
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Micelle structure in a deep eutectic solvent: a small-angle scattering study†
A.
Sanchez-Fernandez
ab,
K. J.
Edler
*a,
T.
Arnold
c,
R. K.
Heenan
d,
L.
Porcar
e,
N. J.
Terrill
c,
A. E.
Terry
d and
A. J.
Jackson
bf
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: K.Edler@bath.ac.uk
bEuropean Spallation Source, Lund, Sweden
cDiamond Light Source, Harwell Campus, Didcot, OX11 0DE, UK
dISIS Spallation Neutron Source, Harwell Campus, Didcot OX11 ODE, UK
eInstitut Laue-Langevin, 71 avenue des Martyrs, 38000, Grenoble, France
fDepartment of Physical Chemistry, Lund University, SE-221 00, Lund, Sweden
First published on 28th April 2016
Abstract
In recent years many studies into green solvents have been undertaken and deep eutectic solvents (DES) have emerged as sustainable and green alternatives to conventional solvents since they may be formed from cheap non-toxic organic precursors. In this study we examine amphiphile behaviour in these novel media to test our understanding of amphiphile self-assembly within environments that have an intermediate polarity between polar and non-polar extremes. We have built on our recently published results to present a more detailed structural characterisation of micelles of sodium dodecylsulfate (SDS) within the eutectic mixture of choline chloride and urea. Here we show that SDS adopts an unusual cylindrical aggregate morphology, unlike that seen in water and other polar solvents. A new morphology transition to shorter aggregates was found with increasing concentration. The self-assembly of SDS was also investigated in the presence of water; which promotes the formation of shorter aggregates.
Introduction
Amphiphile self-assembly is driven by interactions between the amphiphile and the surrounding media. Understanding how novel media influence such self-assembly remains a relatively unexplored area. Characteristics such as solvent polarity and surfactant/solvent interactions are important in terms of self-assembly and, from a physicochemical point of view, are needed to understand how they influence the behaviour of amphiphiles. Traditional solvents have been widely evaluated in terms of their ability to allow self-assembly, in particular surfactant self-assembly, microemulsion formation, protein folding, membrane modelling and polymer conformation.1–4Traditional organic solvents are formed of uncharged molecules bonded by relatively weak intermolecular (van der Waals) interactions. They have not generally been useful for amphiphile self-assembly, with only a limited number of solvents other than water showing such behaviour. The emergence of ionic liquids (ILs)5 has significantly expanded the range of solvents exhibiting amphiphile self-assembly behaviour. Ionic liquids are defined as materials, entirely composed of ions, with a melting point below 100 °C.6,7 These liquids can be used as solvents and the first study of amphiphile self-assembly in ionic liquids was in 1982. This concerned cationic and non-ionic surfactant micellization in protic ionic liquids.8 This work has been recently expanded to a wide range of ILs, including aprotic ILs,9 and a reasonably wide range of amphiphiles, demonstrating micellization of surfactants at low concentrations. There have been several studies into the formation of lyotropic phases at high surfactant concentrations and into the formation of microemulsions with IL as the continuous phase.10–12
As in water, amphiphile self-assembly in ionic liquids is generally governed by the solvophobic effect. The driving force in ethylammonium nitrate appears to be similar to that in water. The main difference is that non-polar compounds are more soluble in the IL than in water, hence the force driving the amphiphiles into micelles is weaker and leads to higher critical micelle concentration (CMC) and lower aggregation numbers.8,13 The solvent structure has been demonstrated to be a key factor in the self-assembly process14 and the use of scattering techniques has allowed a better understanding of the solvation and micellization processes occurring in these ILs.15,16
Deep eutectic solvents (DES) have some common properties with ILs such as a high thermal and chemical stability. However, unlike ILs, DES are formed from an eutectic mixture of Lewis or Brønsted acids or bases instead of discrete anions and cations.17 Many DES can be simply described as a mixture of an ionic entity and a hydrogen bond donor following the formula [Cation]+X−zY, where the cation can be an ammonium or phosphonium salt, X is a Lewis base capable of hydrogen bonding with a hydrogen bond donor or a metal salt Y at the ratio z.18,19 The resulting melting point of the binary mixture is substantially lower than the melting points of the individual components and this depression ensures that the solution is liquid at room temperature and therefore useful as a solvent.20
DES are considered a new class of designer solvents due to the wide variety of possible molecular combinations. They can be made from non-toxic, readily available and biodegradable species and therefore avoid the use of the toxic compounds found in many ionic liquids. Natural products are an ideal source due to their enormous chemical diversity and low toxicity profile. As such attention has been directed towards components such as organic acids, alcohols, sugars and other organic compounds.18,21,22
The wide variety of combinations forming DES present a high variety of physicochemical properties.23 However an important disadvantage is that they often have high viscosity which could limit usage in some potential applications. The addition of small amounts of water can reduce the viscosity, increase the electrical conductivity and modify the solvent polarity, and thereby offers a controllable way to modify the properties of the solvent to suit different applications.24
Some work related to self-assembly in eutectic solvents has been published recently. In 2009 a novel technique to incorporate amphiphiles through freeze-drying in DES was reported, highlighting the potential interest of amphiphile behaviour in this media.25 Self-assembled DNA-based microgels were also found in ethylene glycol DES.26 Self-assembly without the presence of amphiphiles has also been demonstrated.27 This study used dynamic light scattering and small-angle X-ray scattering (SAXS) to demonstrate the presence of the aggregates in the monophasic region of ternary eutectic mixtures without surfactants. The results led to a new approach for creating surfactant and water-free microemulsions using DES as the continuous medium.27 The formation of phospholipids vesicles in DES has been recently reported highlighting that DES promote the spontaneous self-assembly of amphiphiles.28 There have also been a few explicit studies of surfactants in DES. Rengstl et al. have shown that the rather uncommon surfactant, choline dodecylsulfate, is soluble in binary mixtures of choline chloride-based DES.29 Meanwhile Pal et al. have shown some evidence of the assembly of sodium dodecylsulfate (SDS) in choline chloride/urea DES containing water.30 These results suggested larger self-assembled structures in DES than those formed in water, unexpected behaviour since this surfactant usually forms smaller micelles in polar solvents other than water.31 More recently Pal et al. have expanded on this to provide some evidence for the solubility of alkyltrimethylammonium bromides in a glycerol-based DES.32
In a recent publication we presented a more detailed structural analysis of the surface adsorption and micelle structure of SDS in pure choline chloride:urea DES.33 The study confirmed the existence of micelles above a critical micelle concentration (approximately 2 mM) that is substantially lower than that seen for the same surfactant in water (8 mM).34 Small-angle neutron scattering (SANS) data suggested the presence of micelles with an elongated shape rather than spherical, considerably different behaviour to that seen in pure water for this surfactant. Unfortunately the low concentration of the CMC means that the signal to noise ratio of that SANS data limits the amount of information that could be extracted from model fitting.
