
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gas phase complexes of H3N⋯CuF and H3N⋯CuI studied by rotational spectroscopy and ab initio calculations: the effect of X (X = F, Cl, Br, I) in OC⋯CuX and H3N⋯CuX†
Dror M.
Bittner
a,
Susanna L.
Stephens‡
a,
Daniel P.
Zaleski§
a,
David P.
Tew
b,
Nicholas R.
Walker
*a and
Anthony C.
Legon
*b
aSchool of Chemistry, Bedson Building, Newcastle University, Newcastle upon Tyne, Tyne and Wear NE1 7RU, UK. E-mail: nick.walker@newcastle.ac.uk
bSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: a.c.legon@bristol.ac.uk
First published on 19th April 2016
Abstract
Complexes of H3N⋯CuF and H3N⋯CuI have been synthesised in the gas phase and characterized by microwave spectroscopy. The rotational spectra of 4 isotopologues of H3N⋯CuF and 5 isotopologues of H3N⋯CuI have been measured in the 6.5–18.5 GHz frequency range using a chirped-pulse Fourier transform microwave spectrometer. Each complex is generated from a gas sample containing NH3 and a halogen-containing precursor diluted in Ar. Copper is introduced by laser ablation of a solid target prior to supersonic expansion of the sample into the vacuum chamber of the microwave spectrometer. The spectrum of each complex is characteristic of a symmetric rotor and a C3v geometry in which the N, Cu and X atoms (where X is F or I) lie on the Ca3 axis. The rotational constant (B0), centrifugal distortion constants (DJ and DJK), nuclear spin-rotation (Cbb(Cu) = Ccc(Cu)) constant (for H3N⋯CuF only) and nuclear quadrupole coupling constants (χaa(X) where (X = N, Cu, I)) are fitted to the observed transition frequencies. Structural parameters are determined from the measured rotational constants and also calculated ab initio at the CCSD(T)(F12*)/AVQZ level of theory. Force constants describing the interaction between ammonia and each metal halide are determined from DJ for each complex. Trends in the interaction strengths and geometries of B⋯CuX (B = NH3, CO) (X = F, Cl, Br, I) are discussed.
1. Introduction
Gerry and co-workers demonstrated that B⋯MX complexes, where B is a Lewis base, M is a coinage metal, and X is a halogen atom, can be generated through laser vaporisation of a metal target in the presence of a gas sample undergoing supersonic expansion, then interrogated by microwave spectroscopy.1 The pure rotational spectra of OC⋯MX (X = F, Cl, Br) were measured and interpreted to determine the geometries of the complexes. An extensive and systematic study of the rotational spectra of B⋯MX complexes, also generated by a combination of laser vaporisation and supersonic expansion, has since been performed. Complexes where the Lewis base is H2,2,3 N2,4 CO,1,5,6 H2O,7 H2S,8 NH3,9,10 C2H2,11,12 C2H4,13,14 or c-C3H615 have been reported. Metal-containing (B⋯MX) complexes have also been investigated by ab initio methods which have explored the nature of bonding interactions and the influence of the halide on the interaction between the metal and the Lewis base.16–19This work presents analyses of the rotational spectra of H3N⋯CuI and H3N⋯CuF. The structures of the complexes as well as the strengths of interactions between NH3 and the different CuX (X = F, I) molecules are determined from measured spectroscopic constants and through ab initio calculations. Unlike H3N⋯CuCl, for which a crystal structure20 was known before its observation in the gas phase, H3N⋯CuI and H3N⋯CuF have not been structurally characterised in the condensed phase. The reaction of copper(II) fluoride and copper metal with ammonia has been observed both in gaseous and in anhydrous liquid ammonia.21 Among the products obtained were copper(I) fluoride and copper(II) fluoride ammine complexes. It has been suggested that copper(I) fluoride ammine complexes may serve as intermediates that allow the isolation of binary copper(I) fluoride which has not yet been characterized in the solid phase. The results of the present work are compared with those of the previously published reports on H3N⋯CuCl10 and OC⋯CuX (X = F, Cl, Br, I).1,22 Clear trends are identified and described. Legon and co-workers earlier performed systematic studies of hydrogen- and halogen-bonded complexes, B⋯HX23 and B⋯XY24 (where Y is a halogen atom). The present work compares the properties of H3N⋯MX with selected H3N⋯HX and H3N⋯XY complexes that have been characterised previously.
2. Experimental and theoretical methods
Broadband microwave spectra were measured using a chirped-pulse Fourier-transform microwave (CP-FTMW) spectrometer fitted with a laser ablation source. Detailed descriptions of the spectrometer and laser ablation source are provided in ref. 25 and 26. A gas sample containing ∼4.0% NH3 and ∼1.5% SF6 (when generating H3N⋯CuF) or CF3I (when generating H3N⋯CuI) is diluted in argon and prepared at a total pressure of 6 bar. The sample is pulsed (from a Parker-Hannifin Series 9 solenoid valve, using pulse length of 960 μs set using Parker-Hannifin Iota One Valve Driver) into the vacuum chamber of the spectrometer and passes over the surface of a copper rod that is ablated by a Nd:YAG laser pulse (λ = 532 nm, pulse duration of 10 ns, pulse energy of 20 mJ) before undergoing supersonic expansion. A repetition rate of ∼1.05 Hz is employed. The copper rod is continually translated and rotated in order to expose a fresh surface to each laser pulse and to ensure shot-to-shot reproducibility of signal intensities. Isotopically-enriched samples of 15NH3 (Sigma-Aldrich, 98% 15N) and ND3 (Sigma-Aldrich, 99% D) were used to detect 15NH3- and ND3-containing complexes respectively.The sequence employed to record broadband microwave spectra involves (i) polarization of the sample by a microwave chirp that sweeps from 6.5 to 18.5 GHz within 1 μs and (ii) recording of the free induction decay of the molecular emission over a subsequent period of 20 μs. The sequence of (i) and (ii) is repeated eight times following each gas sample introduction pulse. The free induction decay (FID) of the polarization is digitized using a 25 Gs s−1 digital oscilloscope after down-mixing against a 19 GHz local oscillator. Frequency domain spectra are obtained through a Fourier transform that uses the high resolution window function (full width at half maximum (FWHM) ∼ 65 Hz) described by Tektronix (details provided as ESI†). The spectra of H3N⋯63/65CuF, H315N⋯CuF and D3N⋯63CuF were averaged for 1500k, 540k and 2520k free induction decays (FIDs) respectively prior to being Fourier transformed. The spectra of H3N⋯63/65CuI, H315N⋯CuI and D3N⋯63/65CuI were averaged for 660k, 180k and 540k FIDs respectively prior to being Fourier transformed.
