
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom dioxin to dioxin congeners: understanding the differences in hydrophobic aggregation in water and absorption into lipid membranes by means of atomistic simulations†
Mosé
Casalegno
a,
Guido
Raos
a and
Guido
Sello
*b
aDipartimento di Chimica, Materiali e Ingegneria Chimica “G. Natta”, Politecnico di Milano, via L. Mancinelli 7, 20131 Milano, Italy
bDipartimento di Chimica, Università degli Studi di Milano, via Golgi 19, I-20133 Milano, Italy. E-mail: guido.sello@unimi.it
First published on 6th June 2016
Abstract
Translocation of small molecules through a cell membrane barrier is a fundamental step to explain the response of cells to foreign molecules. Investigating the mechanisms through which this complex process takes place is especially important in the study of the adverse effects of toxicants. In this work, we start from the results of a previous simulation study of the mechanism of dioxin (2,3,7,8-tetrachlorodibenzo-p-dioxin) absorption into a model membrane, and extend it to four structural congeners of dioxin. The new molecules have been chosen taking into consideration the structural features that characterize dioxin: aromaticity, planarity, the presence of chlorine and oxygen atoms, and hydrophobicity. Our results for the absorption mechanism confirm our expectations based on the chemical structures, but also reveal some interesting differences in single-molecules and especially in cooperative actions underlying cluster absorption. The analysis of key parameters, such as free energies of transfer and translocation times, supports the idea that dioxin, more than its congeners investigated here, likely accumulates in cell membranes.
1 Introduction
The term “dioxin” is commonly used to refer to a family of toxic chemicals that share key structural features and cause harm through similar mechanisms of action.1 Overall, the dioxin family has more than two hundred members, based on polichlorinated dibenzo-p-dioxins (PCDDs), and polychlorinated dibenzofurans (PCDFs). Although not strictly dioxins, polychlorinated biphenyls (PCBs) also are included within this broad class. Only about thirty dioxins are of primary concern in risk assessment, with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, hereafter) recognized as the most toxic.Dioxins are unintentionally produced as by-products in a number of human activities. These include combustion processes such as waste incineration, backyard trash burning, as well as some industrial processes such as chlorination processes,2 plastic and herbicide manufacturing. Once released in the environment, dioxins accumulate preferentially in sediments and soils, and, to a lesser extent, in air and groundwater.3–5 Depending on the exposure level, the toxic effects related to dioxin contamination in animals include immunological and hormonal dysfunctions, reproductive diseases, and teratogenesis. Studies of humans exposed to dioxins6,7 suggest that dioxins can cause chloracne, metabolic disorders, impaired reproduction, abnormal development, suppression of the immune system, and other systemic problems, including cancer.8–11
The toxic potency of dioxins is related to their chemical stability, and tendency to accumulate in fat tissues, where they can reach high concentrations prior to excretion. After the Seveso accident,12 the mode of action of TCDD and related dioxins has been quite extensively studied, and involves binding to the aryl hydrocarbon receptor (AhR).13,14 The AhR is a ligand-dependent transcription factor that induces expression of a number of genes encoding drug metabolizing enzymes.15 In the absence of a ligand the dioxin receptor is present in a latent conformation in the cytoplasmic compartment of the cell16 associated with the molecular chaperone hsp90.17 Binding of dioxins to the PAS-B domain of AhR triggers the translocation of AhR to the nucleus and its heterodimerization with the AhR nuclear translocator (Arnt).13,18,19 Finally, the AhR–Arnt complex binds xenobiotic response elements that encode the transcription of metabolizing enzymes such as Cyp1a1, Cyp1a2, and Cyp1b1.20
The very first step of this route is represented by the absorption and diffusion of the toxic molecules into cell membranes. Several observations in both humans and animals have indicated that, although present in many tissues and organs, dioxins primarily accumulate in fat tissues because of their hydrophobic character.21,22 Phospholipids represent the major components of all cell membranes. Their lipophilic tails likely provide an ideal environment for dioxin storage and accumulation. Although not necessarily the final target of toxic action, cell membranes may behave as storage depots for dioxins, thus becoming an internal source of chronic exposure to these pollutants.21 Understanding dioxin absorption and distribution in cell membranes is therefore essential to elucidate dioxin toxicokinetics. The complexity of biological membranes and the lack of experimental data make this a challenging task.
Membranes are dynamic assemblies of molecules,23 characterized by the capability of self ordering in a water environment. The absence of covalent bonds between molecules allows subtle structural variations in response to the environmental changes. Atomistic molecular dynamics (MD) approaches have become standard numerical tools to study these variations at the molecular level.24–26 So far, these methods have been successfully applied to passive membrane transport,27–34 with the aim of characterizing the dynamics of the absorption process,33,34 as well as the effect of solute molecules on membrane properties.30,32 Current MD simulations cannot deal with the tremendous complexity of real cell membranes.23 Nonetheless, the use of simplified model membranes, essentially consisting of phospholipid bilayers, provides a good starting point to answer some fundamental questions and can help in the development of more refined models. Recently,35 we have applied MD to the study of TCDD translocation from water into a 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) bilayer membrane. Through the calculation of the free energy of transfer, we have shown that TCDD absorption is a spontaneous process, with a free energy minimum at about −40 kJ mol−1, with respect to the aqueous phase. Our simulations have also revealed the tendency of TCDD clusters to quickly penetrate the membrane, and soon disaggregate once the absorption process completes. Although the concentration of TCDD in our simulations was higher than that usually observed in risk assessment studies,36,37 this finding suggested that local clustering does not prevent dioxin storage in cell membranes. Given this possibility, it is reasonable to ask whether the hydrophobic character of TCDD is the main factor responsible for its behavior, or other structural factors might also be involved. To answer this question, in this work, we compare the absorption dynamics of TCDD to that of four of its congeners, with the aim of finding possible correlations between their molecular structure and interaction with the membrane. The congeners, to be discussed in the next section, were selected on the basis of their structural similarities with TCDD. To ensure consistency with our previous work, we used the same 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) model membrane. Besides, the model system was improved by replacing pure water with a physiological (0.15 M NaCl) solution. MD simulations were performed for both single molecules and their aggregates.