In the present study, we expand on our previous work to investigate a broader range of concentrations and provide greater detail on the morphological behaviour of SDS in choline chloride:urea DES. In addition, we have included measurements aimed at understanding the effect of moderate water content on the self-assembly in this system. This is a particularly important aspect of this work since the hydroscopic nature of this DES means that it is unlikely that any potential applications would use completely dry solutions.
Experimental
Materials
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 choline chloride:urea was prepared by mixing and heating at 80 °C one mole equivalent of choline chloride (ChCl, >98%, Sigma) and two mole equivalents of urea (>99.5%, Sigma) until an homogeneous and transparent liquid was obtained. After the synthesis the liquid was equilibrated for at least for 24 h in an oven at 40 °C. The deuterated version of the DES was prepared following the same procedure. d9-choline chloride (N,N,N-trimethyl-d9, 99% atom D, 99% purity) and d4-urea (98% atom D, 99% purity) were supplied by QMX Laboratories and manufactured by CDN Isotopes.
:2 choline chloride:urea was prepared by mixing and heating at 80 °C one mole equivalent of choline chloride (ChCl, >98%, Sigma) and two mole equivalents of urea (>99.5%, Sigma) until an homogeneous and transparent liquid was obtained. After the synthesis the liquid was equilibrated for at least for 24 h in an oven at 40 °C. The deuterated version of the DES was prepared following the same procedure. d9-choline chloride (N,N,N-trimethyl-d9, 99% atom D, 99% purity) and d4-urea (98% atom D, 99% purity) were supplied by QMX Laboratories and manufactured by CDN Isotopes.
Sodium dodecylsulfate (SDS, >98.5%) was purchased from Sigma-Aldrich and used without further purification. d25-SDS was supplied by the ISIS Deuteration Facility.
The high concentrations of protonated DES with surfactant were prepared as a large stock solution. The lower concentrations were prepared by subsequent dilution except for the deuterated samples which were all prepared directly in order to reduce the quantity of deuterated compounds required.
The pure DES was dried on a vacuum line and freeze-dried to reduce the water content before each experiment. The water content was determined to be below 0.25 wt% by Karl–Fischer titration (Mettler Toledo DL32 Karl–Fischer Coulometer Aqualine Electrolyte A (Fisher Scientific) Aqualine Catholyte CG A (Fisher Scientific)). As in our previous study, in order to confirm that this water content does not vary substantially during the whole experimental procedure we have repeatedly measured the water content for a set of samples that were stored under the same conditions as the samples used in our scattering experiments. Each measurement was taken 3 times using masses between 0.2 and 0.5 g. After synthesis the samples containing water were prepared by simply adding water to the DES samples as prepared above prior to the addition of surfactant. Such samples were prepared with water content in the molar ratios (choline chloride:urea:water) of 1:2:1, 1:2:2 and 1:2:4 and equilibrated for at least for 24 h before use. All the samples were sealed, stored and equilibrated in an oven for at least 1 h at 40 °C before use in the experiments described below.
Methods
In order to obtain a set of different contrasts that could be simultaneously modelled, samples were prepared in three different isotopic mixtures. We will refer to these contrasts as follows: h-SDS in d-ChCl:d-urea, h-SDS in h-ChCl:d-urea and d-SDS in h-ChCl:h-urea. It is worth noting that the “fully deuterated” solvents referred to here are in fact partially deuterated since the choline chloride used was only partially deuterated (d9). Solutions were prepared at different concentrations above the CMC (approximately at 2 mM): 8, 20, 41, 80, 200, 300 and 400 mM.
The procedure for each experiment was the same. The samples were loaded in 1 mm path length, 1 cm wide, quartz Hellma cells and placed in a temperature controlled sample changer at 30 °C. The samples were allowed to equilibrate in the cells at this temperature for a minimum of 2 hr prior to measurement. The data was reduced following the standard routines using Mantid,36 on Sans2d, and GRASP,37 on D22. The empty cell scattering was subtracted from each run and data was normalised to the sample transmission, empty beam flux and detector efficiency.
The scattering from the solvent (measured without surfactant) was subtracted as a background accounting for the scaled contribution to the incoherent scattering of each sample using SasView.38 The data was analysed with the indirect Fourier transformation tool (p(r) inversion) and fitted to analytical inverse-space models using SasView.
The solvent scattering was subtracted and the data analysed with the same procedures as for the SANS data.
Data analysis
Two general approaches are widely used for the treatment of small-angle scattering data. Indirect Fourier transformation (IFT) is a model-free numerical method to fit data in real space and allows one to obtain the radius of gyration of particles in solution.40 The IFT method uses an a priori value of the maximum dimension of the scatterer to obtain the pair distance distribution function (PDDF, p(r)) assuming monodisperse particles in the system. The interpretation is limited to non-periodic structures at low concentrations, where the interparticle interactions are negligible. This method can be applied to any scattering curve regardless the actual structure of the particles. The information obtained from the p(r) function can be used to develop a suitable model for the particles, which can then be fitted to the experimental scattering curve. The p(r) function differs from zero in a limited region of real space, between 0 and Dmax (the largest dimension of the scattering particles). The shape of the function directly enables an assessment of the particle shape, such as globular or elongated particles, and also provides an approximate value of the radius of gyration of these particles.41,42Our analysis, used the IFT procedure in SasView.43 The input parameters were Dmax, the number of terms (the number of base functions used to build the p(r) expansion) and the regularisation constant (used to set the smoothness of the resultant function, where higher values lead to smoother curves, but with a worse fit to the original scattering pattern). The value of Dmax was optimised in order to reduce the chi-squared parameter, while the initial values for the number of terms and regularisation constant were suggested by the software and recalculated with each new value of Dmax. The p(r) was found by fitting these base functions of the IFT procedure to the I(q)/q experimental data. The pair distance distribution function is scaled to unity in order to make the results comparable between contrast and techniques.
The value of Dmax obtained for a scattering pattern provides an approximate value of the length of elongated particles. The region around the maximum value of the p(r) function corresponds to the scattering of the particle cross-section. In this region, the inflection point in the decreasing part of the curve, rI, provides an approximate size of this cross-section. This approach also allows us to calculate a first approximation to the radius of gyration of the scatterers, Rg (related to the second moment of the pair distance distribution function of the particles).
The second approach consists of direct modelling of reciprocal space data using shape-dependent models. The experimental scattering curve is directly fitted to a mathematical model. These models can be used with both dilute and concentrated solutions, hence giving the possibility of evaluating both the shape of the particles and their interparticle interactions.44,45 However this method requires the fitting of several parameters, therefore some preliminary information is required to obtain reliable results. In these models, the total scattered intensity of a monodisperse, homogeneous and isotropic centrosymmetric particle dispersion can be written as a product:
| I(q) = NP(q)S(q) |
This is illustrated in Fig. 1, which shows the theoretical scattering pattern of a system of monodisperse, elongated particles with and without a significant structure factor.
 | ||
| Fig. 1 An illustration of the influence of the structure factor in the measured scattering. (I) Theoretical reciprocal space scattering intensity for a dispersion of rigid cylinders with a non-constrained effective hard sphere structure factor S(q) (inset, same axes). I(q) corresponds to the theoretical scattered intensity of a system with interacting particles and P(q) corresponds to the scattering of such cylinders without interparticle interactions. (II) p(r) shows the pair distance distribution function resulting from P(q) and i(r) includes the oscillations due to the interparticle interactions present in I(q). The quantities rI and Dmax are defined in the text. The parameters used to create these simulations were: length 250 Å, core radius 15 Å, shell thickness 5 Å, effective radius 35 Å, particle volume fraction 0.05 and effective scale 0.15. | ||
Different reciprocal space models were tested for our data in order to optimise the fitting and choose the most appropriate option (see ESI† for details). Of the models tested, the core–shell cylinder model was chosen as the best fit to this data set.
