
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogenated earth abundant metalloporphyrins as photostable sensitizers for visible-light-driven water oxidation in a neutral phosphate buffer solution†
Hung-Cheng
Chen
,
Joost N. H.
Reek
,
René M.
Williams
and
Albert M.
Brouwer
 *
*
van't Hoff Institute for Molecular Sciences, Universiteit van Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: A.M.Brouwer@uva.nl
First published on 10th May 2016
Abstract
Very photostable tetrachloro-metalloporphyrins were developed as sensitizers for visible-light-driven water oxidation coupled to cobalt based water-oxidation catalysts in concentrated (0.1 M) phosphate buffer solution. Potassium persulfate (K2S2O8) acts as a sacrificial electron acceptor to oxidize the metalloporphyrin photosensitizers in their excited states. The radical cations thus produced drive the cobalt based water-oxidation catalysts: Co4O4-cubane and Co(NO3)2 as pre-catalyst for cobalt-oxide (CoOx) nanoparticles. Two different metalloporphyrins (Cu(II) and Ni(II)) both showed very high photostability in the photocatalytic reaction, as compared to non-halogenated analogues. This indicates that photostability primarily depends on the substitution of the porphyrin macrocycle, not on the central metal. Furthermore, our molecular design strategy not only positively increases the electrochemical potential by 120–140 mV but also extends the absorption spectrum up to ∼600 nm. As a result, the solar photon capturing abilities of halogenated metalloporphyrins (Cu(II) and Ni(II)) are comparable to that of the natural photosynthetic pigment, chlorophyll a. We successfully demonstrate long-term (>3 h) visible-light-driven water oxidation using our molecular system based on earth-abundant (first-row transition) metals in concentrated phosphate buffer solution.
Introduction
One of the options to create energy in a sustainable way is the solar-to-fuel approach.1 Solar-driven splitting of water to produce O2 and H2via artificial photosynthesis is the key challenge in this field.2 The water splitting reaction consists of two half reactions that are frequently studied as independent reactions: a water oxidation reaction in which water is oxidized to give oxygen and protons and a proton reduction reaction that gives molecular hydrogen. In homogeneous solution studies on light-driven water oxidation, three components are required to achieve photocatalytic activity: a sacrificial electron acceptor, a photosensitizer (PS) and a water oxidation catalyst (WOC).3 A significant number of water oxidation catalysts has been reported in the last decade including Ru,4 Ir,5 Mn,6 Co,7 Fe,8 and Cu9 complexes10 as well as related micro- or nano-scale materials such as iridium oxide, and cobalt oxide.11,12 Less effort has been invested in the development of photosensitizers for homogenous photocatalytic water oxidation and photoanode devices. Currently, mainly Ru polypyridine complexes are used as sensitizers.13–16 On the other hand, metalloporphyrins could be powerful alternatives.17 The excited photosensitizer reacts with the sacrificial electron acceptor to produce the photosensitizer radical cation PS˙+ (Scheme 1).18 This then oxidizes the WOC, provided that the redox potential E(PS˙+/PS) is