1.1 Simulated systems
Fig. 1 shows the molecular sketches of TCDD and the four congeners chosen for the present investigation. These are anthracene (ANTH), tetrahydrodibenzo-p-dioxin (THDD), 3,5,3′,5′-tetrachlorobiphenyl (TCBP), and 1,2-dihydroxytetrahydrodibenzo-p-dioxin (THDO). The structure of TCDD is characterized by the presence of aromatic rings. The ether linkages between the two benzene rings formally make the compound not completely aromatic because the central ring does not conform to the Hückel rule. However, the two oxygen atoms participate in the conjugation of the benzene rings, increasing the electron availability. In addition, the four chlorine atoms contribute to enhance the benzene activity. These heteroatoms also make the compound polar and able to form hydrogen bonds with hydrogen donors. The molecule is fundamentally flat and does not possess rotatable dihedral bonds. | ||
| Fig. 1 Molecular structures of the selected compounds. The arrows connect closely-related chemical structures. | ||
In order to study the importance of these structural features with respect to the interaction with the membrane, we chose four congeners whose structures can be seen as variations of the TCDD structure. ANTH is a polycyclic aromatic hydrocarbon (PAH). Compared to TCDD it can be considered its parent hydrocarbon structure. Due to its hydrophobic character and the absence of heteroatoms, ANTH is structurally the most diverse compound from TCDD. The first step from this structure to the TCDD one is the substitution of the two central CH groups with oxygen atoms. This leads to THDD, which has the same structure as that of TCDD, but no chlorine atoms. It is also hydrophobic but can accept H-bonds from water molecules. If we consider the possibility of structural variations introduced by cell metabolism, we can hypothesize the introduction of two hydroxyl groups in the 1,2 positions of the tricyclic moiety on the THDD structure, thus generating THDO. This is the simplest metabolic transformation of aromatic compounds made by oxidative enzymes, and its all-hydrocarbon version is a well known metabolite of PAHs.38 THDO is still hydrophobic, but can accept and donate H-bonds both intramolecularly and intermolecularly. Thus, it can be expected to interact with water molecules and with DPPC headgroups more strongly than THDD and TCDD.
Flat molecules can have a privileged interaction with phospholipid assemblies because, if properly oriented, they can be expected to more easily pass through the phospholipidic barrier. TCBP is an example of non-flat compounds belonging to the PCB class. This toxic molecule has four chlorine atoms (as TCDD), but no oxygen atom. TCBP is characterized by the presence of a single rotatable C–C bond between the two benzene rings. This feature makes the rotation around the biphenyl C–C bond possible, introducing a new degree of freedom. The free rotation of this bond may hinder membrane permeation. TCBP is also hydrophobic, but cannot form stable hydrogen bonds.
1.2 Numerical methods
We have performed both conventional and z-constrained39–42 MD simulations. All simulations were carried out using the GROMACS-5.0 program suite.43 Pressure and temperature were maintained at 1 atm and 325 K. For the DPPC molecules, we adopted the force field by Ulmschneider et al.,44 whereas the TIP3P one was used for water molecules.45 Suitable OPLS-AA parameters were used to develop the force fields for the solute molecules.46–52 Plain simulations were carried out for systems containing 1 (N1) or 10 (N10) solute molecules. In all cases, the MD input was assembled starting from a pre-equilibrated bilayer containing 128 DPPC molecules in water (3655 molecules).53 In order to give more room to the solute molecules, and prevent the bilayer to interact with its own top and bottom periodic images, the size of the simulation box along the z axis was doubled and filled with water molecules. Na+ and Cl− were inserted afterwards in order to obtain a physiological (0.15 M NaCl) solution. The simulation box was equilibrated within the NPT ensemble at 325 K, for 20 ns, in order to adjust the box size and correct the density. The final box size was about 6.5 × 6.5 × 11.0 nm3. A program written by our group was then used to insert the contaminants in the aqueous phase. This was accomplished by removing the minimum number of water molecules surrounding them.After this step, NPT production runs were performed for each system. For N1 systems, 3 to 5 independent NPT simulations were carried out in order to investigate the absorption dynamics. The resulting trajectories were used both to calculate structural and orientational parameters, and to generate the configurations to be used in z-constrained simulations (see below). For N10 systems, 3 production runs were performed to assess the dynamics of cluster formation and absorption and calculate the relevant descriptors. The overall duration of these simulations was between 50 and 200 ns, although in some cases (TCBP), runs longer than 500 ns were carried out.