Evidence exists for cylindrical SDS micelles in the literature, so we believe that this model is more than empirically justified. A spherical core–shell structure was initially used to model SDS aggregates in pure water,44 and SDS micelles have also been described as ellipsoids in pure water and other polar organic solvents.31,46 However structural changes in surfactant aggregates have been observed by the addition of salt to screen charge interactions47 or where the counterion penetrates into the head group layer and promotes micellar growth.48,49 These changes can be easily understood through the packing parameter.50 This parameter can be calculated as v/al, where v is the volume of the lyophobic tail, l is the length of the lyophobic tail and a is the effective area per monomer at the headgroup–tail interface. The penetration of counterions into the charged head group layer screens the charge between the neighbouring monomers, thus decreasing the repulsion forces. This reduces the effective area per head group, if steric factors are not important, and leads to a different packing parameter. Given the ionic nature of the solvent used in this study, we expect that such salt effects are potentially important for our understanding of surfactant behaviour in DES. Thus we believe that the presence of an ionic media is one factor in promoting the micelle growth observed here.
Elongated SDS micelles formed in the presence of salt have been described using a cylindrical shape with an ellipsoidal cross section instead of circular.51,52 Furthermore a different approach has considered the presence of flexible, wormlike shaped micelles formed by SDS in the presence of NaBr.47 Such models could improve the chi-squared quality of the fitting in our case, but they represent a significant increase in complexity that is not justified for our data. Instead we take the approach of minimising the number of parameters used and hence prefer a core–shell circular cylinder model. In our previous study33 we used a simple cylinder model because the low concentrations meant that the signal-to-noise ratio was not sufficient to support the use of a more complex model.
The higher concentrations, multiple contrasts and the higher flux instruments used in this work enable us to increase the complexity of the model used and thereby to extract more information from this data.
As explained above, at higher concentrations the IFT and model-based methods require inclusion of the structure factor in order to account for interparticle interactions. However, evaluating the interparticle contribution to the scattering is especially difficult for non-spherical charged particles and such an analytical evaluation remains a challenge to colloid science. Unfortunately previous approaches used to evaluate interparticle interactions in similarly complex systems4,45 are not appropriate in this case because we have limited information regarding the physicochemical properties of the solvent.
In order to evaluate the data we have, therefore, used a custom model that combines the form factor described above and a hard-sphere structure factor (Percus–Yevick) that is not constrained to the dimensions of the form factor. The Percus–Yevick hard-sphere structure factor represents an interparticle interaction defined as an excluded volume repulsion interaction.53 This approach reflects the expected charge screening effect of the high concentration of choline chloride. We would expect that such charge screening will effectively remove any longer range repulsive interactions between the negatively charged surfactant micelles. The Percus–Yevick hard-sphere approximation uses the following approach to solve the interparticle potential, w(r), in the Ornstein–Zernike equation.54

Results
We have performed SAXS and SANS experiments to study the SDS micelle structure within the choline chloride:urea DES and its mixtures with water. Our previous study demonstrated the presence of elongated micelles above the CMC point at low concentrations (up to 25 mM).33 Here we expand that study and analyse the system for a range of concentrations between the maximum concentration of the previous study and the limit of solubility of the system.The available SAXS data covers a limited q-range (0.04 to 0.3 Å−1) due to scattering from the beam-stop at low q. This low-q limitation meant that the SAXS data is not appropriate to determine the length of the micelles. Instead we have used this data to constrain the micelle cross-section in the SANS fits rather than to attempt a simultaneous fit. The SAXS data has significantly better resolution at high-q and better contrast between the shell-head group and the solvent than we can achieve with SANS. We believe that this approach produces a reliable overall fit since the head group sensitivity (and therefore tight cross-sectional constraint) of the SAXS data is complementary to the wider q-range and variable contrasts provided by SANS.
Indirect Fourier transformation
We begin the data analysis by using the IFT method without considering interparticle interactions. Fig. 2 shows the pair distance distribution function of different concentrations of SDS in pure DES and DES containing water. The parameters extracted from this data are shown in Fig. 3. | ||
| Fig. 2 Pair distribution functions, p(r), obtained from the SANS patterns at the contrast d-SDS in h-ChCl:h-urea:H2O without considering interparticle interactions for (I) different concentrations of SDS (with no water) and (II) a fixed concentration of surfactant, 190 mM of SDS, in solvents containing different molar proportions of water. The error bars come from the calculation of the p(r) function through the IFT procedure. | ||
 | ||
| Fig. 3 Parameters from the IFT fitting, Dmax and rI (left) as a function of SDS concentration in 1:2 choline chloride:urea and (right) as a function of water content for three different SDS concentrations: 81.4 (blue), 204 (green), and 319 mM (black). These parameters and the error bars were obtained from the IFT results for all of the contrasts. The solid lines show the trend followed by the parameters. | ||
At an SDS concentration of 20.8 mM, the shape of the p(r) function is similar to that expected from cylindrical micelles.42 At this concentration the p(r) function is above zero for all values between r = 0 and r = Dmax (cf.Fig. 1). This pair distance distribution function could also resemble a multimodal system with diversity of sizes and/or shapes, however this is unlikely given the thermodynamics of micellization.
Fig. 3 shows how the parameters of the IFT fits vary with surfactant concentration and water content. In the absence of water the position of the point of inflection, rI, and therefore the approximate cross-sectional size of the micelles, remains constant at 39 ± 1 Å with increasing concentration.
Given the invariance of the cross-sectional size, the surfactant concentration dependence of Dmax and Rg suggest a change in the length of the particles. Although an increase in the micelle length at low concentrations with increasing surfactant concentration was observed in our previous work, the data here, at higher concentrations, show a reversal in that behaviour from around 42 mM of SDS. Above this concentration the aggregate length decreases as more surfactant is added to the system from a maximum length of 232 ± 11 Å at 42.5 mM to 71 ± 2 Å at 424 mM. The p(r) functions for our measurements at higher concentrations show the presence of weak oscillations. This probably comes from a weak intermicellar contribution to the scattering. The error bars in p(r) also increase at these concentrations (particularly obvious for the data at 424 mM), showing a variability that probably comes from this intermicellar interaction. The highest concentration also shows a minimum below zero, which is a clear indication of a non-negligible structure factor, hence a strong influence of the interactions. Since the IFT assumes no intermicellar interactions, we have not used this to draw further conclusions regarding these concentrations.