Structure optimizations and counter-poise corrected dissociation energies were calculated using the molpro package27 at the CCSD(T)(F12*) level of theory,28 a coupled-cluster method with single and double excitations, explicit correlation,29 and a perturbative treatment of triple excitations.30 Only valence electrons are included in the correlation treatment. A basis set combination consisting of aug-cc-pVQZ on H, N and F atoms and aug-cc-pVQZ-PP on Cu and I atoms was used and will be referred to by AVQZ. ECP-10-MDF31,32 and ECP-28-MDF33,34 were used on Cu and I respectively to account for scalar relativistic effects. For the density fitting approximation used to accelerate the CCSD(T)(F12*) calculation, the respective def2-QZVPP basis sets were employed for the MP235 and Fock36 terms. For the complementary auxiliary basis required for the F12 treatment,37 the def2-TZVPP MP2 density fitting basis sets were used.38 Force constants were calculated from the second derivative of the energy with respect to the internal coordinates using the GAUSSIAN 09 package39 at the MP2 level of theory. A basis set combination consisting of aug-cc-pVTZ on H, N and F atoms, aug-cc-pVTZ-PP on Cu and I atoms was used.
3. Results
3.1 Spectral analysis and assignment
Fig. 1 displays sections of the broadband spectrum measured when probing a gas sample containing CF3I, NH3 and argon that interacts with the plasma plume produced by ablation of the copper rod. The most intense transitions are observed for CF3I40 while those of NH3⋯Ar,41 CF3I⋯NH3,42 IF43 and CuI44 are also strong. The spectrum of H3N⋯CuI was identified through the analysis of groups of transitions, significantly weaker in intensity than those of the species listed above, which are separated by frequency increments of ∼2.2 GHz. Distinctive and extensive hyperfine splittings are present in each J′ → J′′ transition as expected for a complex that contains copper and iodine nuclei (which have I = 3/2 and 5/2 respectively). A different range of chemical products was observed, including NH3⋯SF6,45 NH3⋯Ar41 and (NH3)246 after exchanging the gas sample for another that contains SF6, NH3 and argon. The frequency of the J′ → J′′ = 1 → 0 transition of CuF lies above the upper limit of the spectrometer. Consecutive J′ → J′′ transitions of H3N⋯CuF, which have lower intensities than those of NH3⋯SF6, NH3⋯Ar and (NH3)2, were observed at intervals of ∼8.0 GHz and display hyperfine structure arising from the presence of the copper nucleus (I = 3/2). The J′ → J′′ = 1 → 0 and 2 → 1 transitions of H3N⋯CuF are shown in Fig. 2. The spectra observed for each of H3N⋯CuF and H3N⋯CuI contain only a-type transitions, consistent with the C3v geometries (Fig. 3) anticipated for each of these complexes and previously observed for each of H3N⋯AgCl9 and H3N⋯CuCl.10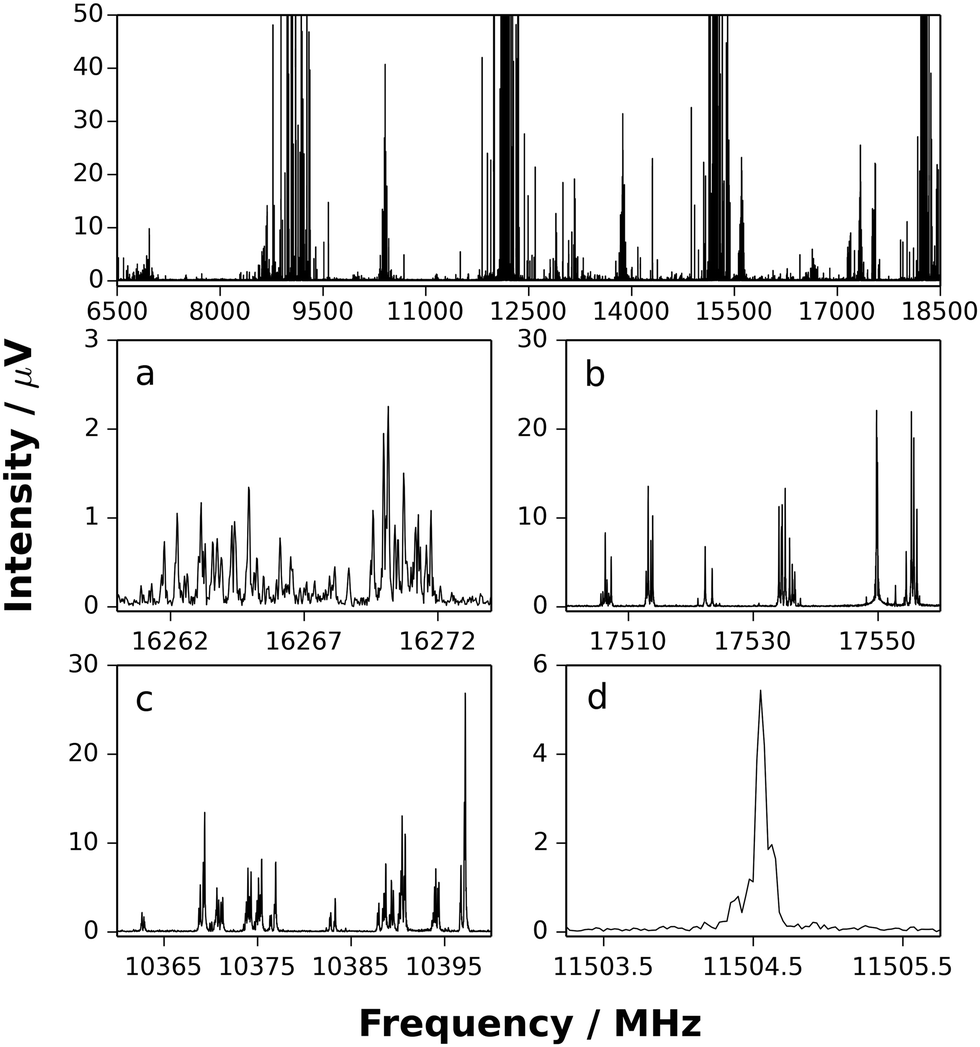 | ||
| Fig. 1 Top panel: The broadband rotational spectrum (660k FIDs) obtained while using a gas sample containing small concentrations of CF3I and NH3 diluted in argon. (a) J′ → J′′ = 7 → 6 transitions of H3N⋯63CuI. (b) J′ → J′′′ = 4 → 3 transitions of 63CuI. (c) J′ → J′′ = 6 → 5 transitions of H3N⋯ICF3. (d) J′ → J′′ = 2 → 1 transition in the Σ0a state of NH3⋯Ar. | ||
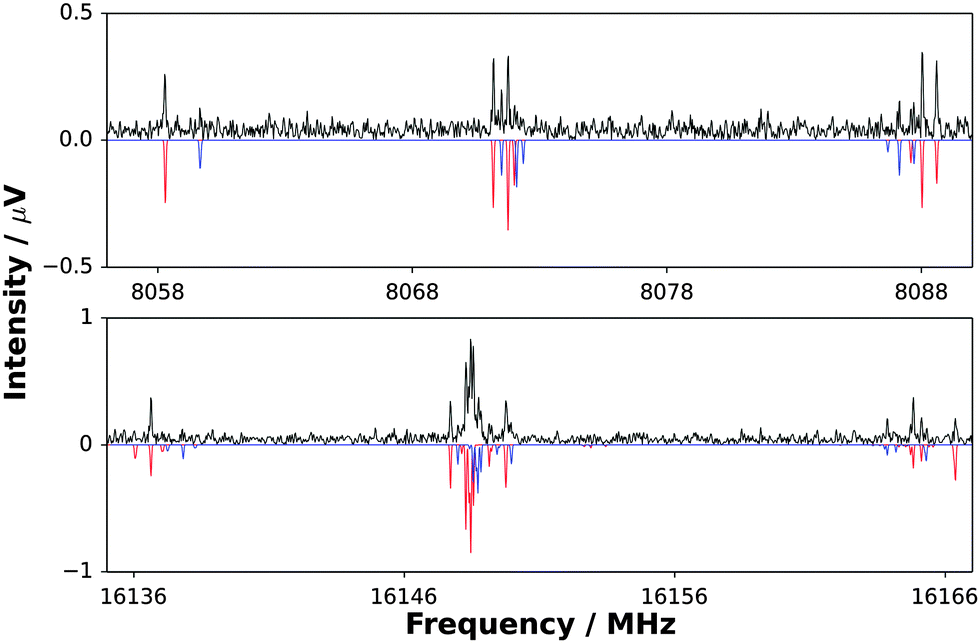 | ||
| Fig. 2 Top panel: A section of the broadband spectrum showing J′ → J′′ = 1 → 0 transitions of H3N⋯CuF (black trace) and a simulation of the transitions of H3N⋯63CuF and H3N⋯65CuF (red and blue traces respectively). Bottom panel: A section of the broadband spectrum showing J′ → J′′ = 2 → 1 transitions of H3N⋯CuF (black trace) and a simulation of the transitions of H3N⋯63CuF and H3N⋯65CuF (red and blue traces respectively). | ||
 | ||