high enough (>1.20 V vs. NHE, considering that the WOC typically function with moderate overpotentials η ≤ 400 mV, under neutral conditions).19 Although the potentials in metalloporphyrins can be varied by changing the central metal atom,20 only few of these porphyrins have a sufficiently high reduction potential of their radical cations. Recently, systems with a highly positive potential >1.40 V vs. NHE were reported, e.g. a Zn(II)-porphyrin with strongly electron withdrawing pentafluorophenyl substituents and a Pt(II)-porphyrin,21,22 and these were successfully used for light-driven water oxidation under neutral conditions. More recently, Mallouk et al. reported a series of free-base porphyrins with a broad range of potentials from 1.23 to 1.50 V vs. NHE, and also successfully demonstrated light driven water splitting using photoelectrodes obtained by co-deposition of these porphyrins and IrO2.23,24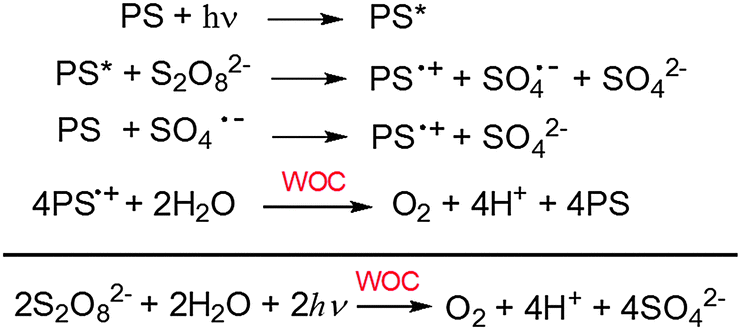 | ||
| Scheme 1 Light-driven water oxidation mechanism for the three-component system used here. | ||
Because proton-coupled electron transfer (PCET) plays a key kinetic role in catalytic water-oxidation mechanisms,25 buffer base and concentration dramatically influence the catalytic activity.26 Moreover, in model studies that focus on catalytic water oxidation progressive acidification of the reaction medium occurs at higher conversion, resulting in less favorable thermodynamics.27 A high buffer capacity is required to assure good proton management. To address these issues, aqueous phosphate salts, such as potassium phosphate (KPi, pKa = 7.21) are ideal buffer systems for studying light-driven water oxidation under neutral conditions. The hydrogen phosphate ion has been proven to act as an efficient proton acceptor in the oxygen-producing reaction under nearly neutral conditions.28 Therefore, concentrated and (nearly) neutral phosphate buffer is the preferred medium for the study of water oxidation.
In order to produce solar fuel on an industrial scale, it is necessary to make large scale and long-term stable artificial photosynthetic devices. Consequently, cost-effective and robust materials are required to build devices based on molecular components.2 Importantly, the usage of earth abundant metals in molecular systems for visible-light-driven water oxidation in near-neutral buffer solution is still a big challenge. The β-halogenated metalloporphyrins and metallocorroles as robust catalysts for water oxidation, proton reduction and other chemical reactions have been reported in the literature.29–33 Moreover, it is known that halogenation at the pyrrole β-positions of porphyrins can positively shift the electrochemical potential for its oxidation.34 Inspired by this molecular design strategy we report here new photostable metalloporphyrin sensitizers with suitable potentials E(PS˙+/PS) using only earth abundant metals for light-driven water oxidation. We combine the photosensitizers with two Co-based water oxidation catalysts (WOC), thus completely avoiding the use of precious metals. The chemical structures are shown in Fig. 1. Our light-driven water oxidation studies were performed in concentrated (0.1 M) phosphate buffer solution containing a sacrificial electron acceptor (K2S2O8).
