below them. In spite of the presence of the accessible Pd atom, the surface Ga atoms make this site unfavourable (Eads = 0.05 eV = 4.82 kJ mol−1) for the adsorption of the CO molecule.
Summarizing, the HSE06 functional predicts that the CO molecule preferentially adsorbs only at the site T1, perpendicular to the substrate, with the C atom pointing towards the topmost Pd atom. Furthermore, no CO–Ga interaction is observed. These results are in perfect agreement with the experimental results obtained with the Fourier transform infrared spectroscopy (FTIRS) and the reflection–absorption infrared spectroscopy (RAIRS).1,4,7,56–59 The CO adsorption on the GaPd() surface at room temperature (50 mbar dosage) resulted in the appearance of only one band that was assigned to CO adsorbed on Pd in the on-top position.1,4,7,56–59 Moreover, the smaller adsorption energy for CO at the site T1 of the Pd1-terminated model of the GaPd() surface in comparison to CO adsorption on the Pd(111) surface (1.8 eV),69 is also in line with the thermal desorption spectroscopy (TDS) results, where the CO desorption from the Pd(111) surface needs a higher energy (desorption at 450 K) than from the GaPd() surface (desorption at 215 K).4 In order to correlate the experimentally obtained desorption temperatures4 with the computed adsorption energies, an estimation of the activation energies of desorption Ed is given in Table 3. Considering the results and conditions of the TDS experiments for CO desorption on the GaPd() and the Pd(111) surfaces given by Rosenthal et al.4 and the equation of Redhead for thermal desorption:70
| Ed ≈ R·TP [ln(ν1·TP/βH) − 3.64], | (2) |
) and on Pd(111) is 0.6 eV, signifying a stronger interaction between CO and the surface for Pd(111) than for GaPd(). The here obtained HSE06 CO adsorption energies for the sites T1 and H1 are somewhat smaller than previous values obtained with semi-local exchange–correlation functionals. Adsorption energies of 1.19 eV (PW91-GGA),6 1.37 eV (PBE-GGA),7 and 1.59 eV (RPBE-GGA)8 have been calculated for the T1 site, and of 0.69 eV (PW91-GGA),6 0.56 eV (PBE-GGA),7 and 0.49 eV (RPBE-GGA)8 for the H1 site. All the previous calculations were performed at the GGA level of theory using the pseudopotential plane-wave method,6,8 and a mixed Gaussian and plane-wave approach.7
| Surface | T P [K] | β H [K s−1] | ν 1 [s−1] | E d [eV] |
|---|---|---|---|---|
| Pd(111) | 450 | 1.4 | 1 × 1013 | 1.2 |
| GaPd() |
215 | 1.5 | 1 × 1013 | 0.6 |
(a) Influence of the exchange–correlation functional on adsorption energies and atomic relaxations
To gain further insight into the influence of the exchange–correlation functional on the description of CO adsorption on the GaPd() surface, LDA- and GGA-based adsorption energies were calculated for CO above the sites T1 and H1. In both cases, the CO molecule was allowed to fully relax starting from the final geometries obtained with the HSE06 functional. Table 4 presents the C–O bond length and the interlayer distances of the topmost layers of the Pd1 surface model before (clean surface, CO in gas phase) and upon CO adsorption on T1 and H1, as well as the respective adsorption energies, obtained with LDA, GGA, and hybrid functionals. The adsorption energies Eads* are calculated at the HSE06 slab geometry, while the adsorption energies Eads are calculated using slabs built with the lattice parameter optimized with each functional (see Table 1). For the here tested LDA and GGA functionals, the differences between Eads and Eads* are smaller than 0.02 eV for CO adsorption on T1, while they can account for differences up to 0.08 eV (LDA) for CO adsorption on H1. The results obtained with the HSE06 functional show that, the main effect of the atomic relaxation of the clean surface is the inward relaxation of the topmost Pd atom (0.18 Å), while lower lying atomic planes exhibit much smaller relaxations (<0.04 Å). Upon adsorption of CO on top of a Pd atom (site T1), the latter relaxes outward again, and the surface interlayer distances become almost the same as the ones from the initial bulk positions (the differences are smaller than 0.02 Å). This indicates a strong interaction between the CO molecule and