| Fig. 3 The structure of H3N⋯CuF. N, Cu and F are the blue, grey and green spheres respectively. The a and b principal axes are shown. | ||
Western's PGOPHER47 was used to fit parameters in the Hamiltonian shown below to the observed transitions of each complex;
| H = HR − ⅙Q(Cu):∇E(Cu) − ⅙Q(I):∇E(I) − ⅙Q(N):∇E(N) + ICu·CCu·J | (1) |
| H3N⋯63CuF | H3N⋯65CuF | D3N⋯63CuF | H315N⋯63CuF | |
|---|---|---|---|---|
| a Numbers in parentheses are one standard deviation in units of the last significant figure. b Value in square brackets is fixed to the result for H3N⋯63CuF. c The value in square brackets has been determined by scaling Cbb = Ccc for H3N⋯63CuF by the ratio of the magnetic moments of the 65Cu and 63Cu nuclei. d N is the number of fitted transitions, σr.m.s. denotes the r.m.s. deviation of the fit. | ||||
| B 0/MHz | 4037.4554(19)a | 4037.513(10) | 3570.3874(16) | 3926.6944(36) |
| D J /kHz | 1.20(25) | 2.9(13) | [1.20]b | 1.04(48) |
| χ aa (Cu)/MHz | 66.196(12) | 61.183(61) | 66.441(68) | 66.212(21) |
| χ aa (N)/MHz | −2.717(15) | −2.787(90) | −2.776(41) | — |
| [Cbb(Cu) = Ccc(Cu)]/kHz | 18.9(12) | [20.2]c | 21.7(54) | 17.3(23) |
| N | 18 | 8 | 8 | 7 |
| σ r.m.s. /kHz | 7.4 | 23.8 | 15.8 | 9.1 |
| H3N⋯63CuI | H3N⋯65CuI | D3N⋯63CuI | D3N⋯65CuI | H315N⋯63CuI | |
|---|---|---|---|---|---|
| a Numbers in parentheses are one standard deviation in units of the last significant figure. b Numbers in square brackets have been fixed. c N is the number of fitted transitions. d σ r.m.s. denotes the r.m.s. deviation of the fit. | |||||
| B 0/MHz | 1162.03613(67)a | 1153.4533(19) | 1064.91348(73) | 1058.27044(50) | 1135.26715(70) |
| D J /kHz | 0.1289(73) | 0.097(17) | 0.0907(72) | [0.097]b | 0.1048(63) |
| D JK /kHz | 12.96(26) | 9.73(80) | 9.66(54) | 10.91(95) | 14.15(47) |
| χ aa (Cu)/MHz | 62.943(95) | 58.70(44) | 63.75(17) | 58.99(41) | 62.55(29) |
| χ aa (N)/MHz | −2.28(11) | −2.18(41) | −2.59(19) | −2.82(18) | — |
| χ aa (I)/MHz | −613.99(28) | −613.62(77) | −612.86(49) | −614.32(77) | −614.25(43) |
| N | 71 | 35 | 46 | 14 | 32 |
| σ r.m.s. /kHz | 13.2 | 20.3 | 21.0 | 14.6 | 12.2 |
All spectra were measured using a copper sample containing 63Cu and 65Cu isotopes in their natural abundances. The intensities of the spectra of all isotopologues are consistent with the natural fractional abundances of 63Cu and 65Cu which are 69% and 31% respectively. Isotopically-enriched samples were used to allow measurement and assignment of the spectra of D3N⋯63/65CuI, H315N⋯63CuI, D3N⋯63CuF and H315N⋯63CuF. Given that only one isotope of each of fluorine and iodine is available, isotopic substitution was not possible at the halogen atom. Confirmation that the molecular carriers of the observed spectra contain copper was obtained through comparison of the nuclear quadrupole coupling constants, χaa(Cu) = eQqaa(Cu), for the various isotopologues. This nuclear quadrupole coupling constant provides a measure of the strength of the coupling of the electric field gradient (qaa) at the Cu nucleus along the axis a with the ‘conventional’ nuclear electric quadrupole moment eQ, defined as eQ = 〈I,I|Qzz|I,I〉. Note that both the dyadic Q and its element Qzz contain the nuclear charge density while the constant Q does not; hence multiplication by the charge of a proton e on the left-hand side of the definition. For any given isotopologue, the electric field gradient is invariant to isotopic exchange (to a very good first approximation) even in the zero-point state so the ratio of the values of χaa(63Cu) in B⋯63CuX and χaa(65Cu) in B⋯65CuX should be equal to the ratio of the nuclear electric quadrupole moments of 63Cu and 65Cu. This condition is satisfied with the required level of precision for each pair of isotopologues