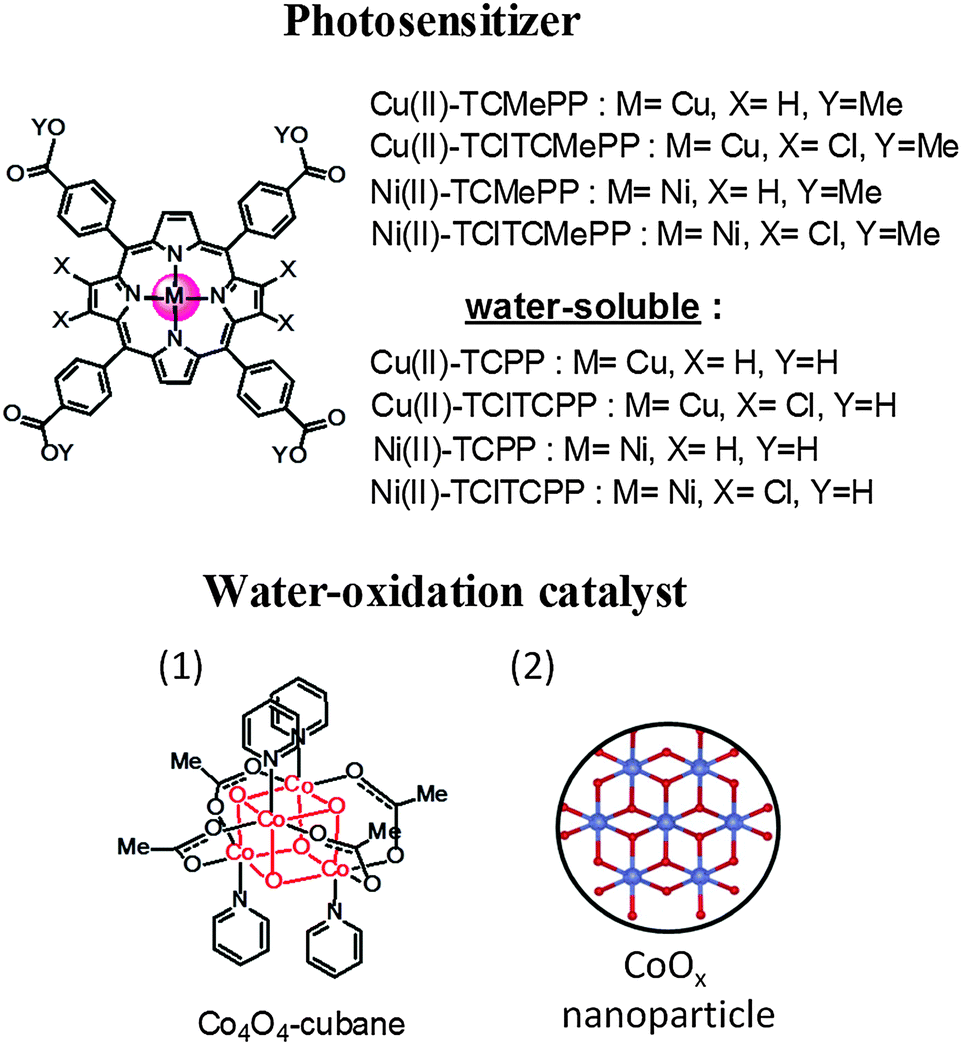 | ||
| Fig. 1 Chemical structures of Cu(II)- and Ni(II)-porphyrinphotosensitizers and water oxidation catalyst: (1) Co4O4-cubane, (2) representation of CoOx nanoparticle formed from Co(NO3)2 pre-catalyst.7,35,36 | ||
Results and discussion
Photophysical and electrochemical studies
The chlorinated Cu(II) and Ni(II) derivatives of 5,10,15,20-tetrakis(4-methoxycarbonylphenyl)-porphyrin, bearing four halogens at the β-pyrrole positions, were prepared in high yield (up to 70%) using a modified literature procedure.34 The UV-Vis absorption spectra and cyclic voltammograms of the compounds were obtained in CH2Cl2 at 298 K. Data on the molar absorption coefficients and the potentials E(PS˙+/PS) for various metalloporphyrins used in this study are collected in Table 1. The absorption spectra of all metalloporphyrins studied are dominated by bands that can be attributed to spin-allowed π–π transitions with an intense Soret band around 400 nm and moderately intense Q bands in the range 500–600 nm, which are shown in Fig. 2.| Molecule | λ max, nm (ε) | ||||
|---|---|---|---|---|---|
| Soret | Q x | Q y | E(T1)c | E(PS˙+/PS) | |
| a In CH2Cl2. b In phosphate buffer. c Derived from the maxima of the emission bands in Fig. S1 (ESI). d Derived from the first peaks in the differential pulse voltammograms (DPV, Fig. S8 (ESI)). | |||||
| Cu(II)-TCMePPa | 417 (38.2) | 539 (2.2) | 574 (sh) | 1.53 | 1.32 |
| Cu(II)-TClTCMePPa | 423 (34.6) | 548 (2.0) | 591 (sh) | 1.73 | 1.44 |
| Cu(II)-TCPPb | 407 (23.2) | 550 (1.6) | 592 (sh) | 1.47 | 1.13d |
| Cu(II)-TClTCPPb | 404 (22.2) | 554 (1.5) | 592 (sh) | 1.46 | 1.22d |
| Ni(II)-TCMePPa | 415 (33.2) | 528 (2.4) | 562 (sh) | — | 1.37 |
| Ni(II)-TClTCMePPa | 425 (29.4) | 542 (2.0) | 583 (sh) | — | 1.51 |
| Ni(II)-TCPPb | 406 (22.2) | 529 (1.6) | 565 (sh) | — | 1.04d |
| Ni(II)-TClTCPPb | 404 (19.5) | 539 (1.6) | 580 (sh) | — | 1.33d |
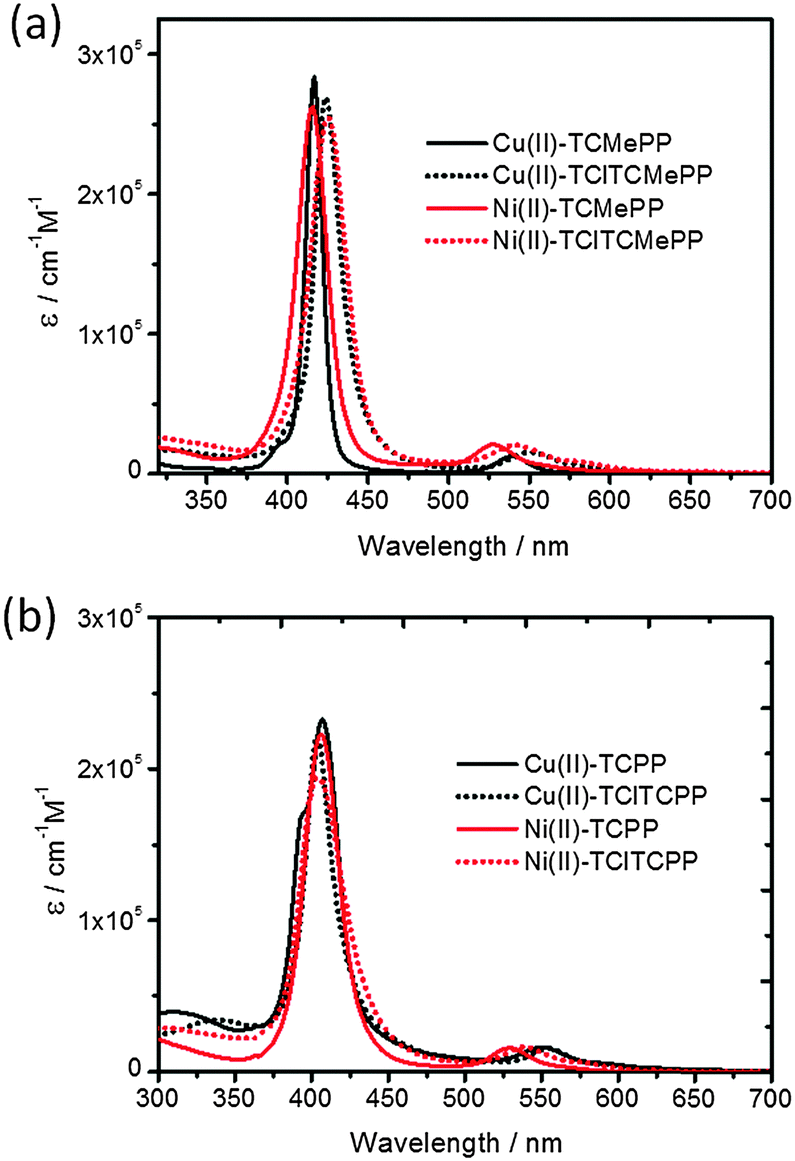 | ||