Beside plain MD simulations, we performed z-constrained simulations by constraining the distance separating the solute and the bilayer center-of-mass along the z-coordinate. Five independent sets were assembled for each species, each made up by twenty-one equally spaced points (from 0 to 4 nm) along the z axis. For each position within each set, a 20 ns NPT simulation was performed to calculate the mean force, ![[F with combining macron]](https://www.rsc.org/images/entities/i_char_0046_0304.gif) (z), acting on the solute molecule. In order to improve the statistical convergence of the force estimates, two strategies were adopted. First, the simulation time of some “critical points” located in the polar headgroup region (between 1.8 and 2.4 nm) was extended from 20 to 40 ns in all the constrained runs. Additionally, the first 10 ns of each constrained run were considered as equilibration and neglected in the calculation of the average force and all related quantities. One additional test was also performed to verify that our z-constrained runs were not biased by the existence of poorly sampled regions along a coordinate orthogonal to the constrained one (see ESI† for details). The free energy of transfer (ΔG(z)) from the bulk water (z = 4 nm) to a given position along the bilayer normal (z) was evaluated as potential of the mean force (PMF):39,40
(z), acting on the solute molecule. In order to improve the statistical convergence of the force estimates, two strategies were adopted. First, the simulation time of some “critical points” located in the polar headgroup region (between 1.8 and 2.4 nm) was extended from 20 to 40 ns in all the constrained runs. Additionally, the first 10 ns of each constrained run were considered as equilibration and neglected in the calculation of the average force and all related quantities. One additional test was also performed to verify that our z-constrained runs were not biased by the existence of poorly sampled regions along a coordinate orthogonal to the constrained one (see ESI† for details). The free energy of transfer (ΔG(z)) from the bulk water (z = 4 nm) to a given position along the bilayer normal (z) was evaluated as potential of the mean force (PMF):39,40
 | (1) |
2 Results and discussion
2.1 Single-molecule simulations
We describe the results of our MD calculations, starting from the results of conventional simulations, e.g. unrestrained single-molecule simulations (N1, hereafter). All molecules were absorbed by the DPPC membrane with capture times varying between 10 and 80 ns. The capture times were different from one run to another. In most cases, the molecules were observed to repeatedly approach the membrane surface before absorption occurred. Once started, the absorption process took about 1–2 ns to complete, regardless of the molecule considered. Afterwards, the molecules were found to reach stable positions within the bilayer, according to their free energy minima (see below). Some simulations were also extended to check for the possibility of membrane crossing. This process, however, was never observed within the maximum simulation time considered in this work (about 100 ns). Numerical estimates of the translocation time will be discussed below. The trajectories extracted from these runs were used to calculate structural as well as orientational parameters, namely the membrane thickness, the area-per-lipid, the solute orientation with respect to the bilayer normal, and the distance between the solute and the bilayer center-of-mass. Regardless of the species considered, the membrane thickness was found to vary between 4.2 and 4.4 nm during the simulations. This result well compares with those published in the literature for pure DPPC membranes.30 No significant variation of this parameter was observed upon solute absorption. Similarly, the area-per-lipid was not significantly affected by solute permeation. Its values ranged between 0.5 and 0.7 nm2 per molecule. Fig. S1–S5 in the ESI† provide an example of the evolution of these descriptors over time (one NPT run for each solute molecule). These plots also display the evolution of the tilt angle (θ) between the solutes and the bilayer surface normal. In all cases, the molecules entering the membrane were preferentially oriented at relatively small θ. On the basis of these observations, we conclude that the differences in the structure of the solute molecules did not appreciably affect the mechanism of the absorption process. | ||
| Fig. 2 Plot of the free energies of transfer along the bilayer normal, with z = 0 corresponding to the bilayer center-of-mass. Standard errors are omitted. | ||
The profiles confirm that absorption is spontaneous for all the species considered. Crossing the polar headgroups was a barrier-free process, always followed by a steep gain in energy, demonstrating that the molecules preferred the hydrophobic part of the membrane to the water environment. As shown in Table 1, each molecule has its own energy minimum at slightly different positions inside the membrane.
| Molecule | ΔG(zmin) [kJ mol−1] | z min [nm] |
|---|---|---|
| a Standard errors are reported in parentheses. | ||
| TCDD | −51.58 (±2.59) | 1.19 |
| TCBP | −41.17 (±5.26) | 1.08 |
| THDD | −36.55 (±1.61) | 1.06 |
| ANTH | −32.84 (±2.48) | 1.10 |
| THDO | −29.96 (±3.17) | 1.17 |
The free energy minimum shows the ascending order: TCDD < TCBP < THDD < ANTH < THDO. These results indicate that the free energy change on transferring these molecules from water was higher for moieties containing chlorine (TCDD and TCBP), than for the less polar hydrocarbons ANTH and THDD. The result for TCDD agrees with our previous estimate (−43 ± 1.8 kJ mol−1), obtained for the TCDD/DPPC/water system, in the absence of NaCl. For THDO, the result is consistent with the presence of two hydroxyl groups. Once inside the membrane the molecules move more or less easily crossing the membrane centre. The energy barriers for this process show small magnitudes for all species (about 5 kJ mol−1), except for THDO (about 20 kJ mol−1). In fact, for this molecule crossing of the membrane centre was never observed during the simulations.
Fig. 3 compares the local diffusion coefficients, D(z), calculated for all species. The differences among the different species are limited. In bulk water, diffusion coefficients of the order of 2.5 × 10−5 cm2 s−1 were obtained. Approaching the membrane heads the diffusion coefficients decreased, reaching the minimum value inside the membrane, with an order of magnitude (10−6 cm2 s−1) consistent with the results of calculations on similar systems.32 The figure inset shows the small variations near the membrane centre: all the compounds show greater D(z) in the membrane center, where the alkyl tails are less dense. In addition, ANTH and THDD, the most lipophilic compounds, have slightly greater values than the other molecules.
 | ||
| Fig. 3 Plot of the diffusion coefficients along the bilayer normal. The inset magnifies the region between 0 and 2 nm. | ||
Fig. 4 shows the solute resistance profiles R(z) along the bilayer normal, calculated by combining the free energy profiles and the local diffusion coefficients:
 | (2) |
 | ||
| Fig. 4 Logarithmic plot of solute resistance profiles along the bilayer normal. | ||
On going from the membrane heads to the interior, eΔG(z)/kBT decreases and so does R(z). As expected, the minima of R(z) are very close to those of ΔG(z). Substantial differences in R(z) values can be noticed across the different species, TCDD showing a minimum value about two orders of magnitude smaller than its congeners. Approaching the membrane center, R(z) moderately increases for all compounds except THDO, for which a thousand-fold increase was observed.