for which the ratio can be calculated. The fitted rotational constant of H3N⋯63CuF is slightly smaller than that of H3N⋯65CuF. This implies that the Cu atom is sufficiently close to the centre of mass of this complex that any decrease in rotational constant (which would be required under the assumption of a rigid molecular framework, if Cu is not exactly at the centre of mass) is compensated by zero point effects. The rotational constant of H3N⋯63CuI is greater than that of H3N⋯65CuI by about 10 MHz. For H3N⋯63CuI, comparing the intensities of transitions having K = 1 with those having K = 0, the former are observed to be significantly more intense than would be expected at a rotational temperature of 2 K. This enhanced population of K = 1 states has been discussed previously10 and is explained by a reduced efficiency of collisional relaxation when the symmetry of a complex allows for hydrogen atoms to be exchanged by a C3 rotation about the inertial a axis. K = 0 and K = 1, 2 transitions belong to different symmetry species with relaxation taking place only within the two stacks, such that K = 0 and K = 1 transitions become equal in intensity because K = 0 transitions have twice the statistical weight of each of K = 1, 2. This is consistent with the ratio of the observed intensities of K = 0 and K = 1 transitions of H3N⋯CuI. Transitions having K = 2 were not observed for either H3N⋯CuF or H3N⋯CuI. Transitions having K = 1 were not observed for H3N⋯CuF for which only J′ → J′′ = 1 → 0 and 2 → 1 transitions are within the frequency range probed by the spectrometer.
3.2 Molecular geometry
By analogy with the geometries previously determined for the H3N⋯AgCl9 and H3N⋯CuCl10 complexes, it will initially be assumed that H3N⋯CuX (X = F, I) adopt a C3v symmetry in which N, Cu and X are located on the inertial a axis while interconnected in the geometry shown in Fig. 3. This assumption is consistent with the observations of a-type transitions and of a copper atom positioned close to the centre of mass in each of H3N⋯CuF and H3N⋯CuI. From this starting point, effective ground state (r0) and substitution (rs) geometries will be determined by fitting parameters in the model geometry (Fig. 3) to the experimentally-measured rotational constants. Ab initio calculations have been performed to establish equilibrium (re) geometries and other molecular properties.The STRFIT48 program is used to fit the r0 geometries. The experimental results do not allow independent determination of the N–H bond length, r(N–H), and the angle defining the positions of the hydrogen atoms, ∠(H–N–Cu). For this reason, r(N–H) is held fixed while the r(Cu–X), r(Cu–N) and ∠(H–N–Cu) structural parameters are fitted. The assumed values of r(N–H) in the r0 geometries of H3N⋯CuF and H3N⋯CuI are established by; (1) calculating the difference between the r0 value of r(N–H) in isolated NH3 and the re value of the same parameter calculated at the CCSD(T)(F12*)/AVQZ level; (2) adding the result of (1) to the re value calculated for r(N–H) in each of H3N⋯CuF and H3N⋯CuI respectively. Large zero point changes occur in isolated NH3 upon substitution of hydrogen atoms for deuterium. A shrinkage of 0.0011 Å in r(N–H) and an enlargement of 0.04 degrees in ∠(H–N–Cu) are observed. These changes are assumed to occur in H3N⋯CuF and H3N⋯CuI and are accounted for when fitting structural parameters. Fixing the r(N–H) distance as described above allows r(Cu–X), r(Cu–N) and ∠(H–N–Cu) to be determined with reasonable accuracy. Given that no isotopic substitution is available at either halogen atom, the uncertainties listed in Table 3 will be underestimated but the results agree satisfactorily with the results of the ab initio calculations.