| Fig. 2 UV-visible absorption spectra (molar absorption coefficient ε) of Cu(II) and Ni(II)-porphyrin photosensitizers (a) esters in CH2Cl2 and (b) carboxylic acids in 0.1 M KPi solution at pH 7.0. | ||
For halogenated meso-tetraarylporphyrins, the electron-withdrawing halogen atoms at the β-positions reduce the energies of both the HOMOs and LUMOs, and cause a disruption of the planarity of the porphyrin macrocycle framework, reducing the HOMO–LUMO gap.34,37,38 The net result is a small lowering of the HOMO energies and a greater stabilization of the LUMOs in halogenated metallo-porphyrins.38 Consequently, spectral red-shifts of the Soret and Q-bands of porphyrins are observed in Cu(II)-TClTCMePP and Ni(II)-TClTCMePP, compared to the nonhalogenated analogs. Moreover, spectral broadening is also usually observed for halogenated porphyrins as a result of the distorted macrocycle.39 The spectral red shifts and broadening improve the light-harvesting properties of the photosensitizers.37 The photon-capture ability under solar irradiation will be discussed later. In order to carry out homogeneous three-component light-driven water oxidation studies, water-soluble metalloporphyrins were prepared by hydrolysis of the ester groups. Small spectral red-shifts of the Q-bands in Cu(II)-TClTCPP and Ni(II)-TClTCPP are observed in 0.1 M KPi solution compared to the corresponding esters in CH2Cl2. The excited state energies were determined with steady state luminescence spectroscopy in solution (deoxygenated by Ar purging) at room temperature (21 °C). The emission spectra are shown in Fig. S1 (ESI†). Cu(II)-TClTCMePP in CH2Cl2 shows emission at shorter wavelengths than Cu(II)-TCMePP (Fig. S1(a), ESI†). This phenomenon was also observed in other Cu(II)-porphyrins with modified 5,10,15,20-tetraphenyl groups.40 Surprisingly, there is only a small shift in wavelength between Cu(II)-TCPP and Cu(II)-TClTCPP in phosphate buffer solution (Fig. S1(b), ESI†). The deduced values of