the topmost Pd atom. On the other hand, when CO relaxes above the site H1, the interlayer distances relax back to the values calculated for the clean surface (before CO adsorption), as if the CO molecule and the surface acted as two independent systems, corroborating the findings discussed above. Similar to the HSE06 results, the LDA and GGA calculations show an inward relaxation of the topmost Pd atom as main effect of the atomic relaxations on the clean surface. However, deviations in the interlayer distances up to 0.08 Å for LDA and 0.04 Å for the GGA functionals are obtained, when compared to the HSE06 results. After CO adsorption above the site T1, all tested functionals predict that the CO molecule adsorbs on top of the Pd atom, with the molecule perpendicular to the surface and the C atom pointing towards the surface. The LDA functional predicts a much stronger CO–surface interaction than the GGA and hybrid functionals. Furthermore, while the C–Pd1 distance d(C–Pd1) calculated with the different GGA functionals is closer to the HSE06 value than the one obtained with the LDA functional, the latter predicts a C–O bond length dC–O in better agreement with the HSE06 result. The obtained differences are more pronounced in the case of CO adsorption above H1. Although for all tested functionals the CO molecule relaxed towards the center of the three Ga atoms of the Ga3 layer (CO perpendicular to the surface, with the C atom pointing towards the surface), the C–Ga3 distance changes significantly as function of the exchange–correlation functional. Here, the obtained LDA and PBE final geometries indicate that the CO molecule adsorbs on the site H1 (d(C–Ga3)LDA = 1.55 Å, d(C–Ga3)PBE = 1.68 Å), i.e. CO does interact with the surface at the site H1. However, the adsorption energies Eads calculated with these two functionals present a difference of 0.55 eV. On the other hand, both the final geometry and the adsorption energy obtained with the revPBE and RPBE functionals indicate that the CO molecule desorbs (in both cases Eads ≈ 0 eV and d(C–Ga3) = 3.49 Å), i.e. does not interact with the surface at the site H1. In line with the “CO-adsorption puzzle”,11,14,15 the obtained results show significant differences between the LDA, GGA and hybrid functionals. Even though the difference in the adsorption energies for the sites T1 and H1 is similar for all tested functionals (≈1 eV), total adsorption energies change significantly with the choice of the functional.
| Distances [Å] | Clean surface & CO in gas phase | CO above T1 | CO above H1 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LDA | PBE | revPBE | RPBE | HSE06 | LDA | PBE | revPBE | RPBE | HSE06 | LDA | PBE | revPBE | RPBE | HSE06 | |
| d C–O | 1.13 | 1.14 | 1.15 | 1.15 | 1.13 | 1.14 | 1.15 | 1.16 | 1.16 | 1.13 | 1.16 | 1.17 | 1.14 | 1.15 | 1.13 |
| d(C–Pd1) | — | — | — | — | — | 1.92 | 1.96 | 1.97 | 1.98 | 1.98 | 1.08 | 1.17 | 2.94 | 2.94 | 2.29 |
| d(Pd1–Ga3) | 0.57 | 0.56 | 0.56 | 0.56 | 0.59 | 0.75 | 0.75 | 0.76 | 0.76 | 0.76 | 0.47 | 0.50 | 0.55 | 0.55 | 0.59 |
| d(Ga3–Pd3) | 0.79 | 0.88 | 0.90 | 0.91 | 0.87 | 0.77 | 0.87 | 0.90 | 0.91 | 0.85 | 0.90 | 0.97 | 0.92 | 0.93 | 0.87 |
| d(Pd3–Ga1) | 1.00 | 1.00 | 1.00 | 1.01 | 0.98 | 0.93 | 0.93 | 0.93 | 0.94 | 0.92 | 0.98 | 0.98 | 1.00 | 1.01 | 0.98 |
| E ads [eV] | — | — | — | — | — | 1.74 | 1.17 | 0.96 | 0.93 | 1.07 | 0.79 | 0.24 | 0.01 | 0.02 | 0.05 |
| E ads* [eV] | — | — | — | — | — | 1.72 | 1.18 | 0.97 | 0.94 | 1.07 | 0.71 | 0.25 | −0.05 | 0.02 | 0.05 |
(b) Vibrational frequencies
The calculated C–O stretching frequency is 2247 cm−1 for the free molecule and 2180 cm−1 for the CO molecule adsorbed at the T1 site, applying the HSE06 functional. The corresponding experimental values are 2143 and 2078 cm−1, respectively.7,56,58 Thus, the experimentally obtained change in the vibrational frequency of CO upon adsorption (65 cm−1) is in an excellent agreement with the HSE06 calculations (67 cm−1). On the other hand, the calculated C–O stretching frequency is 2137 cm−1 for the free molecule and 1989 (2032) cm−1 for the CO molecule adsorbed at the T1 site, applying the RPBE (PBE) functional.7,8 The calculated red shift with the RPBE functional is 148 cm−1, much larger than the experimental value (65 cm−1). Furthermore, the calculated C–O stretching vibration frequency for CO above H1 is 2241 cm−1, applying the HSE06 functional. Thus, the vibrational frequency difference between the C–O stretching mode of the free molecule and the CO molecule above H1 is only 6 cm−1, consistent with the nearly-unbounded behavior of the CO molecule above the site H1 predicted by the HSE06 functional.(c) Electronic structure of the adsorbate–substrate system