| r 0 | r s | r e(CCSD(T)(F12*)/AVQZ) | ||||
|---|---|---|---|---|---|---|
| H3N⋯CuF | H3N⋯CuI | H3N⋯CuF | H3N⋯CuI | H3N⋯CuF | H3N⋯CuI | |
| a Numbers in parentheses are one standard deviation in units of the last significant figure. b Numbers in square brackets have been fixed at values determined as described under “Molecular Geometry”. c An imaginary number was obtained as the Kraitchman coordinate. | ||||||
| r(Cu–X)/Å | 1.74919(55)a | 2.35525(46) | — | — | 1.7372 | 2.3574 |
| r(Cu–N)/Å | 1.89276(61) | 1.9357(13) | 1.89(5) | 1.9361(13) | 1.8804 | 1.9226 |
| r(H–N)/Å | [1.0187]b | [1.0185] | [1.0180] | [1.0178] | 1.0145 | 1.0143 |
| ∠(H–N–Cu)/° | 111.462(26) | 111.430(54) | 111.500(50) | 111.535(30) | 111.746 | 111.811 |
| a Cu/Å | −0.00124(20) | 1.28011(37) | 0.00(5)c | 1.27874(121) | 0.0014945 | 1.2825 |
| a X/Å | −1.75043(35) | −1.07511(9) | — | — | −1.7387 | −1.0750 |
| a N/Å | 1.89152(41) | 3.21585(88) | 1.89128(79) | 3.21480(47) | 1.8789 | 3.2051 |
| a H/Å | 2.26426(12) | 3.58797(26) | — | — | 2.2548 | 3.5819 |
| b H/Å | 0.94806(17) | 0.94809(34) | — | — | 0.94230 | 0.94170 |
The a-axis coordinates of Cu and N can also be determined by a (rs) substitution method.49 The equation shown below is appropriate for an atom located on the symmetry axis of a symmetric rotor;
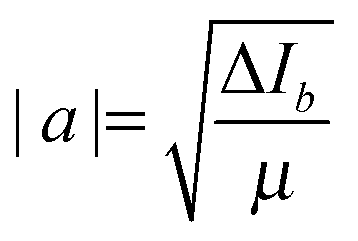 | (2) |
 where M is the mass of the isotopologue selected as parent and Δm is the change in mass upon substitution. The signs assigned to coordinates are those yielding bond lengths consistent with the ab initio calculations within acceptable precision limits.
where M is the mass of the isotopologue selected as parent and Δm is the change in mass upon substitution. The signs assigned to coordinates are those yielding bond lengths consistent with the ab initio calculations within acceptable precision limits.
When using eqn (2) the a coordinate is defined with respect to the principal axis system of the parent isotopologue and ∠(H–N–Cu) and r(N–H) cannot be independently determined for either H3N⋯CuF or H3N⋯CuI. The scaling procedure applied earlier to fix r(N–H) when fitting an r0 geometry is now used to fix an rs value. In this case, the difference between the rs and re values of r(N–H) in free NH3 is added to the re value calculated for r(N–H) in H3N⋯CuX to establish the value of r(N–H) in the rs geometry. Thus, a value of ∠(H–N–Cu) is determined using the equation provided in ref. 50. The uncertainties of rs coordinates are calculated using δa = 0.0015/|a| as recommended by Costain.51 All structural parameters and nuclear coordinates are provided in Table 3 alongside those of H3N⋯CuCl. The lack of an isotopic substitution at the halogen atom prevents the determination of an rs coordinate for the halogen atom. rs coordinates determined from Kraitchman's equations52 are typically closer to re structural parameters than r0 values owing to partial cancellation of zero point effects when rs coordinates are calculated.53 From Table 3 it can be seen that r(N–Cu) increases with increasing mass of the halogen atom of H3N⋯CuX. The value of ∠(H–N–Cu) is essentially independent of the halogen atom.
3.3 Interaction strength and ionicity
The strength of the interaction between NH3 and CuX can be assessed with reference to the dissociation energy for the H3N⋯CuX → NH3 + CuX reaction and also the force constant of the N⋯Cu bond. Ab initio calculations of both these quantities have been performed. As shown in Table 4, the dissociation energy of H3N⋯CuX decreases with increasing mass of the halogen atom with the highest result being 197 kJ mol−1 for the H3N⋯CuF complex.| F 22 = kσ(exp.)/[N m−1] | k σ(MP2/AVTZ)/[N m−1] | D e(CCSD(T)(F12*)/AVQZ)/[kJ mol−1] | |
|---|---|---|---|
| a Determined from DJ of the H3N⋯63CuF isotopologue. b Determined from DJ of the D3N⋯63Cu35Cl isotopologue. c Determined from DJ of the H3N⋯63CuI isotopologue. | |||
| H3N⋯CuF | 210(70)a | 242 | 197 |
| H3N⋯CuCl | 170(60)b | 219 | 183 |
| H3N⋯CuI | 111(33)c | 201 | 168 |
The results of the ab initio calculations of the force constants, kσ, follow the same trend with kσ(N⋯Cu) of H3N⋯CuF found to be 242 N m−1. The ab initio results for the kσ(N⋯Cu) can be compared with others determined from the experimental data as shown in Table 4. The experimentally-determined force constants are calculated by applying a model54 which accounts for contributions to the centrifugal distortion constants from both the Cu–X and N⋯Cu bonds. The force constants appropriate to the Cu–X and N⋯Cu bonds are denoted by F11 and F22 respectively in eqn (3);
 | (3) |
For each of H3N⋯CuF, H3N⋯CuCl and H3N⋯CuI, it can initially be assumed that F11 is equal to kσ of the appropriate, isolated metal halide diatomic.44,55,56 The r0 coordinates and moments of inertia (Ibb) calculated from the ground state rotational constants (B0) of the various complexes are then used to determine the F22 values. In the limit of a rigid Cu–X bond, F11 tends to infinity and F22 approaches an asymptotic value. Where F11 ≫ F22, any change in the assumed value of F11 induces only a very small change in the determined value of F22. As shown in Fig. 4, the assumption that the various F11 are reasonably approximated by kσ for the isolated metal halides (kσ(Cu–F) = 333 N m−1; kσ(Cu–I) = 172 N m−1), implies that the bond between the metal and halogen atoms is significantly more rigid than that between the metal and the nitrogen atoms, in each of H3N⋯CuF and H3N⋯CuI. After propagating the uncertainty in the measured value of DJ, the assumption that F11 of H3N⋯CuI = 172 ± 5 N m−1 implies F22 = 111(33) N m−1 for H3N⋯CuI where the dominant contribution is the uncertainty in DJ. An assumed value of F11 for H3N⋯CuF of 333 ± 10 N m−1 would imply that F22 falls within a narrow range between 202 and 213 N m−1. However, the uncertainty in the measured value of DJ again makes the dominant contribution and F22 = 210(70) N m−1 after propagation of the uncertainty in DJ. The level of agreement between the experimentally-determined results and those calculated ab initio is closest for H3N⋯CuF and H3N⋯CuCl which possess the most rigid MX bonds. It is apparent from both the force constants and the dissociation energies that the strengths of N⋯Cu bonds formed between NH3 and MX are in the order H3N⋯CuF > H3N⋯CuCl > H3N⋯CuI.