triplet state energies E(T1) are shown in Table 1. For none of the Ni(II)-porphyrins emission could be detected.41 Therefore, we used the value of E(T1) = 1.18 eV (1051 nm) reported in the literature for Ni(II)-TPP for all nickel-porphyrins.42 Excited state lifetimes of the metalloporphyrins were determined with nanosecond transient absorption (ns-TA) and time-resolved emission spectroscopy (Fig. S2–S5, ESI†).43,44 Emissive lifetimes of copper porphyrins have been reported to show a large variation (from 300 to 15 ns) and to be influenced by penta-coordination by e.g. the solvent. Triplet-quartet and triplet-doublet states (and fast sub-ns components related to their equilibration) play a role as well as vibronic distortions.45,46 This complex behaviour is reflected in the multi-exponential emission decays and ns-TA data observed here. Cu(II)-TCMePP and Cu(II)-TCPP show the longest lifetimes (28 ns and 12 ns respectively) next to one or two shorter components. The tetrachloro compounds (Cu(II)-TClTCMePP and Cu(II)-TClTCPP) display only short (sub ns) lifetimes (see Table S1, ESI†).
Cyclic voltammetry was performed with all metalloporphyrins in CH2Cl2, containing 0.1 M tetrabutyl-ammonium hexafluorophosphate, (NBu4PF6). In addition, the water-soluble compounds were studied in 0.2 M KPi as electrolyte (see ESI†). In order to unite all potentials for the water oxidation reaction, all the oxidation potentials of metalloporphyrins are referenced to NHE. The potential was determined using the ferrocene/ferrocenium redox couple as an internal standard and the half-wave potential (E1/2) was taken as 0.690 V vs. NHE in dichloromethane.47,48
For the nonhalogenated derivatives of the metallo-porphyrins in dichloromethane two quasi-reversible one-electron redox processes are observed with midpoint potentials E1/2(PS˙+/PS) = 1.32 and E1/2(PS2+/PS˙+) = 1.69 V vs. NHE for Cu(II)-TCMePP (Fig. 3(a)) and E1/2 of 1.37 and 1.66 V vs. NHE in Ni(II)-TCMePP (Fig. 3(b)).