Although rl is unaffected by the presence of water and is about 41 ± 1 Å, Dmax is seen to decrease with increasing water content except at the highest SDS concentration where the change is not significant within the errors. This behaviour suggests a uniaxial decrease in the length of the micelles. The DES containing 199 mM SDS fits to a model with a Dmax of 149 ± 17 Å while this value decreases to 114 ± 3 Å for the same concentration of SDS in the DES with a water content of 4 mole equivalents (see Fig. 3). These values were found to be much higher than those calculated for SDS in pure water: 42 Å, using the generalised indirect Fourier transformation method.55
The p(r) corresponding to the DES mixed with water also shows some weak oscillations at lower SDS concentrations than in the SDS solutions without water. This suggests a weak structure factor and might imply that increased water content leads to an increase in intermicellar interactions.
Model-based fitting
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2 choline chloride:urea.
As explained above, SAXS data was used to determine the dimensions of the cross-section. Fig. 4 shows the SAXS data and the resulting fits for different concentrations of SDS. The parameters of the cross section of the micelles are presented in the Table 1.
:2 choline chloride:urea.
As explained above, SAXS data was used to determine the dimensions of the cross-section. Fig. 4 shows the SAXS data and the resulting fits for different concentrations of SDS. The parameters of the cross section of the micelles are presented in the Table 1.
 | ||
| Fig. 4 SAXS data together with best fits (black solid lines) for (I) different concentrations of SDS in 1:2 choline chloride:urea and (II) an averaged concentration of 85.0 mM of SDS in 1:2:1, 1:2:2 and 1:2:4 choline chloride:urea:water. | ||
| SDS concentration (mM) | Core radius (Å) | Shell thickness (Å) | Total radius (Å) |
|---|---|---|---|
| 1:2 choline chloride:urea |
|||
| 12.8 | 14.2 ± 0.1 | 6.4 ± 0.1 | 20.6 ± 0.1 |
| 18.4 | 14.3 ± 0.1 | 6.5 ± 0.1 | 20.8 ± 0.1 |
| 41.2 | 13.8 ± 0.1 | 6.0 ± 0.1 | 19.8 ± 0.1 |
| 82.4 | 13.7 ± 0.1 | 5.7 ± 0.1 | 19.4 ± 0.1 |
| 212 | 14.0 ± 0.1 | 5.9 ± 0.1 | 19.9 ± 0.2 |
| 326 | 14.3 ± 0.1 | 6.2 ± 0.1 | 18.9 ± 0.1 |
| 449 | 14.7 ± 0.2 | 7.0 ± 0.1 | 19.8 ± 0.1 |
| 1:2:1 choline chloride:urea:water |
|||
| 84.5 | 14.1 ± 0.2 | 6.3 ± 0.1 | 20.4 ± 0.1 |
| 216 | 13.9 ± 0.2 | 5.9 ± 0.1 | 19.8 ± 0.1 |
| 314 | 14.1 ± 0.2 | 6.0 ± 0.1 | 20.1 ± 0.2 |
| 1:2:2 choline chloride:urea:water |
|||
| 85.1 | 14.5 ± 0.2 | 6.7 ± 0.1 | 21.2 ± 0.1 |
| 217 | 14.0 ± 0.1 | 6.1 ± 0.1 | 20.1 ± 0.1 |
| 345 | 14.1 ± 0.2 | 6.0 ± 0.1 | 20.1 ± 0.1 |
| 1:2:4 choline chloride:urea:water |
|||
| 85.3 | 15.0 ± 0.1 | 6.8 ± 0.1 | 21.8 ± 0.1 |
| 217 | 14.5 ± 0.1 | 6.4 ± 0.1 | 20.9 ± 0.1 |
| 330 | 14.4 ± 0.1 | 6.3 ± 0.1 | 20.7 ± 0.1 |
The scattering length densities (SLD) for the surfactant tails and the solvents (1:2:n choline chloride:urea:water, n = 0, 1, 2, 4) were calculated and kept constant during fitting. The SLD of the micelle core, corresponding to the tail of the surfactants, was considered to not be affected by solvent penetration.
The shell SLD was fixed to 12.6 × 10−6 Å−2, considering solvent penetration into the shell.33 This value is an arbitrary choice and was used to allow the determination of the radius of the micelle core, which was rather invariant with small changes in the shell SLD and/or thickness. Therefore we have not drawn conclusions about the shell dimensions or solvation from the X-Ray data.
The length was first included in the X-Ray fitting as a parameter to fit. However, the limited q-range did not allow to obtain accurate values for this parameter. Although variances in this parameter have not shown major impact in the size of the cross-section, the results from the neutron analysis were afterwards used to refine the X-Ray fitting and shown to be consistent between both techniques.
Fig. 5 shows the normalised-to-concentration SANS data for the h-SDS in d-ChCl:d-urea contrast and fits of different concentrations of SDS. The parameters used for these fits are shown in Table 2. Three different contrasts were simultaneously fitted to a core–shell cylinder model with a circular cross section. The scattering length density of the micelle core and solvent were held constant during the fitting procedure. The radius of the core was held to a value of 14.5 Å (from the SAXS data described above). This value is consistent with our previous study.33 The length, thickness and volume fraction parameters were linked between contrasts. The scattering length density of the shell was allowed to vary for each contrast, expecting a change with solvent penetration. Two different approaches to the fitting were compared: simultaneous fitting and averaged independent fitting. Both approaches were found to be in good agreement (see ESI†) with the former selected as the most appropriate option.