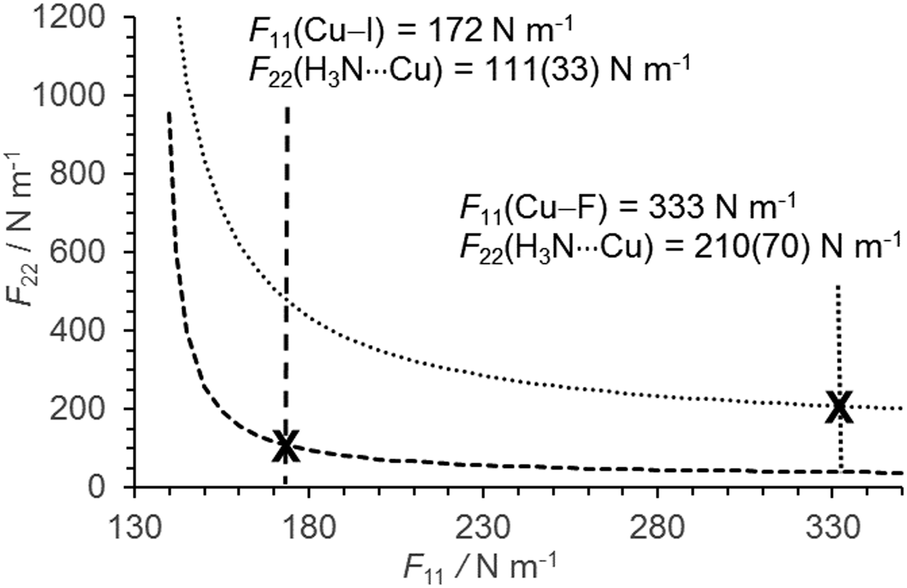 | ||
| Fig. 4 Plot to illustrate the variation of F22 as a function of the assumed value of F11 for each of H3N⋯CuF and H3N⋯CuI. The values of kσ for the isolated CuF and CuI molecules, assumed equal to F11 in the respective H3N⋯CuF and H3N⋯CuI complexes, are marked with a cross (X). | ||
Two types of hyperfine interaction have been included in the Hamiltonian. These are the nuclear quadrupole coupling and nuclear spin-rotation interactions. The former allows the determination of the nuclear quadrupole coupling constant, eQq(X), for X = 14N, 63/65Cu and I as appropriate for each isotopologue. The term  in eQqaa(X) represents the electric field gradient with respect to the symmetry axis at atom X. Applying the Townes–Dailey model57,58 to interpret the eQq(I) value of H3N⋯CuI, the ionicity (ic) of the Cu–I bond is calculated to be 0.73 from
in eQqaa(X) represents the electric field gradient with respect to the symmetry axis at atom X. Applying the Townes–Dailey model57,58 to interpret the eQq(I) value of H3N⋯CuI, the ionicity (ic) of the Cu–I bond is calculated to be 0.73 from  , where eQq(5,1,0)(I) is the coupling constant that would result from the presence of an unpaired electron in the 5pz orbital of the isolated atom and has a value of 2292.71 MHz.59 The Townes–Dailey model cannot be readily applied to the analysis of eQq values of Cu and N.
, where eQq(5,1,0)(I) is the coupling constant that would result from the presence of an unpaired electron in the 5pz orbital of the isolated atom and has a value of 2292.71 MHz.59 The Townes–Dailey model cannot be readily applied to the analysis of eQq values of Cu and N.
4. Conclusions
The spectra of H3N⋯CuF and H3N⋯CuI are each consistent with the C3v geometry illustrated in Fig. 3. Quantitative details of the geometries and measured nuclear quadrupole coupling constants of the H3N⋯CuX series are compared with results from other B⋯CuX in Table 5. The r(Cu–N) distances determined for H3N⋯CuF and H3N⋯CuI are consistent with expectations following the earlier study of H3N⋯CuCl and also with the trend established for OC⋯CuX. r(Cu–N) lengthens on substituting a lighter by a heavier halogen atom and is longer in H3N⋯CuI than in H3N⋯CuF by 0.043(2) Å. A very similar difference is observed between r(Cu–C) in OC⋯CuF and the same parameter in OC⋯CuI. The trend observed earlier for the Ar⋯CuX60 series involved greater incremental changes on substitution of the halogen atom. r(Cu–Ar) in Ar⋯CuF is 0.077(3) Å shorter than the same parameter in Ar⋯CuBr. The attachment of an isolated CuX species to either CO or NH3 to form B⋯CuX causes a change in r(Cu–X). The Cu–F bonds of H3N⋯CuF and OC⋯CuF shrink slightly on formation of these complexes from the isolated NH3/CO and CuX sub-units. In contrast, r(Cu–X) in the analogous B⋯CuCl, B⋯CuBr and B⋯CuI species extend slightly on complex formation. The difference between r(Cu–X) in B⋯CuX and in the corresponding CuX diatomic is denoted by Δr in Table 5 for H3N⋯CuX and OC⋯CuX. For both B = CO and B = NH3, Δr is greatest for the iodide. Recent works have described significant changes in the geometry of C2H2 when it attaches to MX to form a T-shaped C2H2⋯MX complex. Much smaller changes in the bond length of carbon monoxide were identified where OC⋯CuX and OC⋯AgX form from their constituent sub-units. At the level of precision of the present experiments and a previous work, the bond angle within NH3 does not change significantly when the molecule attaches to MX to form either H3N⋯CuX or H3N⋯AgX.| H3N⋯MX | OC⋯MX | MXa | ||||
|---|---|---|---|---|---|---|
| χ aa (M)/MHz | χ aa (X)/MHz | χ aa (M)/MHz | χ aa (X)/MHz | χ aa (M)/MHz | χ aa (X)/MHz | |
| a Ref. 44, 55, 56 and 61–64. b Ref. 24 and references therein. c Δr denotes the change in r(Cu–X) when B⋯MX form from isolated B and MX units. | ||||||
| CuF | 66.196(12) | — | 75.406(19) | — | 21.956 | — |
| CuCl | 66.629(24) | −23.041(34) | 70.8323(210) | −21.4735(22) | 16.169 | −32.127 |
| CuBr | — | — | 67.534(12) | 171.600(18) | 12.851 | 261.180 |
| CuI | 62.943(95) | −613.99(28) | 64.504(3) | −593.465(9) | 7.901(1) | −938.377(1) |
| HCl | — | −47.607(9)b | — | −52.086(9)b | — | −67.6189(5) |
| HI | — | −1324.891(8)b | — | −1346.238(18)b | — | −1828.286(9) |
| ICl | −3073.118(6)b | −68.927(3)b | −2953.798(10)b | −79.837(6)b | −2927.859(2) | −85.887(3) |