 | ||
| Fig. 3 Cyclic voltammograms of (a) Cu(II)-TCMePP, Cu(II)-TClTCMePP and (b) Ni(II)-TClTCMePP, Ni(II)-TClTCMePP; concentrations 1.0 mM in CH2Cl2 solution with 0.1 M NBu4PF6 electrolyte, scan rate 100 mV s−1. Fc/Fc+ used as internal standard and NHE = Fc/Fc+ +0.69 V.47,48 | ||
The electron withdrawing character of the chlorine substituents is expected to influence significantly the electrochemical properties of the porphyrin derivatives.34,49 Higher potentials for oxidation can clearly be observed for the tetrachloro-metalloporphyrins as compared to the nonhalogenated analogs. E1/2(PS˙+/PS) values of 1.44 V vs. NHE for Cu(II)-TClTCMePP and 1.51 V vs. NHE for Ni(II)-TClTCMePP were obtained. These more positive potentials for oxidation of the tetrachloro-metalloporphyrins imply that their radical cations could provide a larger driving force to activate the water oxidation catalysts. For water-soluble metalloporphyrins, the presence of four negatively charged peripheral benzoate substituents can alter the redox potentials in KPi solution. All potentials of the water-soluble metalloporphyrin are 150–200 mV lower than those of the water-insoluble analogs with methyl benzoate substituents. However, also in phosphate buffer, there is a clear difference between tetrachloro-metalloporphyrins and the non-halogenated analogs. Importantly, the highest E(PS˙+/PS) of 1.33 V vs. NHE was obtained for Ni(II)-TClTCPP in KPi solution (see Table 1). This indicates that Ni(II)-TClTCPP has enough electron transfer driving force to activate WOCs with moderate overpotentials (∼400 mV) at neutral pH with standard potential of 0.82 V vs. NHE for water oxidation.
Electrocatalytic water oxidation by Co-based catalysts
Co4O4-cubane and Co(NO3)2 as pre-catalyst for cobalt oxide nanoparticle are known active catalysts for water oxidation reactions in phosphate buffer solution.7,35 Electrochemistry was used to characterize these Co-based catalytic systems. Cyclic voltammetry of a solution containing 1 mM Co4O4-cubane in 0.2 M KPi buffer as electrolyte at pH 7 to pH 10 is shown in Fig. 4. Considering the onset of the anodic current due to water oxidation (assumed as the potential at which the current intensity reaches 40 μA), the