 | ||
| Fig. 5 (a) Normalised-to-concentration SANS patterns and (b) best fits for samples in d-ChCl:d-urea. The concentrations quoted are the average from the three contrasts. Data was fitted with a core-shell cylinder model with the hard-sphere structure factor. The fits are plotted as black-dashed lines over the data points. An additional plot of the other contrasts is included in the ESI.† | ||
:2 choline chloride:urea: structural parameters, shell SLD, fitted volume fraction of micelles (ϕfit), calculated volume fraction of micelles (ϕcalc), aggregation number (Nagg) and structure factor volume fraction were obtained from the model-based fitting. The shell SLD was included following the format shell SLD in h-choline chloride:h-urea (hh), shell SLD in d-choline chloride:d-urea, shell SLD in h-choline chloride:d-urea
| SDS concentration (mM) | Choline:SDS molar ratio |
Length (Å) | Shell thickness (Å) | Shell SLD hh, dd, hd (±0.1, × 10−6 Å−2) | ϕ fit (×10−2) | ϕ calc (×10−2) | S(q) volume fraction (×10−2) | N agg |
|---|---|---|---|---|---|---|---|---|
| 8.71 ± 1.16 | 553 ± 2 | 414 ± 39 | 5.6 ± 0.4 | 1.4, 6.0, 3.0 | 0.10 ± 0.04 | 0.20 ± 0.04 | 0.1 ± 0.5 | 781 ± 74 |
| 20.8 ± 0.4 | 220 ± 4 | 568 ± 81 | 6.1 ± 0.8 | 2.0, 5.3, 2.6 | 0.43 ± 0.02 | 0.68 ± 0.02 | 0.1 ± 0.5 | 1071 ± 153 |
| 42.5 ± 1.7 | 109 ± 1 | 668 ± 28 | 7.4 ± 0.4 | 2.2, 5.0, 2.3 | 1.3 ± 0.1 | 1.2 ± 0.1 | 0.2 ± 0.5 | 1260 ± 53 |
| 81.3 ± 9.2 | 55 ± 8 | 328 ± 12 | 10 ± 1 | 1.6, 6.1, 3.1 | 3.5 ± 0.1 | 2.3 ± 0.2 | 2.8 ± 0.1 | 619 ± 23 |
| 194 ± 10 | 22 ± 2 | 176 ± 4 | 8.4 ± 0.2 | 2.5, 5.8, 3.0 | 6.7 ± 0.1 | 5.5 ± 0.1 | 5.1 ± 0.1 | 332 ± 8 |
| 315 ± 24 | 14 ± 1 | 119 ± 1 | 6.5 ± 0.1 | 2.9, 5.6, 2.6 | 8.2 ± 0.1 | 8.7 ± 0.1 | 8.1 ± 0.2 | 224 ± 2 |
| 424 ± 21 | 10 ± 1 | 106 ± 8 | 4.7 ± 0.1 | 3.2, 5.4, 1.9 | 9.6 ± 0.1 | 11 ± 1 | 12 ± 1 | 206 ± 2 |
A summary of the results from fits is included in Table 2 and Fig. 6. In good agreement with the IFT analysis, these fits show that the micelles are elongated at both low and high concentrations. The head group is situated at the micelle–solvent interface, whereas the lyophobic tails are at the core of the structure. As explained above, the thickness and SLD of the shell layer were allowed to vary since the presence of solvent may affect both parameters. The expected volume fraction of micelles (ϕcalc) was calculated from the actual amounts of each component in the system, surfactant and solvent, minus the equivalent amounts at the CMC. The aggregation number (Nagg) was calculated considering the volume of the lyophobic core and the volume of a surfactant tail.56
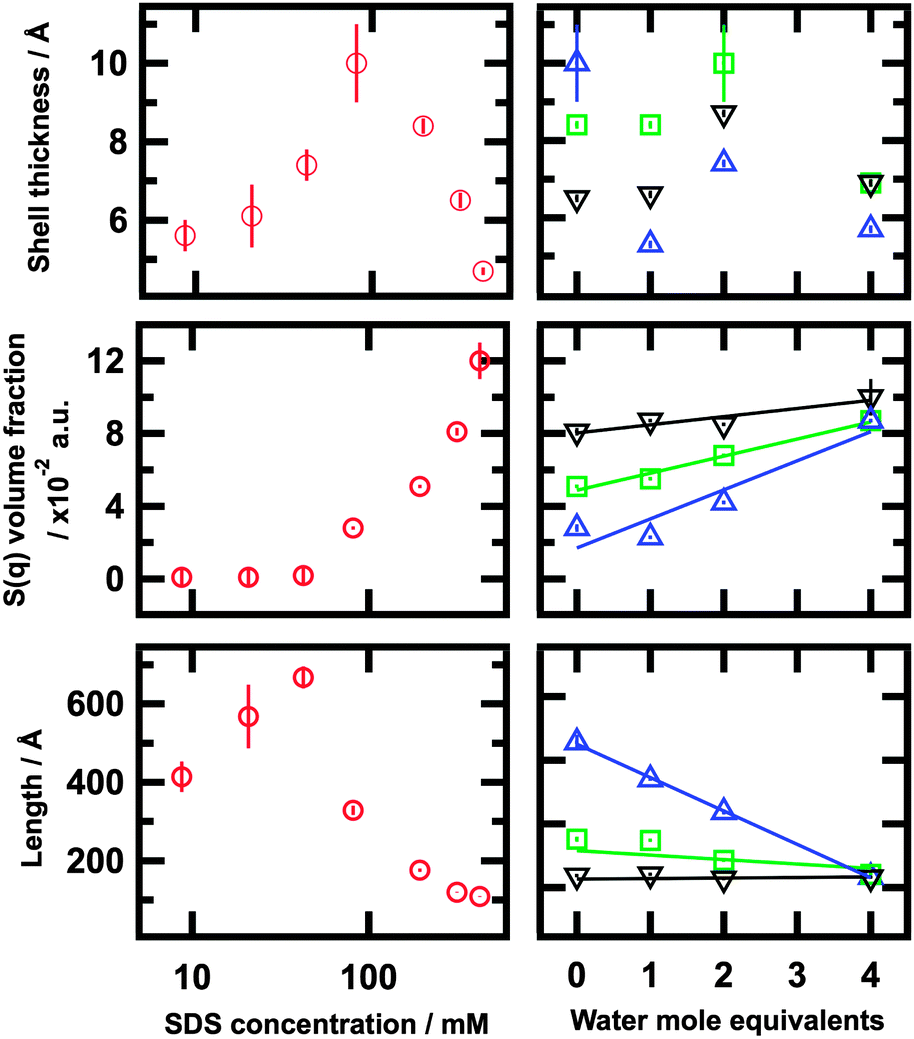 | ||
| Fig. 6 Radius, S(q) volume fraction and length (left) as a function of SDS concentration in 1:2 choline chloride:urea and (right) as a function of water content for three different SDS concentrations: 81.4 (blue), 204 (green), and 319 mM (black) | ||
The possibility of small differences in the particle size and fluctuations due to interchange of surfactant molecules between micelles are accounted by means of a polydispersity term. Polydispersity terms for length, core-radius and shell-thickness were tested as an extra parameter for the optimum fits. However, the implementation of these did not show an improvement in the fits, hence were not included during the fitting procedure.
Micelles were found to be larger than seen in pure water and other polar solvents.31,44 In agreement with the IFT analysis, the axial length of the micelles increases from 414 ± 39 Å at 8.16 mM to 668 ± 28 Å at 42.5 mM. The presence of an interaction peak, commonly found in SDS micelles in water due to repulsive electrostatic interactions, seems to vanish at low concentrations.
Although we were careful to evaluate the possible structure factor, it was found to be negligible below a SDS concentration of 71 mM. Therefore the fitting of the structure factor model leads to zero values. Above 81.3 mM of SDS, the structure factor increases to values above 0 and the length gradually decreases with increasing concentration up to 108 ± 1 Å at 424 mM of surfactant. The trend regarding the length of the aggregates was found to be in good agreement with our previous work, though the utilisation of a different model and limited q-range of the previous data do cause some differences.
The apparent size of the head group appears to be bigger than determined from SAXS data. We believe that this is because of the insignificant X-Ray contrast between the two solvent molecules. Unlike with neutrons, this means that solvation of specific components within the head group does not have a significant impact on the SLD.