The permeability coefficient of a membrane, hereafter abbreviated as P, is generally defined as the proportionality factor linking the steady-state flux of molecules per unit area (J) to the concentration difference between its two sides: J = P(Cout − Cin). Using this equation to estimate P in atomistic simulations is unfeasible, due to the limited time scale accessible to MD. According to the inhomogeneous solubility-diffusion model,40,55–57P can be more conveniently obtained from R(z) as
 | (3) |
| Molecule | P [cm s−1] |
|---|---|
| a Standard errors are reported in parentheses. | |
| TCBP | 12.4 (±4.3) |
| ANTH | 17.0 (±3.6) |
| THDD | 23.8 (±4.2) |
| THDO | 26.8 (±5.3) |
| TCDD | 29.6 (±3.5) |
The values of P follow the ascending order: TCBP < ANTH < THDD < THDO < TCDD. For TCBP the result is dominated by the high solute resistance in the polar headgroup region. For the remaining molecules, the differences in R(z) are less relevant; ANTH and THDD have similar values. The position of THDO, intermediate between THDD and TCDD, can be rationalized considering the presence of two hydroxyl groups. In an apolar environment, such as the lipophilic tails of DPPC, the two hydroxyl groups make an intramolecular H-bond that decreases the lipophobicity of the compound; this effect is well-known in the literature.32 It should be noted that, especially for THDO and TCDD, significant differences in P were observed on going from one set to another, as testified by the standard deviations. In any case, our results suggest that, among the compounds considered, TCDD is the one with the strongest interactions with the membrane.
As mentioned above, no species was observed to cross the DPPC bilayer within the time scale explored by our simulations. In order to get a measure of the time required for this process to occur, we used the average force to estimate the translocation time (τ) assuming Brownian motion. It should be noted that the assumption of Brownian motion not necessarily hold in cell membranes, and therefore, this calculation only provides an order-of-magnitude estimate of τ. Table 3 collects the translocation times for all species, calculated according to the following equation:34
 | (4) |
represents the average force, while ![[D with combining macron]](https://www.rsc.org/images/entities/i_char_0044_0304.gif) the average value of D(z), calculated taking into account the location of the molecules after the translocation process (see the ESI† for details).
the average value of D(z), calculated taking into account the location of the molecules after the translocation process (see the ESI† for details).
| Molecule | τ [s] |
|---|---|
| TCDD | 6.21 |
| TCBP | 0.29 |
| THDD | 1.38 × 10−2 |
| ANTH | 2.97 × 10−3 |
| THDO | 1.44 × 10−3 |
TCDD and TCBP show the highest τ values, and therefore, tend to reside within the membrane longer than the other species considered. This is consistent with the fact these species are more stabilized by the interactions with the membrane tails (see the free energy minima in Fig. 2). On going from these molecules to the chlorine-free ones, the translocation times become two or three orders of magnitude smaller. For THDO, the translocation time is the smallest. This apparently contrasts with the increase of R(z) approaching the membrane centre. At the same time, however, the THDO resistance maximum is lower than the other molecules, finally resulting in a faster transport. We should also note that the translocation time (1440 μs) is anyhow far beyond the time scale that we reached with unconstrained MD simulations.
2.2 Many-molecule simulations
In our previous paper, we reported the results of MD studies performed on TCDD clusters made up of 1, 2, 5, and 10 molecules. In all cases, we observed TCDD aggregation to form molecular clusters. The smaller clusters could form and break up many times during an NPT simulation. Clusters consisting of 10 molecules were long lasting and very stable. In addition, we found that the absorption of one molecule belonging to the cluster immediately resulted in the sequential absorption of all other molecules.Taking into account these outcomes, in the present study we considered N10 systems, namely systems consisting of 10 solute molecules in a physiological solution with the DPPC membrane. We performed three independent NPT runs for each species, including TCDD. The solute molecules were initially placed far apart, in order to test the possibility of spontaneous aggregation. Since, except for THDO, all molecules were hydrophobic, we expected the clusters to show the same behavior as TCDD ones, in terms of cluster formation and cooperative absorption. The results we obtained were partly unexpected.
Both TCDD and TCBP rapidly formed clusters. Once formed, the clusters were stable and we did not observe any occurrence of cluster breakdown. The clusters entered the membrane at different times. For TCDD the dynamics of cluster absorption was similar to that observed in our previous work: the molecules belonging to the cluster were absorbed sequentially, within a relatively short time (about 60 ns in two runs). TCBP clusters, in contrast, required much more time to enter the membrane. In fact, the TCBP cluster was absorbed by the membrane only in one simulation. In both remaining runs, each extended well beyond 500 ns, we never observed TCBP absorption. The solute resistance profile of the isolated molecule provides a qualitative rationale for this outcome, although it does not account for the interactions among TCBP molecules. An analysis of the geometrical properties of TCBP clusters has evidenced the tendency of TCBP clusters to keep an average distance from the membrane heads larger than that of the other molecules. We shall return to this point below.
ANTH, THDD, and THDO also formed clusters. These, however, were characterized by a highly variable composition. As a consequence, the molecules entered the membrane at different times, often isolated, or sequentially like TCDD. ANTH and THDD had similar behaviours. We observed the clusters to form and separate repeatedly during the simulations. Often, the molecules were absorbed individually, sometimes leaving the cluster in proximity of the membrane. As mentioned above, THDO can form H-bonds, and has strong interactions with the membrane headgroups. Once formed, the whole THDO cluster was observed to spend a long time at the bilayer surface, before entering the membrane. Fig. 5 compares THDO (top) and TCDD absorption (bottom).
 | ||
| Fig. 5 Distances of the cluster components as a function of MD time. | ||
Regardless of the species considered, all clusters rapidly broke down into separate molecules after absorption. Fig. 6 gives an example of the final distribution of the molecules for ANTH (left), and THDO (right). ANTH molecules were widely distributed all over the membrane interior. The same was also observed for TCDD, THDD and TCBP. THDO molecules, conversely, were preferentially located close to the membrane polar heads. In this figure we note that THDO molecules entered the membrane from both sides due to system periodicity. However, as for the N1 system, crossing of the membrane centre was never observed during the simulations.