| BrCl | 915.55(2)b | −86.05(1)b | 875.835(5)b | −97.615(3)b | 875.309(1) | −102.450(2) |
Trends in the OC⋯CuX and H3N⋯CuX series can also be examined from the perspective of the measured nuclear quadrupole coupling constants. Changes in χaa for the metal and halogen atoms provide insight into the extent of electric charge redistribution when a complex forms from its component units. The determined χaa of OC⋯CuX and H3N⋯CuX are compared with those for isolated MX species and for selected hydrogen- and halogen-bonded complexes in Table 5. The fractional changes in χaa(M) and χaa(X) on formation of the complex from the isolated sub-units, B and MX, are greater for OC⋯CuX than for H3N⋯CuX or Ar⋯CuX. The opposite trend is observed for the hydrogen-bonded complexes, OC⋯HX and H3N⋯HX. The described differences between the B⋯MX, B⋯HX23 and B⋯XY24 series arise because the properties of hydrogen- and halogen-bonded complexes are governed mainly by electrostatic interactions whereas the binding between OC or NH3 and CuX has a significant covalent component. It has been shown that the ionicity of the Cu–X bond of OC⋯CuX decreases with increasing mass of the halogen atom. An ionicity of 0.73 is determined for H3N⋯CuI during the present work. This is lower than the ionicity of 0.79 determined for H3N⋯CuCl and therefore consistent with the trend identified earlier for the OC⋯CuX series.
Acknowledgements
The authors thank the EPSRC and the School of Chemistry at Newcastle University for the award of a DTA postgraduate studentship to D. M. B., the European Research Council for the postdoctoral fellowships awarded to S. L. S. and D. P. Z., and for project funding (Grant No. CPFTMW-307000). D. P. Z. also thanks Newcastle University for a SAgE Research Fellowship. A. C. L. thanks the University of Bristol for a Senior Research Fellowship and Newcastle University for a Visiting Professor award. D. P. T. is pleased to acknowledge the Royal Society for the award of a University Research Fellowship. We are also grateful to the EPSRC UK National Service for Computational Chemistry Software (NSCCS) at Imperial College London.References
- N. R. Walker and M. C. L. Gerry, Inorg. Chem., 2001, 40, 6158–6166 CrossRef CAS PubMed
.
- D. J. Frohman, G. S. Grubbs, Z. Yu and S. E. Novick, Inorg. Chem., 2013, 52, 816–822 CrossRef CAS PubMed
- G. S. Grubbs, D. A. Obenchain, H. M. Pickett and S. E. Novick, J. Chem. Phys., 2014, 141, 114306 CrossRef PubMed
- S. G. Francis, S. L. Matthews, O. K. Poleshchuk, N. R. Walker and A. C. Legon, Angew. Chem., Int. Ed., 2006, 45, 6341–6343 CrossRef CAS PubMed
- N. R. Walker and M. C. L. Gerry, Inorg. Chem., 2002, 41, 1236–1244 CrossRef CAS PubMed
- C. J. Evans, L. M. Reynard and M. C. L. Gerry, Inorg. Chem., 2001, 40, 6123–6131 CrossRef CAS PubMed
- V. A. Mikhailov, F. J. Roberts, S. L. Stephens, S. J. Harris, D. P. Tew, J. N. Harvey, N. R. Walker and A. C. Legon, J. Chem. Phys., 2011, 134, 134305 CrossRef PubMed
- N. R. Walker, D. P. Tew, S. J. Harris, D. E. Wheatley and A. C. Legon, J. Chem. Phys., 2011, 135, 014307 CrossRef PubMed
- V. A. Mikhailov, D. P. Tew, N. R. Walker and A. C. Legon, Chem. Phys. Lett., 2010, 499, 16–20 CrossRef CAS
- D. M. Bittner, D. P. Zaleski, S. L. Stephens, D. P. Tew, N. R. Walker and A. C. Legon, J. Chem. Phys., 2015, 142, 144302 CrossRef PubMed
- S. L. Stephens, W. Mizukami, D. P. Tew, N. R. Walker and A. C. Legon, J. Chem. Phys., 2012, 137, 174302 CrossRef PubMed
- D. P. Zaleski, S. L. Stephens, D. P. Tew, D. M. Bittner, N. R. Walker and A. C. Legon, Phys. Chem. Chem. Phys., 2015, 17, 19230–19237 RSC
- S. L. Stephens, D. M. Bittner, V. A. Mikhailov, W. Mizukami, D. P. Tew, N. R. Walker and A. C. Legon, Inorg. Chem., 2014, 53, 10722–10730 CrossRef CAS PubMed
- S. L. Stephens, D. P. Tew, V. A. Mikhailov, N. R. Walker and A. C. Legon, J. Chem. Phys., 2011, 135, 024315 CrossRef PubMed
- D. P. Zaleski, J. C. Mullaney, D. M. Bittner, D. P. Tew, N. R. Walker and A. C. Legon, J. Chem. Phys., 2015, 143, 164314 CrossRef PubMed
- Q. Wang, B. Zhang and Z. Huang, Chem. Phys. Lett., 2014, 614, 5–9 CrossRef CAS
- G. Zhang, H. Yue, F. Weinhold, H. Wang, H. Li and D. Chen, ChemPhysChem, 2015, 16, 2424–2431 CrossRef CAS PubMed
- G. Zhang, X. Zhao and D. Chen, J. Phys. Chem. A, 2013, 117, 10944–10950 CrossRef CAS PubMed