The volume fraction of micelles (ϕfit) increases with concentration and the fitting results are comparable with the calculated values. Since the radius of the core remains unchanged over the whole range of concentrations, the aggregation number shows a similar trend to that of the length of the particles. The values for Nagg are comparable to those found in another system composed by SDS and a hydrotropic salt in the limiting situation where the charge on the SDS micelles is totally screened by the presence of the salt.55
:2:n choline chloride:urea:water, n = 1, 2 and 4.
The scattering data and the fits corresponding to SDS micelles in the mixtures of the DES with water are presented in Fig. 4 (X-Ray) and Fig. 7 (neutron). The neutron data was simultaneously fitted using two contrasts for the core–shell cylinder model with the non-constrained hard sphere structure factor. The same fitting procedure was as for the SDS in dry DES. The presence of interparticle interactions is suggested by the oscillations found in the IFT analysis (Fig. 2), and a structure factor is required to fit this data. Fig. 6 and Table 3 compare the results from these fits (Fig. 7) with the same concentrations in pure DES (Fig. 5).
 | ||
| Fig. 7 SANS patterns and fits for SDS in ChCl:urea:H2O in the ratio 1:2:n where n = 1 (1a and b), 2 (2a and b) and 4 (3a and b). Data for three SDS concentrations is shown; average concentrations of 81.4 (red), 204 (blue), and 319 mM (green), for two isotopic contrasts d-SDS in h-ChCl:h-urea:H2O (1a, 2a, 3a) and h-SDS in d-ChCl:d-urea:D2O (1b, 2b, 3b) The black-dashed lines represent the best fit for each pattern. | ||
:2:0, 1:2:1, 1:2:2 and 1:2:4 choline chloride:urea:water
| Water mole equivalents | Length (Å) | Shell-thickness (Å) | Shell SLD hhh, ddd (±0.1, × 10−6 Å−2) | ϕ fit (×10−2) | ϕ calc (×10−2) | S(q) volume fraction (×10−2) | N agg |
|---|---|---|---|---|---|---|---|
| 81.4 ± 10.8 mM | |||||||
| 0 | 328 ± 12 | 10 ± 1 | 1.6, 6.1 | 3.5 ± 0.1 | 2.3 ± 0.2 | 2.8 ± 0.1 | 619 ± 23 |
| 1 | 270 ± 1 | 5.3 ± 0.2 | 2.9, 5.7 | 2.1 ± 0.1 | 2.1 ± 0.1 | 2.3 ± 0.1 | 509 ± 2 |
| 2 | 219 ± 4 | 7.4 ± 0.1 | 2.1, 5.8 | 2.1 ± 0.1 | 2.2 ± 0.3 | 4.2 ± 0.1 | 413 ± 8 |
| 4 | 117 ± 1 | 6.7 ± 0.1 | 2.1, 5.7 | 2.1 ± 0.1 | 2.3 ± 0.1 | 8.7 ± 0.1 | 221 ± 2 |
| 204 ± 15 mM | |||||||
| 0 | 176 ± 4 | 8.4 ± 0.2 | 2.5, 5.8 | 6.7 ± 0.1 | 5.5 ± 0.1 | 5.1 ± 0.1 | 332 ± 8 |
| 1 | 174 ± 1 | 8.4 ± 0.1 | 1.6, 5.9 | 7.7 ± 0.1 | 5.3 ± 0.1 | 5.5 ± 0.1 | 328 ± 2 |
| 2 | 143 ± 1 | 10 ± 1 | 2.1, 5.7 | 6.5 ± 0.1 | 5.6 ± 0.1 | 6.8 ± 0.1 | 270 ± 2 |
| 4 | 121 ± 1 | 6.9 ± 0.1 | 2.2, 5.6 | 6.0 ± 0.1 | 5.3 ± 0.1 | 8.7 ± 0.1 | 228 ± 2 |
| 319 ± 22 mM | |||||||
| 0 | 119 ± 1 | 6.6 ± 0.1 | 2.9, 5.6 | 8.2 ± 0.1 | 8.7 ± 0.1 | 8.1 ± 0.2 | 224 ± 2 |
| 1 | 121 ± 1 | 6.6 ± 0.1 | 2.1, 5.9 | 10 ± 1 | 8.3 ± 0.3 | 8.7 ± 0.1 | 228 ± 2 |
| 2 | 114 ± 1 | 8.7 ± 0.1 | 2.2, 5.8 | 8.8 ± 0.1 | 8.4 ± 0.1 | 8.7 ± 0.1 | 215 ± 2 |
| 4 | 116 ± 1 | 7.0 ± 0.1 | 2.1, 5.8 | 9.1 ± 0.1 | 8.0 ± 0.2 | 10 ± 1 | 219 ± 2 |
As for the dry DES system, the radius of the micelle core was held to 14.5 Å during the fitting. The total radius was found to vary since the head group thickness of the micelle varies with both surfactant concentration and water content. Despite variations with surfactant concentration and water content, the total radius for the micelles was found to be in the same order of magnitude as SDS in water (27.3 Å at 100 mM) and formamide (∼18 Å at 80 mM).31,44
This data demonstrates some interesting changes in self-assembly behaviour relative to that seen in the dry DES. In each case the self-assembled structures tend towards shorter aggregates with increasing water content, but the effect is not of the same magnitude for all concentrations. The length of the micelles at the lowest SDS concentration, 81.4 mM, was found to decrease by 64% at the highest water content compared to the dry solvent. However this effect is less pronounced at higher concentrations, decreasing by 31% and remaining rather unchanged for the 204 and 319 mM SDS solutions respectively. This decreasing effect of water on the length appears to correlate with an increase in the influence of S(q). The aggregation number shows a similar trend to the length since the change is mainly governed by the change in the length. This value decreases with both increasing surfactant concentration and increasing water content.
Discussion
The shape and size of SDS micelles in DES is dependent on surfactant concentration and on water content. Increasing the concentration we found that the length increases to a maximum before decreasing. Such behaviour is unusual and suggests that intermicellar interactions within the system become more important with increasing SDS concentration and so modify the molecular packing. The evolution of the packing parameter can be understood through changes in the effective area of the surfactant head group, which is affected by interaction of the head group with solvent counterions. With increasing the concentration above 42.5 mM or increasing the water content, the packing parameter evolves and leads to shorter elongated micelles.In pure water the surfactant forms a structure in which the counterions, Na+, act as a non-penetrating counterion and remain highly solvated.57 However soft ions, such as the choline cation, may show greater affinity for the head group of the surfactant and possibly directly bind to this layer.
A recent study presented by Dolan et al. has shown remarkable differences between amphiphile behaviour in water and ionic liquids. Ionic liquids have shown the ability to exchange counterions from the bulk solvent with the surfactant and the surfactant counterions can be incorporated to some extent into the hydrogen bond network.14 The DES studied here could behave in a similar way with the choline cation interacting with the negatively charged head-group of SDS. At low concentrations, the charge neutralisation provided by the choline in the DES allows a closer packing of the SDS head groups than in water making elongated micelles more energetically favourable and resulting in uniaxial growth of the micelles.50,58 In this scenario, as the ratio of SDS to choline chloride increases, the availability of choline counterions decreases and results in an increase in the average area occupied by the sulfate head group, changing the molecular packing and leading to a shape transition between elongated to globular micelles.