The Blyholder model is generally applied to describe CO adsorption on surfaces containing transition metal (TM) elements.71–81 According to this model, there is (i) a CO-5σ interaction with the empty TM states of the surface (molecule-to-surface transfer of electrons), that is compensated by (ii) a π-bonding interaction of the filled TM states of the surface with the CO-2π* antibonding states (surface-to-molecule back-donation). The analysis and discussion of the electronic structure of the CO molecule adsorbed at the GaPd() surface calculated with the HSE06 functional, in the light of the Blyholder model, is given below.
 | ||
| Fig. 7 Layer-resolved sDOS of the Pd1 model before and after CO adsorption on T1. The zero of the energy scale is set to the Fermi level (EF). | ||
 | ||
| Fig. 8 Layer-resolved sDOS of the Pd1 model before and after CO adsorption on H1. The zero of the energy scale is set to the Fermi level (EF). | ||
(d) Chemical bonding analysis of the adsorbate–substrate system
Topological analysis of the electron density according to Bader's quantum theory of atoms in molecules (QTAIM)82 is used in combination with the electron localizability indicator (ELI-D)44–46 to characterize the CO–GaPd() system. The ELI-D is an efficient quantum chemical tool for the analysis of chemical bonding in real space, particularly, in the understanding of the properties of intermetallic compounds and the modeling of the adsorption of atoms and molecules where both covalent and ionic interactions may occur.83
The three known ELI-D basins for the free CO molecule in the valence region83 are retrieved in the surface–adsorbate system: two representing lone pairs – one located at the oxygen atom (4.0 electrons in the free molecule) and one located at the carbon atom (2.5 electrons) – and one basin accounting for the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O triple bond with an electron count of 3.3 electrons. Upon adsorption of CO on the site T1, 0.2 electrons are transferred from the CO bonding basin to the oxygen lone pair and the lone-pair at the carbon atom turns into a two-center C–Pd bond with an electron count of 2.8. Here, carbon contributes 2.4 electrons, while 0.4 electrons are coming from the palladium atom. In agreement with the analysis of the electronic structure given above, the formation of a C–Pd σ-bond is also confirmed by the ELI analysis, as proposed in the Blyholder model. In the same line, the back donation of electrons from the metal into the empty C–O antibonding 2π* states does not take place (see Fig. 7). Instead, the negatively charged palladium atom leads to a shift of the electron density to the oxygen lone pair, thus weakening the C–O bond. In this context, the clean GaPd() surface resembles a charged egg carton, where the “dimples” correspond to the unshielded negatively charged Pd centers, while the positively charged Ga species – shielded by partially dangling bonds – form the “spacers”. Due to the very stable, closed-shell nature of carbon monoxide, strong Pauli repulsion occurs when the CO molecule approaches a dangling bond. Since the dangling bonds shield most of the surface, only two potential adsorption sites out of 15 crystallographically possible (see Fig. 6) are left for the CO molecule: on-top of the Pd1 atom (T1 site) and the hollow site at the center of the triangle formed by the Ga3 atoms (H1 site). In addition, the orientation of the CO molecule (carbon oriented to palladium) fits the expectations from the charged egg carton model, resulting from the attractive Coulomb interactions of the positively charged C atom (+1.2) and the negative charge on the palladium (−0.4) according to the QTAIM analysis.