- H. Li, Q. Li, R. Li, W. Li and J. Cheng, J. Chem. Phys., 2011, 135, 074304 CrossRef PubMed
- G. Margraf, J. W. Bats, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2003, 956–957 RSC
- P. Woidy, A. J. Karttunen, M. Widenmeyer, R. Niewa and F. Kraus, Chem. – Eur. J., 2015, 21, 3290–3303 CrossRef CAS PubMed
- S. G. Batten and A. C. Legon, Chem. Phys. Lett., 2006, 422, 192–197 CrossRef CAS
- A. C. Legon and D. J. Millen, Faraday Discuss. Chem. Soc., 1982, 73, 71–87 RSC
- A. C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686–2714 CrossRef
- S. L. Stephens, W. Mizukami, D. P. Tew, N. R. Walker and A. C. Legon, J. Chem. Phys., 2012, 136, 064306 CrossRef PubMed
- D. P. Zaleski, S. L. Stephens and N. R. Walker, Phys. Chem. Chem. Phys., 2014, 16, 25221–25228 RSC
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253, DOI:10.1002/wcms.82
- C. Hättig, D. P. Tew and A. Köhn, J. Chem. Phys., 2010, 132, 231102 CrossRef PubMed
- C. Hättig, W. Klopper, A. Köhn and D. P. Tew, Chem. Rev., 2012, 112, 4–74 CrossRef PubMed
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS
- A. K. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283–296 CrossRef
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef CAS
- I. S. Lim, P. Schwerdtfeger, B. Metz and H. Stoll, J. Chem. Phys., 2005, 122, 104103 CrossRef PubMed
- C. Hattig, Phys. Chem. Chem. Phys., 2005, 7, 59–66 RSC
- F. Weigend, J. Comput. Chem., 2008, 29, 167–175 CrossRef CAS PubMed
- E. F. Valeev, Chem. Phys. Lett., 2004, 395, 190–195 CrossRef CAS
- A. Hellweg, C. Hättig, S. Höfener and W. Klopper, Theor. Chem. Acc., 2007, 117, 587–597 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed
- S. L. Stephens and N. R. Walker, J. Mol. Spectrosc., 2010, 263, 27–33 CrossRef CAS
- D. D. Nelson, G. T. Fraser, K. I. Peterson, K. Zhao, W. Klemperer, F. J. Lovas and R. D. Suenram, J. Chem. Phys., 1986, 85, 5512–5518 CrossRef CAS
- S. L. Stephens, N. R. Walker and A. C. Legon, Phys. Chem. Chem. Phys., 2011, 13, 20736–20744 RSC
- H. S. P. Müller and M. C. L. Gerry, J. Chem. Phys., 1995, 103, 577–583 CrossRef
- S. G. Batten, A. G. Ward and A. C. Legon, J. Mol. Struct., 2006, 780–781, 300–305 CrossRef CAS
- D. M. Bittner, D. P. Zaleski, S. L. Stephens, N. R. Walker and A. C. Legon, ChemPhysChem, 2015, 16, 2630–2634 CrossRef CAS PubMed
- D. D. Nelson, G. T. Fraser and W. Klemperer, J. Chem. Phys., 1985, 83, 6201–6208 CrossRef CAS
-
C. M. Western, PGOPHER, a Program for Simulating Rotational, Vibrational and Electronic Structure, University of Bristol, http://pgopher.chm.bris.ac.uk Search PubMed
- Z. Kisiel, J. Mol. Spectrosc., 2003, 218, 58–67 CrossRef CAS
- C. C. Costain, J. Chem. Phys., 1958, 29, 864–874 CrossRef CAS
- A. Chutjian, J. Mol. Spectrosc., 1964, 14, 361–370 CrossRef CAS
- C. Costain, Trans. Am. Crystallogr. Assoc., 1966, 2, 157–164 CAS
- J. Kraitchman, Am. J. Phys., 1953, 21, 17–24 CrossRef CAS
- J. K. G. Watson, A. Roytburg and W. Ulrich, J. Mol. Spectrosc., 1999, 196, 102–119 CrossRef CAS PubMed
- D. M. Bittner, N. R. Walker and A. C. Legon, J. Chem. Phys., 2016, 144, 074308 CrossRef PubMed
- R. J. Low, T. D. Varberg, J. P. Connelly, A. R. Auty, B. J. Howard and J. M. Brown, J. Mol. Spectrosc., 1993, 161, 499–510 CrossRef CAS
- J. Hoeft, F. J. Lovas, E. Tiemann and T. Törring, Z. Naturforsch., A: Phys. Sci., 1970, 25, 35–39 CAS
- C. H. Townes and B. P. Dailey, J. Chem. Phys., 1949, 17, 782–796 CrossRef CAS
- B. P. Dailey and C. H. Townes, J. Chem. Phys., 1955, 23, 118–123 CrossRef CAS
-
W. Gordy and R. L. Cook, Microwave Molecular Spectra, Wiley, New York, 1984 Search PubMed
- C. J. Evans and M. C. L. Gerry, J. Chem. Phys., 2000, 112, 9363–9374 CrossRef CAS
- F. H. de Leeuw and A. Dymanus, J. Mol. Spectrosc., 1973, 48, 427–445 CrossRef CAS
- E. Herbst and W. Steinmetz, J. Chem. Phys., 1972, 56, 5342–5346 CrossRef CAS
- R. E. Willis and W. W. Clark, J. Chem. Phys., 1980, 72, 4946–4950 CrossRef CAS
- F. A. Van Dijk and A. Dymanus, Chem. Phys. Lett., 1968, 2, 235–236 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp01368f |
| ‡ Current address: Chemistry Department, University of Manitoba, Winnipeg, MB R3T 2N2, Canada. |
| § Current address: Argonne National Laboratory, Chemical Sciences & Engineering, 9700 S. Cass Ave., Bldg. 200, Lemont, IL 60439, USA. |
| This journal is © the Owner Societies 2016 |