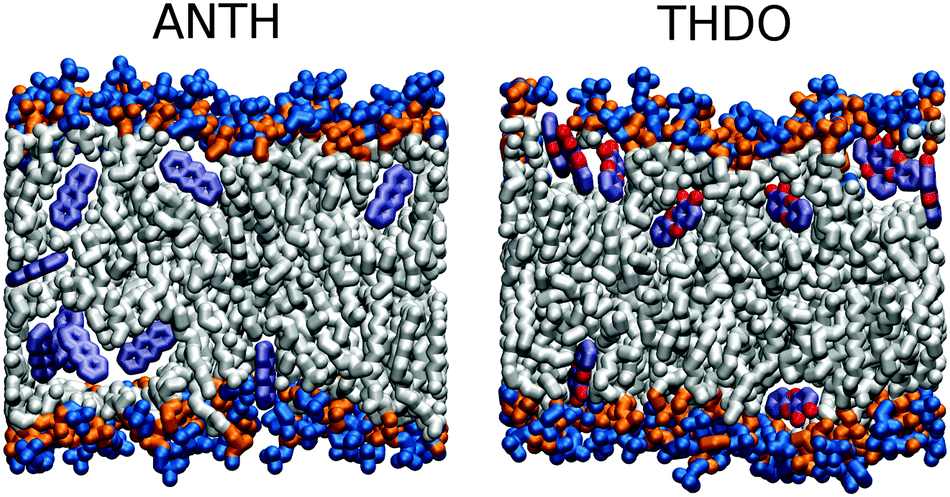 | ||
| Fig. 6 Example snapshots of the MD trajectories of ANTH and THDO. Some atoms in the phospholipid tails have been removed to improve visualization. | ||
To better understand the different behaviour of the clusters in the water phase, we performed an analysis of their stability. In order to qualitatively judge cluster stability, we calculated the Coulomb and Lennard-Jones intermolecular energies associated with the removal of one molecule from the largest cluster. In order to simulate the effect of this process, for a cluster comprising N molecules, we calculated the difference between two intermolecular energies, that of the cluster, namely EN, and that obtained by averaging the energies of the N sub-clusters (EN−1, hereafter), containing N − 1 molecules. The sub-clusters were obtained by deleting in turn each molecule from the cluster. DPPC, water, and ions, were not considered in the calculation. The calculation was performed on 500 MD frames extracted from the simulations, where the clusters were present as whole aggregates (10 molecules), with the exception of ANTH where the maximum cluster size was 9. See the ESI† for details of the selection algorithm. For each species, and each MD frame, the energy difference ΔEN, was calculated as
 | (5) |
| Molecule | ΔEN(L) [kJ mol−1] | ΔEN(C) [kJ mol−1] | ΔEN(T) [kJ mol−1] |
|---|---|---|---|
| a Standard errors are reported in parentheses. | |||
| TCDD | −120.5 (±34.4) | −9.8 (±3.9) | −130.3 (±34.9) |
| TCBP | −91.3 (±25.5) | 5.4 (±2.9) | −85.9 (±25.9) |
| THDO | −83.1 (±22.2) | −77.6 (±20.7) | −160.7 (±31.8) |
| THDD | −72.1 (±21.3) | −0.5 (±2.2) | −72.5 (±21.4) |
| ANTH | −64.4 (±23.0) | −0.2 (±5.0) | −64.6 (±26.4) |
Lennard-Jones interactions always increased cluster stability. The stability order according to ΔEN(L) is: TCDD < TCBP < THDO < THDD < ANTH. This order is consistent with the outcome of the MD simulations: TCBP and TCDD formed stable clusters. THDO exhibits a similar behaviour, however, the interactions with water and with the polar headgroups promoted cluster break-down. ANTH and THDD clusters are much less stable, and their size changed continuously during the simulation. The Coulomb energy contribution decreases in the following order: THDO > TCDD > THDD > ANTH > TCBP. As expected, only for THDO this type of interaction had a role in cluster stability. For all the remaining compounds, the contribution of electrostatic interactions to cluster stability was far less appreciable.
Even considering that the calculation is not a quantitative measure of the cluster stability, these results well agree with the inspection of the MD trajectories. Hydrophobicity is not the sole factor behind cluster formation. Intermolecular interactions between the clustered molecules also play an important role. To propose a rationale to the difficulty encountered by TCBP clusters in entering the membrane, we analyzed the values of the distance between the cluster geometrical centre and the bilayer centre. As a representative for this distance we considered the distance of each molecule from the membrane center along the z coordinate. For each species, 500 MD frames were considered, the same used in the calculation of the stabilization energies (see above). As mentioned above, in all these frames, the clusters were present as whole aggregates and were located in the water phase.
Fig. 7 shows the distribution of distances for each compound. It is easy to note that the minimum distance of TCBP from the membrane center rarely falls below 3 nm. As discussed above, both ΔG(z) and R(z) show that the interaction between the molecules and the membrane just takes place starting at ∼2.8 nm. Hence, the TCBP cluster has statistically less chances to penetrate the membrane.