electrocatalytic potential depends on the pH: it is +1.33 V vs. NHE at pH 7, +1.29 V vs. NHE at pH 8, +1.23 V vs. NHE at pH 9 and +1.15 V vs. NHE at pH 10. This is in agreement with the pH dependence of the water oxidation potential.50 In addition, the increasing anodic current intensity observed by increasing the pH of the solution indicates that the buffer anions promote the proton-coupled electron transfer in the water oxidation reaction.26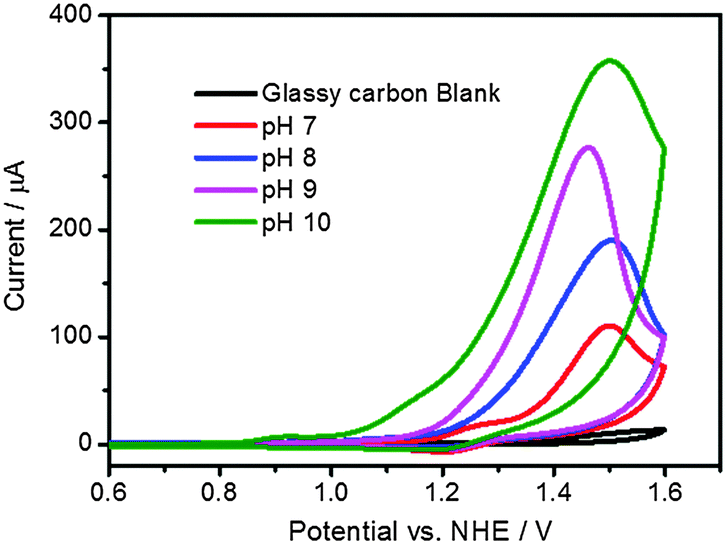 | ||
| Fig. 4 Cyclic voltammograms of water oxidation by Co4O4-cubane (2 mM) in 0.2 M KPi at pH 7 to pH 10. | ||
For the heterogeneous cobalt oxide nanoparticle water oxidation catalyst, cyclic voltammetry of a solution containing 2 mM Co(NO3)2 in 0.2 M phosphate buffer as electrolyte at pH 7 showed a small oxidation peak at 1.21 V (vs. NHE) that is due to CoIII/II oxidation, followed by a rapidly growing catalytic oxidation wave as shown in Fig. S9 (ESI†). Moreover, an appreciable onset catalytic current at ∼1.1 V (vs. NHE) corresponding to η = 280 mV at pH 7.0 can be observed. This result is consistent with recent work on CoPi.28,51
The photophysical and electrochemical data were used to construct energy level diagrams of PS, cobalt based WOCs and sacrificial electron acceptor (K2S2O8) in phosphate buffer at pH 7.0 (Fig. 5).52
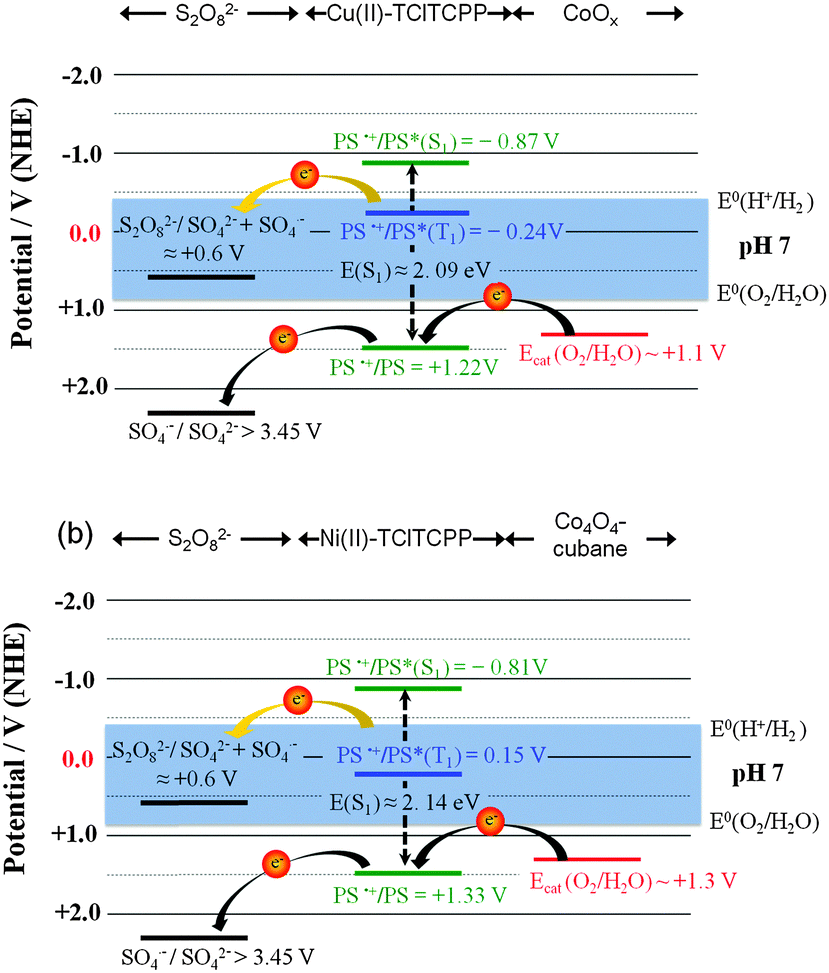 | ||
| Fig. 5 Energy schemes of halogenated metalloporphyrin photosensitizer (PS) and water oxidation catalysts. (a) Cu(II)-TClTCPP with CoOx nanoparticles and (b) Ni(II)-TClTCPP with Co4O4–cubane in phosphate buffer (pH 7.0) solution. Herein, E0(O2/H2O) = +0.82 V and E0(H+/H2) = −0.41 V. | ||
The singlet excited state energies of Cu(II)-TClTCPP and Ni(II)-TClTCPP are estimated by their Qy absorption wavelength. Thermodynamically favorable one-electron transfer from photogenerated triplet-state to S2O82− in buffer solution could be observed in both photocatalytic systems. In Cu(II)-TClTCPP case (Fig. 5(a)), the Cu(II)-TClTCPP radical cation with E(PS˙+/PS) = +1.22 V (vs. NHE) is thermodynamically capable of driving CoOx nanoparticle to oxidize water to O2. In Ni(II)-TClTCPP case (Fig. 5(b)), the Ni(II)-TClTCPP radical cation with E(PS˙+/PS) = +1.33 V (vs. NHE) is capable to activate Co4O4-cubane. The sulfate radical released from S2O82− can further oxidize the ground state of the halogenated metalloporphyrin, or the WOC if present in sufficiently high concentration.53