The variation of the scattering length density of the shell shows the adsorption of solvent to the surfactant head group (Tables 2 and 3). Changes of this value between contrasts have provided qualitative information about the role of the solvent. The fully protonated and fully deuterated solvents modify the SLD of the shell reducing and increasing the SLD respectively. Using the intermediate contrast for the pure solvent, h-choline chloride:d-urea, a decrease in the SLD of the shell suggests a higher affinity of the protonated entity for the head groups, in this case the positively charged choline chloride. This observation confirms the suggestion that there is a specific interaction between the choline chloride and the sulfate headgroups. This behaviour is not as clear in the presence of water, because the additional component complicates matters and the composition changes in the headgroup layer cannot be disentangled from changes observed in the SLD.
A recent investigation has demonstrated the internal structure of the choline chloride:urea DES using neutron diffraction.59 This research shows the presence of a hydrogen bond network, which maintains the components in a slightly ordered structure. The introduction of surfactant to the system may disrupt this hydrogen-bonding network by competing for interaction with the choline cation. In such a case, the self-assembly would be driven by the competition between the counterion affinities for the head group interface and the solvent structure. At low concentrations the ratio between choline ions and SDS is relatively high, so the head group layer can be screened by the adsorption of choline cations effectively without changing the concentration of choline ions in the solvent network. However, with increasing concentration of surfactant a competition between the surfactant and the solvent hydrogen bond network becomes more important. In such a case, the strong hydrogen bond interaction within the solvent may limit the displacement of choline ions to the micelle–solvent interface and result in insufficient screening, therefore reducing the size of the aggregates. The contribution of intermicellar electrostatic forces is observed in the S(q) volume fraction parameter. At low concentrations of SDS, up to 42.5 mM, the presence of these forces appears to be negligible, but above this concentration, the intermicellar interactions increase as more surfactant is added to the system.
Similar behaviour was found with the addition of water to the system. The presence of water seems to modify the molecular packing and promote formation of shorter aggregates. This behaviour is accompanied by an increase in the repulsive forces between micelles, as observed through changes in the structure factor; S(q) volume fraction was found to increase with the addition of water. The introduction of water must substantially modify the competition between micelle interface and hydrogen bond network. However a more detailed understanding of the effect of water content is not possible from this data and we have already begun further studies on the effect of water on the choline chloride:urea solvent structure.
Whilst the SAXS and contrast variation SANS data allow us to identify the specific interaction between the choline chloride and the surfactant, the structural details are beyond the resolution of those techniques. We have therefore already begun some further studies with neutron liquid diffraction in order to examine the interactions of the solvent with the head group layer in more detail. These further investigations will help to develop an accurate atomistic model of the micelle and the surrounding environment.60
Conclusions
We have shown that surfactant aggregation in DES is possible and that the properties and composition of these solvents have significant effects on the structural properties of the surfactant aggregates in these systems. Control of these properties offers new possibilities and potential for future applications. Furthermore DES can play a key role in understanding the fundamentals of the self-assembly process, since they have different characteristics from water and polar solvents. SDS has been shown to form highly tunable micelles with peculiar characteristics such as a cylindrical morphology at low concentrations, undergoing an unusual shape transition as the surfactant or water content is increased.SAXS and SANS measurements consistently show the presence of cylindrical micelles with the polar head groups situated at the interface between the micelle and the solvent and the non-polar tails in the core. At low concentrations, the micelle interaction peak characteristic of SDS in water disappears due to the high charge screening by the solvent, probably due to the presence of the positive charged choline chloride. This charge screening undergoes a reduction with increasing concentration, which favours shorter aggregates, while keeping the cross section constant. The implementation of a model with an effective hard sphere S(q) structure factor, without constraints, incorporating an effective radius and effective volume fraction, allows us to account for the effect of the intermicellar interactions.
The contrast variation SANS results show a specific interaction between the choline chloride component of the DES and the surfactant headgroups. We propose that this interaction is driving the morphological transitions through changes in charge screening.
These results also demonstrate the potential for modifying the self-assembly process, allowing control of the micellar structures by altering the solvent. The measurements reported here were accomplished for up to 4 mole equivalents of water, however extrapolating the behaviour, the addition of greater amounts of water will enable formation of different aggregates becoming more similar to those found in water as the proportion of water is increased.
Acknowledgements
The authors would like to thank DLS, ILL and ISIS for the awarded beamtime (experiment numbers SM11392, 9-12-39961 and RB1510328). We are also thankful to University of Bath Alumni Fund and European Spallation Source for the funding for A. S.-F. This work has benefited from the use of SasView (NSF award DMR-0520547)References
- S. J. Singer and G. L. Nicolson, Science, 1972, 175, 720–731 CAS.
- M. J. Lawrence and G. D. Rees, Adv. Drug Delivery Rev., 2000, 45, 89–121 CrossRef CAS PubMed.
- O. Ikkala and G. ten Brinke, Science, 2002, 295, 2407–2409 CrossRef CAS PubMed.
- J. B. Hayter and J. Penfold, Mol. Phys., 1981, 42, 109–118 CrossRef CAS.
- T. L. Greaves and C. J. Drummond, Chem. Soc. Rev., 2008, 37, 1709–1726 RSC.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- J. D. Holbrey, W. M. Reichert, M. Nieuwenhuyzen, S. Johnston, K. R. Seddon and R. D. Rogers, Chem. Commun., 2003, 1636–1637, 10.1039/b304543a.
- D. F. Evans, A. Yamauchi, R. Roman and E. Z. Casassa, J. Colloid Interface Sci., 1982, 88, 89–96 CrossRef CAS.
- S. C. Sharma, R. Atkin and G. G. Warr, J. Phys. Chem. B, 2013, 117, 14568–14575 CrossRef CAS PubMed.
- D. F. Evans, E. W. Kaler and W. J. Benton, J. Phys. Chem., 1983, 87, 533–535 CrossRef CAS.
- M. U. Araos and G. G. Warr, J. Phys. Chem. B, 2005, 109, 14275–14277 CrossRef CAS PubMed.
- R. Atkin and G. G. Warr, J. Phys. Chem. B, 2007, 111, 9309–9316 CrossRef CAS PubMed.
- M. U. Araos and G. G. Warr, Langmuir, 2008, 24, 9354–9360 CrossRef CAS PubMed.
- A. Dolan, R. Atkin and G. G. Warr, Chem. Sci., 2015, 6, 6189–6198 RSC.
- H. J. Jiang, P. A. FitzGerald, A. Dolan, R. Atkin and G. G. Warr, J. Phys. Chem. B, 2014, 118, 9983–9990 CrossRef CAS PubMed.