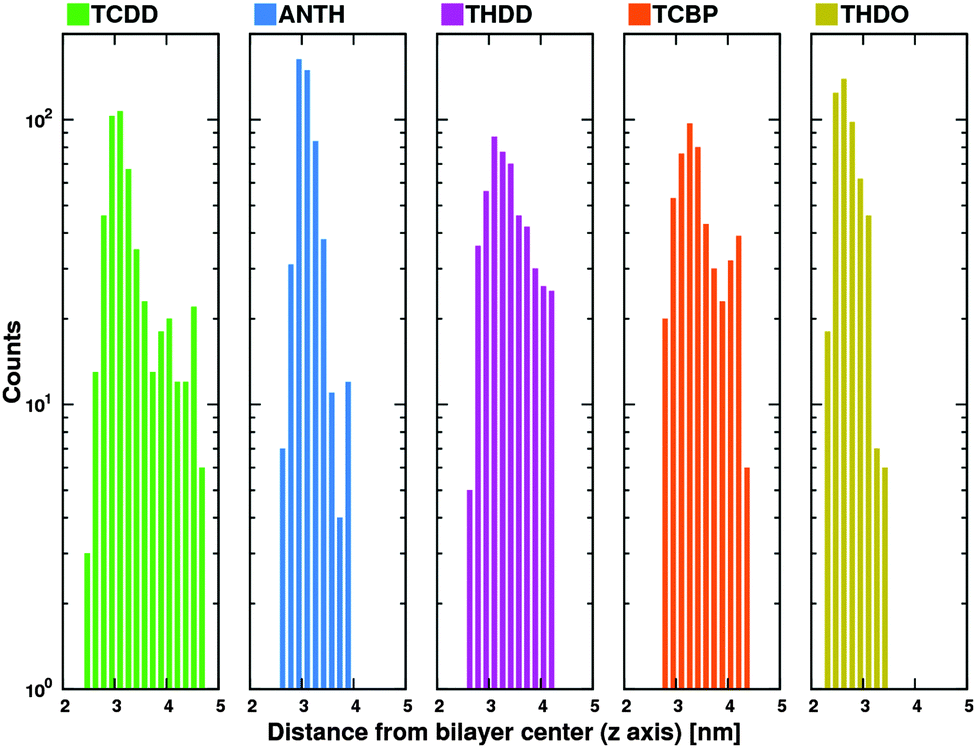 | ||
| Fig. 7 Distribution of distances between cluster and membrane centers along the z axis. The line height represents the number of occurrences. | ||
3 Summary and conclusions
In this work, we used molecular dynamics to compare the absorption dynamics of TCDD in DPPC bilayers with a group of four structurally similar congeners. These were obtained by modifications of the TCDD structure with respect to its key structural features: aromatic character, planarity, presence of chlorine atoms, and hydrophobicity.The results of single-molecule simulations have evidenced some aspects of the interaction between the solute molecules and the membrane that can be rationalized on the basis of the structural properties. We have shown that TCDD and its congeners share some features: they spontaneously enter the membrane, their free energies of transfer indicate the presence of energy minima inside the membrane. Once inside the membrane, the molecules settle preferentially at 1 nm from the membrane center, although wide displacements may occasionally occur, leading to membrane center crossing. Beside the analogies, some important differences also emerge from our results. ANTH and THDD are hydrophobic molecules that similarly interact with the membrane. The presence of chlorine atoms has a different impact on the absorption behavior. Both TCDD and TCBP show deep energy minima and freely move inside the membrane, in agreement with their translocation times. However, the other calculated variables discriminate the two compounds: TCDD has the highest permeability coefficient and TCBP the lowest. TCBP has the highest solute resistance upon crossing the polar headgroups. These differences are likely related to the rotational freedom about the biphenyl dihedral. Due to the presence of two hydroxyl groups, THDO is the least lipophilic compound, the sole compound that can accept and donate hydrogen bonds, which can stabilize intermolecular interactions. This is confirmed by the significant interactions with the membrane heads and the difficulty in crossing the membrane centre. Although not affecting the absorption mechanism, the presence of hydroxyl groups increases the permeability and decreases the translocation time.
The results obtained for clusters revealed some unexpected outcomes, often related to cluster formation in bulk water. Despite the hydrophobicity of all compounds, only TCDD and TCBP formed large and stable clusters made up of all solute molecules. Consistently with our previous study, TCDD clusters were absorbed quickly. By contrast, the absorption of TCBP clusters required longer times and was less frequently observed. The clusters formed by the other species, especially ANTH, were characterized by highly variable compositions. The behaviour in the water phase had important consequences on the absorption dynamics, since poorly clustered molecules were often individually absorbed. The importance of intermolecular interactions in the cluster formation and absorption was also evidenced by the analysis of cluster stability.
Eventually, compared to its congeners, TCDD emerged as the species with the highest tendency to penetrate the DPPC bilayer and accumulate in its interior. Due to the high complexity of real biological membranes in comparison with our model system, this result is only a first step toward a better understanding of the absorption and transport mechanism of TCDD into cells that represent a fundamental requisite for its toxic action.
It is clear that the compounds considered in this study only cover a fraction of the chemical space spanned by the structural moieties of TCDD. A thorough study on the effect of some functional groups on membrane absorption would have required more congeners. In this respect, however, we note that the calculation of the free energy of transfer and the related properties via z-constrained MD simulations was very CPU demanding (about 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hours on a Tier-1 supercomputer58), despite the limited number of compounds considered. Besides, z-constrained MD offers some advantages with respect to other methods, in that it has been validated,31,40,59,60 and successfully applied to quite many solute/membrane systems.31,32,60–63 In view of the extension to more complex systems, we are currently testing other approaches, such as meta-dynamics64–68 and adaptive biasing force.69–71 The inclusion of polarization effects72,73 would additionally be considered as a major step toward a more realistic description of the molecular interactions.
000 hours on a Tier-1 supercomputer58), despite the limited number of compounds considered. Besides, z-constrained MD offers some advantages with respect to other methods, in that it has been validated,31,40,59,60 and successfully applied to quite many solute/membrane systems.31,32,60–63 In view of the extension to more complex systems, we are currently testing other approaches, such as meta-dynamics64–68 and adaptive biasing force.69–71 The inclusion of polarization effects72,73 would additionally be considered as a major step toward a more realistic description of the molecular interactions.