O triple bond with an electron count of 3.3 electrons. Upon adsorption of CO on the site T1, 0.2 electrons are transferred from the CO bonding basin to the oxygen lone pair and the lone-pair at the carbon atom turns into a two-center C–Pd bond with an electron count of 2.8. Here, carbon contributes 2.4 electrons, while 0.4 electrons are coming from the palladium atom. In agreement with the analysis of the electronic structure given above, the formation of a C–Pd σ-bond is also confirmed by the ELI analysis, as proposed in the Blyholder model. In the same line, the back donation of electrons from the metal into the empty C–O antibonding 2π* states does not take place (see Fig. 7). Instead, the negatively charged palladium atom leads to a shift of the electron density to the oxygen lone pair, thus weakening the C–O bond. In this context, the clean GaPd() surface resembles a charged egg carton, where the “dimples” correspond to the unshielded negatively charged Pd centers, while the positively charged Ga species – shielded by partially dangling bonds – form the “spacers”. Due to the very stable, closed-shell nature of carbon monoxide, strong Pauli repulsion occurs when the CO molecule approaches a dangling bond. Since the dangling bonds shield most of the surface, only two potential adsorption sites out of 15 crystallographically possible (see Fig. 6) are left for the CO molecule: on-top of the Pd1 atom (T1 site) and the hollow site at the center of the triangle formed by the Ga3 atoms (H1 site). In addition, the orientation of the CO molecule (carbon oriented to palladium) fits the expectations from the charged egg carton model, resulting from the attractive Coulomb interactions of the positively charged C atom (+1.2) and the negative charge on the palladium (−0.4) according to the QTAIM analysis.
In agreement with the much smaller calculated adsorption energy for the H1 site, no significant chemical interactions between the substrate and the CO molecule are found by ELI-D analysis upon CO adsorption.
VI. Conclusions
The CO adsorption on the () surface of the intermetallic compound GaPd has been examined using a quantum-chemical all-electron full-potential approach. Comparison of the GaPd bulk, clean surface, and CO adsorption obtained with LDA-, GGA-, and hybrid Hartree–Fock/GGA-based functionals was considered. The choice of the functional was found to have a decisive influence on the bulk and surface electronic structure of GaPd. In particular, bulk calculations using the different functionals lead to differences in the optimized lattice parameter resulting in different bulk electronic structure. Despite the fact that all the here tested methods find the Pd on-top site as the most preferred one for CO adsorption on the GaPd() surface when compared to the H1 site, the obtained results demonstrate that classic LDA and GGA functionals cannot retrieve a correct description of the energies of the substrate–adsorbate electronic states, leading to the wrong description of the CO adsorption on a surface containing Pd atoms, including important differences in adsorption energies and surface–adsorbate interaction as reflected in the calculated vibrational frequencies. On the other hand, experimental findings and the results obtained with the hybrid functional HSE06 are in excellent agreement. This holds for the predicted adsorption sites, adsorption energies, as well as vibrational frequencies. This study thus reveals the computational level necessary to derive reliable predictions from DFT calculations regarding the chemical properties of intermetallic surfaces.
Acknowledgements
We thank A. Ormeci for valuable discussions.References
- K. Kovnir, M. Armbrüster, D. Teschner, T. V. Venkov, F. C. Jentoft, A. Knop-Gericke, Yu. Grin and R. Schlögl, Sci. Technol. Adv. Mater., 2007, 8, 420–427 CrossRef CAS.