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, J. Phys. Chem. C, 2014, 118, 13998–14008 CAS.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, JACS, 2004, 126, 9142–9147 CrossRef CAS PubMed.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- M. A. Kareem, F. S. Mjalli, M. A. Hashim and I. M. AlNashef, J. Chem. Eng. Data, 2010, 55, 4632–4637 CrossRef CAS.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- A. P. Abbott, R. C. Harris, K. S. Ryder, C. D’Agostino, L. F. Gladden and M. D. Mantle, Green Chem., 2011, 13, 82–90 RSC.
- A. Hayyan, F. S. Mjalli, I. M. AlNashef, T. Al-Wahaibi, Y. M. Al-Wahaibi and M. A. Hashim, Thermochim. Acta, 2012, 541, 70–75 CrossRef CAS.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jerome, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- Y. Dai, G.-J. Witkamp, R. Verpoorte and Y. H. Choi, Food Chem., 2015, 187, 14–19 CrossRef CAS PubMed.
- M. C. Gutiérrez, M. L. Ferrer, C. R. Mateo and F. del Monte, Langmuir, 2009, 25, 5509–5515 CrossRef PubMed.
- C. Mukesh and K. Prasad, Macromol. Chem. Phys., 2015, 216, 1061–1066 CrossRef CAS.
- V. Fischer, J. Marcus, D. Touraud, O. Diat and W. Kunz, J. Colloid Interface Sci., 2015, 453, 186–193 CrossRef CAS PubMed.
- S. J. Bryant, R. Atkin and G. G. Warr, Soft Matter, 2016, 12, 1645–1648 RSC.
- D. Rengstl, V. Fischer and W. Kunz, Phys. Chem. Chem. Phys., 2014, 16, 22815–22822 RSC.
- M. Pal, R. Rai, A. Yadav, R. Khanna, G. A. Baker and S. Pandey, Langmuir, 2014, 30, 13191–13198 CrossRef CAS PubMed.
- T. Perche, X. Auvray, C. Petipas and R. Anthore, Langmuir, 1997, 13, 1475–1480 CrossRef CAS.
- M. Pal, R. K. Singh and S. Pandey, ChemPhysChem, 2015, 16, 2538–2542 CrossRef CAS PubMed.
- T. Arnold, A. J. Jackson, A. Sanchez-Fernandez, D. Magnone, A. E. Terry and K. J. Edler, Langmuir, 2015, 31, 12894–12902 CrossRef CAS PubMed.
- P. H. Elworthy and K. J. Mysels, J. Colloid Interface Sci., 1966, 21, 331–347 CrossRef CAS.
- R. K. Heenan, S. E. Rogers, D. Turner, A. E. Terry, J. Treadgold and S. M. King, Neutron News, 2011, 22, 3 CrossRef.
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal, M. A. Gigg, V. E. Lynch, A. Markvardsen, D. J. Mikkelson, R. L. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. G. Perring, P. F. Peterson, S. Ren, M. A. Reuter, A. T. Savici, J. W. Taylor, R. J. Taylor, R. Tolchenov, W. Zhou and J. Zikovsky, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- C. Dewhurst, GRASP, http://www.ill.eu/instruments-support/instruments-groups/groups/lss/grasp/home/.
- M. Doucet, S. King, P. Butler, P. Kienzle, P. Parker, J. Krzywon, A. Jackson, T. Richter, M. Gonzales, T. Nielsen, R. Ferraz Leal, A. Markvardsen, R. Heenan, J. Bakker and G. Alina, SasView version 3.1.2, http://www.sasview.org, DOI: http://10.5282/zenodo.35065.
- M. Basham, J. Filik, M. T. Wharmby, P. C. Y. Chang, B. El Kassaby, M. Gerring, J. Aishima, K. Levik, B. C. A. Pulford, I. Sikharulidze, D. Sneddon, M. Webber, S. S. Dhesi, F. Maccherozzi, O. Svensson, S. Brockhauser, G. Naray and A. W. Ashton, J. Synchrotron Radiat., 2015, 22, 853–858 Search PubMed.
- O. Glatter, J. Appl. Crystallogr., 1977, 10, 415–421 CrossRef.
- O. Glatter, J. Appl. Crystallogr., 1979, 12, 166–175 CrossRef CAS.
- O. Glatter, G. Fritz, H. Lindner, J. Brunner-Popela, R. Mittelbach, R. Strey and S. U. Egelhaaf, Langmuir, 2000, 16, 8692–8701 CrossRef CAS.
- P. B. Moore, J. Appl. Crystallogr., 1980, 13, 168–175 CrossRef CAS.
- J. B. Hayter and J. Penfold, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 1851–1863 RSC.
- H. Pilsl, H. Hoffmann, S. Hofmann, J. Kalus, A. W. Kencono, P. Lindner and W. Ulbricht, J. Phys. Chem., 1993, 97, 2745–2754 CrossRef CAS.
- C. C. Ruiz, L. Diaz-Lopez and J. Aguiar, J. Dispersion Sci. Technol., 2008, 29, 266–273 CrossRef CAS.
- L. Arleth, M. Bergström and J. S. Pedersen, Langmuir, 2002, 18, 5343–5353 CrossRef CAS.
- L. J. Magid, Z. Li and P. D. Butler, Langmuir, 2000, 16, 10028–10036 CrossRef CAS.
- P. A. Hassan, S. R. Raghavan and E. W. Kaler, Langmuir, 2002, 18, 2543–2548 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- M. Bergstrom and J. S. Pedersen, Phys. Chem. Chem. Phys., 1999, 1, 4437–4446 RSC.
- C. A. Dreiss, Soft Matter, 2007, 3, 956–970 RSC.
- J. K. Percus and G. J. Yevick, Phys. Rev., 1958, 110, 1–13 CrossRef CAS.
- L. S. Ornstein and F. Zernike, Proc. – Acad. Sci. Amsterdam, 1914, 15 Search PubMed.
- P. A. Hassan, G. Fritz and E. W. Kaler, J. Colloid Interface Sci., 2003, 257, 154–162 CrossRef CAS PubMed.
- C. Tanford, J. Phys. Chem., 1972, 76, 3020–3024 CrossRef CAS.
- L. J. Magid, Z. Han, Z. Li and P. D. Butler, Langmuir, 2000, 16, 149–156 CrossRef CAS.
- R. Nagarajan and E. Ruckenstein, Langmuir, 1991, 7, 2934–2969 CrossRef CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Green Chem., 2016, 18, 2736–2744 RSC.
- R. Hargreaves, D. T. Bowron and K. Edler, JACS, 2011, 133, 16524–16536 CrossRef CAS PubMed.
- K. J. Edler, A. J. Jackson, T. Arnold, L. Porcar and A. Sanchez-Fernandez, 2015, DOI:10.5291/ILL-DATA.9-12-399.
Footnote |
| † Electronic supplementary information (ESI) available: Examples of alternative reciprocal space models compared to the experimental data and plots of additional contrasts not shown in the main text. See DOI: 10.1039/c6cp01757f |
| This journal is © the Owner Societies 2016 |