Acknowledgements
G. S. received partial funding through computational time at CINECA (IscraC project MASCRADD) and the Università degli Studi di Milano.References
- D. E.-C. U. S. Environmental Protection Agency, Washington, U.S. EPA. Exposure and Human Health Reassessment of 2,3,7,8-Tetrachlorodibenzo-P-Dioxin (TCDD) and Related Compounds National Academy Sciences (External Review Draft) (2004), 2004.
- L. Zhang, W. Yang, L. Zhang and X. Li, Chemosphere, 2015, 133, 1–5 CrossRef CAS PubMed.
- E. N. Sappington, A. Balasubramani and H. S. Rifai, Chemosphere, 2015, 133, 82–89 CrossRef CAS PubMed.
- F. Jeanneret, D. Tonoli, D. Hochstrasser, J.-H. Saurat, O. Sorg, J. Boccard and S. Rudaz, Toxicol. Lett., 2016, 240, 22–31 CrossRef CAS PubMed.
- R. Hoogenboom, W. Traag, A. Fernandes and M. Rose, Food Control, 2015, 50, 670–683 CrossRef CAS.
- P. A. Bertazzi, I. Bernucci, G. Brambilla, D. Consonni and A. C. Pesatori, Environ. Health Perspect., 1998, 106, 625–633 CrossRef CAS PubMed.
- A. C. Pesatori, D. Consonni, M. Rubagotti, P. Grillo and P. A. Bertazzi, Environ. Health, 2009, 8(39), 1–11 Search PubMed.
- M. Warner, P. Mocarelli, S. Samuels, L. Needham, P. Brambilla and B. Eskenazi, Environ. Health Perspect., 2011, 119, 1700–1705 CrossRef CAS PubMed.
- P. Boffetta, K. A. Mundt, H.-O. Adami, P. Cole and J. S. Mandel, Crit. Rev. Toxicol., 2011, 41, 622–636 CrossRef PubMed.
- K. Steenland, P. Bertazzi, A. Baccarelli and M. Kogevinas, Environ. Health Perspect., 2004, 112, 1265–1268 CrossRef CAS PubMed.
- G. Biswas, S. Srinivasan, H. K. Anandatheerthavarada and N. G. Avadhani, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 186–191 CrossRef CAS PubMed.
- A. Baccarelli, A. C. Pesatori, S. A. Masten, D. G. Patterson Jr., L. L. Needham, P. Mocarelli, N. E. Caporaso, D. Consonni, J. A. Grassman, P. A. Bertazzi and M. T. Landi, Toxicol. Lett., 2004, 149, 287–293 CrossRef CAS PubMed.
- J. P. Whitlock, Chem. Res. Toxicol., 1993, 6, 754–763 CrossRef CAS PubMed.
- S. B. Tavakoly Sany, R. Hashim, A. Salleh, M. Rezayi, D. J. Karlen, B. B. M. Razavizadeh and E. Abouzari-lotf, Environ. Sci. Pollut. Res., 2015, 22, 19434–19450 CrossRef CAS PubMed.
- A. Kazlauskas, L. Poellinger and I. Pongratz, J. Biol. Chem., 1999, 274, 13519–13524 CrossRef CAS PubMed.
- T. Ikuta, H. Eguchi, T. Tachibana, Y. Yoneda and K. Kawajiri, J. Biol. Chem., 1998, 273, 2895–2904 CrossRef CAS PubMed.
- H. Saibil, Nat. Rev. Mol. Cell Biol., 2013, 14, 630–642 CrossRef CAS PubMed.
- J. Mimura and Y. Fujii-Kuriyama, Biochim. Biophys. Acta, Gen. Subj., 2003, 1619, 263–268 CrossRef CAS.
- M. S. Denison, A. A. Soshilov, G. He, D. E. DeGroot and B. Zhao, Toxicol. Sci., 2011, 124, 1–22 CrossRef CAS PubMed.
- M. Nukaya, B. C. Lin, E. Glover, S. M. Moran, G. D. Kennedy and C. A. Bradfield, J. Biol. Chem., 2010, 285, 35599–35605 CrossRef CAS PubMed.
- M. L. Merrill, C. Emond, M. J. Kimi, J.-P. Antignaci, B. L. Bizeci, K. C. L. S. Birnbaum and R. Barouki, Environ. Health Perspect., 2013, 121, 162–169 Search PubMed.
- S. M. Regnier and R. M. Sargis, Biochim. Biophys. Acta, Mol. Basis Dis., 2014, 1842, 520–533 CrossRef CAS PubMed.
- L. Daniel and S. Kai, Science, 2010, 327, 46–50 CrossRef PubMed.
- A. A. Gurtovenko, J. Anwar and I. Vattulainen, Chem. Rev., 2010, 110, 6077–6103 CrossRef CAS PubMed.
- M. L. Berkowitz, D. L. Bostick and S. Pandit, Chem. Rev., 2006, 106, 1527–1539 CrossRef CAS PubMed.
- S. M. Loverde, J. Phys. Chem. Lett., 2014, 5, 1659–1665 CrossRef CAS PubMed.
- S. Riahi and C. N. Rowley, J. Am. Chem. Soc., 2014, 136, 15111–15113 CrossRef CAS PubMed.
- M. Paloncýová, K. Berka and M. Otyepka, J. Phys. Chem. B, 2013, 117, 2403–2410 CrossRef PubMed.
- N. A. Krylov, V. M. Pentkovsky and R. G. Efremov, ACS Nano, 2013, 7, 9428–9442 CrossRef CAS PubMed.
- W.-E. Jirasak, B. Svetlana, T. Wannapong, T. I-Ming, T. D. Peter and M. Luca, Nat. Nanotechnol., 2008, 3, 363–368 CrossRef PubMed.
- M. Orsi, W. E. Sanderson and J. W. Essex, J. Phys. Chem. B, 2009, 113, 12019–12029 CrossRef CAS PubMed.