- J. Osswald, R. Giedigkeit, R. E. Jentoft, M. Armbrüster, F. Girgsdies, K. Kovnir, T. Ressler, Yu. Grin and R. Schlögl, J. Catal., 2008, 258, 210–218 CrossRef CAS.
- J. Osswald, K. Kovnir, M. Armbrüster, R. Giedigkeit, R. E. Jentoft, U. Wild, Yu. Grin and R. Schlögl, J. Catal., 2008, 258, 219–227 CrossRef CAS.
- D. Rosenthal, R. Widmer, R. Wagner, P. Gille, M. Armbrüster, Yu. Grin, R. Schlögl and O. Gröning, Langmuir, 2012, 28, 6848–6856 CrossRef CAS PubMed.
- J. Prinz, R. Gaspari, C. A. Pignedoli, J. Vogt, P. Gille, M. Armbrüster, H. Brune, O. Gröning, D. Passerone and R. Widmer, Angew. Chem., Int. Ed., 2012, 51, 9339–9343 CrossRef CAS PubMed.
- M. Krajčí and J. Hafner, J. Chem. Phys., 2013, 138, 124703 CrossRef PubMed.
- J. Prinz, R. Gaspari, Q. S. Stöck, P. Gille, M. Armbrüster, H. Brune, O. Gröning, C. A. Pignedoli, D. Passerone and R. Widmer, J. Phys. Chem. C, 2014, 118, 12260–12265 CAS.
- P. Bechthold, J. S. Ardhengi, A. Juan, E. A. González and P. V. Jasen, Appl. Surf. Sci., 2014, 315, 467–474 CrossRef CAS.
- J. Prinz, C. A. Pignedoli, Q. S. Stöckl, M. Armbrüster, H. Brune, O. Gröning, R. Widmer and D. Passerone, J. Am. Chem. Soc., 2014, 136, 11792–11798 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. J. Feibelman, B. Hammer, J. K. Nørskov, F. Wagner, M. Scheffler, R. Stumpf, R. Watwe and J. Dumesic, J. Phys. Chem. B, 2001, 105, 4018–4025 CrossRef CAS.
- A. Gil, A. Clotet, J. M. Ricart, G. Kresse, M. García-Hernández, N. Rösch and P. Sautet, Surf. Sci., 2003, 530, 71–87 CrossRef CAS.
- M. Gajdoš, A. Eichler and J. Hafner, J. Phys.: Condens. Matter, 2004, 16, 1141–1164 CrossRef.
- X. Ren, P. Rinke and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 045402 CrossRef.
- L. Schimka, J. Harl, A. Stroppa, A. Grüneis, M. Marsman, F. Mittendorfer and G. Kresse, Nat. Mater., 2010, 9, 741–744 CrossRef CAS PubMed.
- W. Kohn, Rev. Mod. Phys., 1999, 71, 1253–1266 CrossRef CAS.
- P. Atkins, Molecular Quantum Mechanics, Oxford University Press, Oxford, 1996 Search PubMed; R. G. Parr and Y. Weitao, Density-Functional Theory of Atoms and Molecules (International Series of Monographs on Chemistry), Oxford University Press, Oxford, 1989 Search PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev. B: Condens. Matter Mater. Phys., 1964, 136, 864–871 Search PubMed.
- W. Kohn and L. J. Sham, Phys. Rev. A: At., Mol., Opt. Phys., 1965, 137, 1697–1705 CAS.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Chem. Rev., 2012, 112, 289–320 CrossRef CAS PubMed.
- M. Grüning, O. V. Gritsenko, S. J. A. Van Gisbergen and E. J. Baerends, J. Phys. Chem. A, 2001, 105, 9211–9218 CrossRef.
- B. Santra, A. Michaelides, M. Fuchs, Al. Tkatchenko, C. Filippi and M. Scheffler, J. Chem. Phys., 2008, 129, 194111 CrossRef PubMed.
- C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009, 11, 10757–10816 RSC.
- K. Yang, J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2010, 132, 164117 CrossRef PubMed.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Science, 2008, 321, 792–794 CrossRef CAS PubMed.
- J. P. Perdew, MRS Bull., 2013, 38, 743–750 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- G. Kresse, A. Gil and P. Sautet, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 073401 CrossRef.
- S. E. Mason, I. Grinberg and A. M. Rappe, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 161401(R) CrossRef.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1998, 80, 890 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS; J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef; A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- T. Auckenthaler, V. Blum, H.-J. Bungartz, T. Huckle, R. Johanni, L. Krämer, B. Lang, H. Lederer and P. R. Willems, Parallel Comput., 2011, 37, 783–794 CrossRef.
- V. Havu, V. Blum, P. Havu and M. Scheffler, J. Comput. Phys., 2009, 228, 8367–8379 CrossRef CAS.
- E. Van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- R. Winkler, Spin-Orbit Coupling Effects in Two-Dimensional Electron and Hole Systems, Springer-Verlag, New York, Berlin, Heildelberg, 2003 Search PubMed.
- W. K. Press, S. A. Teukolsky, W. T. Vetterlin and B. T. Flannery, Numerical Recipes, Cambridge University Press, Cambridge, 2007 Search PubMed.
- J. Nocedal and S. J. Wright, Numerical optimization, Springer-Verlag, New York, Berlin, Heildelberg, 2006 Search PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef.
- V. G. Ruiz, W. Liu and A. Tkatchenko, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 035118 CrossRef.
- V. G. Ruiz, W. Liu, E. Zojer, M. Scheffler and A. Tkatchenko, Phys. Rev. Lett., 2012, 108, 146103 CrossRef PubMed.
- G.-X. Zhang, A. Tkatchenko, J. Paier, H. Appel and M. Scheffler, Phys. Rev. Lett., 2011, 107, 245501 CrossRef PubMed.
- M. Kohout, Int. J. Quantum Chem., 2004, 97, 651 CrossRef CAS.
- M. Kohout, Faraday Discuss., 2007, 135, 43 RSC.
- F. R. Wagner, V. Bezugly, M. Kohout and Yu. Grin, Chem. – Eur. J., 2007, 13, 5724 CrossRef CAS PubMed.
- M. Kohout, Program DGRID, version, 4.6, Radebeul, Germany, 2011. http://www.cpfs.mpg.de/˜kohout/dgrid.html.
- M. Armbrüster, H. Borrmann, M. Wedel, Y. Prots, R. Giedigkeit and P. Gille, Z. Kristallogr. - New Cryst. Struct., 2010, 225, 617–618 Search PubMed.
- J. L. F. Da Silva, C. Stampfl and M. Scheffler, Surf. Sci., 2006, 600, 703–715 CrossRef CAS.
- S. Müller, J. Phys.: Condens. Matter, 2003, 15, R1429 CrossRef.
- B. D. Sharma and J. A. Donohue, Z. Kristallogr., 1962, 117, 293–300 CrossRef CAS.
- X. F. Li, G. T. Fei, X. M. Chen, Y. Zhang, K. Zheng, X. L. Liu and L. D. Zhang, EPL, 2011, 94, 16001 CrossRef.
- C. N. Rao and K. K. Rao, Can. J. Phys., 1964, 42, 1336–1342 CrossRef CAS.
- P. Gille, T. Ziemer, M. Schmidt, K. Kovnir, U. Burkhardt and M. Armbrüster, Intermetallics, 2010, 18, 1663–1668 CrossRef CAS.
- S. V. Meschel and O. J. Kleppa, Thermochim. Acta, 1997, 292, 13–17 CrossRef CAS.
- M. Armbrüster, R. Schlögl and Yu. Grin, Sci. Technol. Adv. Mater., 2014, 15, 034803 CrossRef.
- Yu. Grin, M. Armbrüster, A. I. Baranov, K. Finzel, M. Kohout, A. Ormeci, H. Rosner and F. R. Wagner, Mol. Phys., 2016, 114, 1250–1259 CrossRef CAS.