- D. Bemporad, C. Luttmann and J. Essex, Biophys. J., 2004, 87, 1–13 CrossRef CAS PubMed.
- R. Guo, J. Mao and L.-T. Yan, Biomaterials, 2013, 34, 4296–4301 CrossRef CAS PubMed.
- R. Qiao, A. P. Roberts, A. S. Mount, S. J. Klaine and P. C. Ke, Nano Lett., 2007, 7, 614–619 CrossRef CAS PubMed.
- M. Casalegno, G. Raos and G. Sello, Phys. Chem. Chem. Phys., 2015, 17, 2344–2348 RSC.
- M. Schiavon, V. Torretta, E. C. Rada and M. Ragazzi, Environ. Monit. Assess., 2015, 188, 1–20 Search PubMed.
- E. N. Sappington, A. Balasubramani and H. S. Rifai, Chemosphere, 2015, 133, 82–89 CrossRef CAS PubMed.
- M. M. Pratt, K. John, A. B. MacLean, S. Afework, D. H. Phillips and M. C. Poirier, Int. J. Environ. Res. Public Health, 2011, 8, 2675–2691 CrossRef CAS PubMed.
- S. J. Marrink and H. J. C. Berendsen, J. Phys. Chem., 1996, 100, 16729–16738 CrossRef CAS.
- S. J. Marrink and H. J. C. Berendsen, J. Phys. Chem., 1994, 98, 4155–4168 CrossRef CAS.
- B. Roux and M. Karplus, Biophys. J., 1991, 59, 961–981 CrossRef CAS PubMed.
- B. Roux and M. Karplus, J. Phys. Chem., 1991, 95, 4856–4868 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- J. P. Ulmschneider and M. B. Ulmschneider, J. Chem. Theory Comput., 2009, 5, 1803–1813 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- W. L. Jorgensen and N. A. McDonald, THEOCHEM, 1998, 424, 145–155 CrossRef CAS.
- N. A. McDonald and W. L. Jorgensen, J. Phys. Chem. B, 1998, 102, 8049–8059 CrossRef CAS.
- R. C. Rizzo and W. L. Jorgensen, J. Am. Chem. Soc., 1999, 121, 4827–4836 CrossRef CAS.
- M. L. P. Price, D. Ostrovsky and W. L. Jorgensen, J. Comput. Chem., 2001, 22, 1340–1352 CrossRef CAS.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- C. Caleman, P. J. van Maaren, M. Hong, J. S. Hub, L. T. Costa and D. van der Spoel, J. Chem. Theory Comput., 2012, 8, 61–74 CrossRef CAS PubMed.
- J. Domański, P. J. Stansfeld, M. S. P. Sansom and O. Beckstein, J. Membr. Biol., 2010, 236, 255–258 CrossRef PubMed.
- R. H. Bartels, J. C. Beatty and B. A. Barsky, An introduction to splines for use in computer graphics and geometric modelling, Morgan Kaufmann, 1998, pp. 211–245 Search PubMed.
- T. X. Xiang and B. D. Anderson, J. Membr. Biol., 1994, 140, 111–122 CrossRef CAS PubMed.
- T. X. Xiang, Y.-H. Xu and B. D. Anderson, J. Membr. Biol., 1998, 165, 77–90 CrossRef CAS PubMed.
- T. X. Xiang and B. D. Anderson, J. Membr. Biol., 2000, 173, 187–201 CrossRef CAS PubMed.
- IBM NeXtScale at CINECA-SCAI, Bologna, Italy, http://www.hpc.cineca.it, last access: 2016-02-29.
- M. Paloncýová, K. Berka and M. Otyepka, J. Chem. Theory Comput., 2012, 8, 1200–1211 CrossRef PubMed.
- C. Neale, W. F. D. Bennett, D. P. Tieleman and R. Pomès, J. Chem. Theory Comput., 2011, 7, 4175–4188 CrossRef CAS PubMed.
- J. J. L. Cascales, S. D. O. Costa and R. D. Porasso, J. Chem. Phys., 2011, 135, 135103 CrossRef PubMed.
- J. L. MacCallum, W. F. D. Bennett and D. P. Tieleman, Biophys. J., 2008, 94, 3393–3404 CrossRef CAS PubMed.
- N. Sapay, W. F. D. Bennett and D. P. Tieleman, Soft Matter, 2009, 5, 3295–3302 RSC.
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2008, 100, 020603 CrossRef PubMed.
- M. Bonomi and M. Parrinello, Phys. Rev. Lett., 2010, 104, 190601 CrossRef CAS PubMed.
- D. Branduardi, F. L. Gervasio and M. Parrinello, J. Chem. Phys., 2007, 126, 054103 CrossRef PubMed.
- C. Micheletti, A. Laio and M. Parrinello, Phys. Rev. Lett., 2004, 92, 170601 CrossRef PubMed.
- J. P. M. Jämbeck and A. P. Lyubartsev, J. Phys. Chem. Lett., 2013, 4, 1781–1787 CrossRef PubMed.
- E. Darve and A. Pohorille, J. Chem. Phys., 2001, 115, 9169–9183 CrossRef CAS.
- E. Darve, D. Rodríguez-Gómez and A. Pohorille, J. Chem. Phys., 2008, 128, 144120 CrossRef PubMed.
- J. Hénin, G. Fiorin, C. Chipot and M. L. Klein, J. Chem. Theory Comput., 2010, 6, 35–47 CrossRef PubMed.
- J. P. M. Jämbeck and A. P. Lyubartsev, Phys. Chem. Chem. Phys., 2013, 15, 4677–4686 RSC.
- I. Vorobyov, W. D. Bennett, D. P. Tieleman, T. W. Allen and S. Noskov, J. Chem. Theory Comput., 2012, 8, 618–628 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp01728b |
| This journal is © the Owner Societies 2016 |