- K. Kovnir, M. Armbrüster, D. Teschner, T. V. Venkov, L. Szentmiklósi, F. C. Jentoft, A. Knop-Gericke, Yu. Grin and R. Schlögl, Surf. Sci., 2009, 603, 1784–1792 CrossRef CAS.
- M. Armbrüster, K. Kovnir, M. Behrens, D. Teschner, Yu. Grin and R. Schlögl, J. Am. Chem. Soc., 2010, 132, 14745–14747 CrossRef PubMed.
- J. Jaramillo, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 1068–1073 CrossRef CAS.
- J. Paier, M. Marsman, K. Hummer, G. Kresse, I. C. Gerber and J. G. Ángyán, J. Chem. Phys., 2006, 124, 154709 CrossRef CAS PubMed.
- A. Stroppa, K. Termentzidis, J. Paier, G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 195440 CrossRef.
- A. Stroppa and G. Kresse, New J. Phys., 2008, 10, 063020 CrossRef.
- K. Doll, Surf. Sci., 2004, 573, 464–473 CrossRef CAS.
- M. Neef and K. Doll, Surf. Sci., 2006, 600, 1085–1092 CrossRef CAS.
- J. J. Markham, Rev. Mod. Phys., 1959, 31, 956–989 CrossRef CAS.
- M. Iwan and C. Kunz, Phys. Lett. A, 1977, 60, 345–347 CrossRef.
- D. Ochs, W. Maus-Friedrichs, M. Brause, J. Günster, V. Kempter, V. Puchin, A. Schluger and L. Kantorovich, Surf. Sci., 1996, 365, 557–571 CrossRef CAS.
- N. M. Martin, M. Van den Bossche, H. Grönbeck, C. Hakanoglu, F. Zhang, T. Li, J. Gustafson, J. F. Weaver and E. Lundgren, J. Phys. Chem. C, 2014, 118, 1118–1128 CAS.
- P. A. Redhead, Vacuum, 1962, 12, 203–211 CrossRef CAS.
- G. Blyholder, J. Phys. Chem., 1964, 68, 2772–2777 CrossRef CAS.
- H. Conrad, G. Ertl, J. Küppers and E. E. Latta, Faraday Discuss. Chem. Soc., 1974, 58, 116–124 RSC.
- R. J. Behm, K. Christmann, M. A. Van Hove and G. Ertl, J. Chem. Phys., 1980, 73, 2984–2995 CrossRef CAS.
- E. Bertel, Appl. Phys. A: Mater. Sci. Process., 1991, 53, 356–368 CrossRef.
- A. Sandell, O. Björneholm, J. N. Andersen, A. Nilsson, E. O. F. Zdansky, B. Hernnäs, U. O. Karlsson, R. Nyholm and N. Mårtensson, J. Phys.: Condens. Matter, 1994, 6, 10659 CrossRef CAS.
- T. Anazawa, I. Kinoshita and Y. Matsumoto, J. Electron. Spectrosc. Relat. Phenom., 1998, 88–91, 585–590 CrossRef CAS.
- G. Tsilimis, J. Kutzner and H. Zacharias, Appl. Phys. A: Mater. Sci. Process., 2003, 76, 743–749 CrossRef CAS.
- J. Rogozik, J. Küppers and V. Dose, Surf. Sci., 1985, 148, L653–L658 Search PubMed.
- B. Hammer, Y. Morikawa and J. K. Nørskov, Phys. Rev. Lett., 1996, 76, 2141–2144 CrossRef CAS.
- M. J. S. Dewar, Bull. Soc. Chim. Fr., 1951, 18, C79 Search PubMed.
- J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939–2947 RSC.
- R. F. W. Bader, Atoms in Molecules – A Quantum Theory, Clarendon Press and Oxford University Press, Oxford, New York, 1994 Search PubMed.
- Yu. Grin, A. Savin and B. Silvi, The ELF perspective of chemical bonding, The Chemical Bond: Chemical Bonding Across the Periodic Table, Wiley-VCH, 2014, pp. 1–53 Search PubMed.
| This journal is © the Owner Societies 2016 |