Visible-light driven water oxidation
To investigate the activities of visible light driven water oxidation we employed a three-component system composed of a water-soluble metalloporphyrin as the photosensitizer, potassium persulfate (K2S2O8) as a sacrificial electron acceptor, and Co4O4-cubane as a catalyst or Co(NO3)2 as pre-catalyst in concentrated phosphate buffer (0.1 M KPi) solution. The photochemical oxygen generation was recorded by means of a Clark oxygen electrode under 120 W halogen lamp illumination. The O2 generation in this photocatalytic system is believed to follow the well-established reaction mechanism presented in Scheme 1.By using the electrochemical potentials of the water-soluble metalloporphyrin photosensitizers and the electrocatalytic water oxidation results in phosphate buffer solution, the energy relationships between photosensitizers and catalysts can be estimated. In principle, the redox potential of the radical cation of the water-soluble photosensitizer should be higher than the onset of the catalytic potential of the catalyst54 in order to get catalysis. However, both the E(PS˙+/PS) value of +1.13 V vs. NHE for Cu(II)-TCPP and +1.22 V vs. NHE observed for Cu(II)-TClTCPP are less than the onset catalytic potential (+1.33 V vs. NHE) of Co4O4-cubane at pH 7. Therefore, the Cu(II)-porphyrin photosensitizers were further studied coupled to Co4O4-cubane at a higher pH. Herein, the turnover number is defined as TON = [O2]/[catalyst]. The slope of the initial oxygen evolution is used to determine the maximum turnover frequency (TOFmax = d[TON]/dt).
Fig. 6(a) and (b) show the time courses of photocatalytic O2 generation by Co4O4-cubane with Cu(II)-TCPP and Cu(II)-TClTCPP as photosensitizers. Clearly, Cu(II)-TCPP shows only very little production of O2, even at pH 10. For Cu(II)-TClTCPP, on the other hand, the O2 yield is substantial for pH > 8. The TOFmax = 1.1 × 10−3 s−1 at pH 9 increased to TOFmax = 3.0 × 10−3 s−1 at pH 10. Fig. 6(c) shows photocatalytic water oxidation activities with Co(NO3)2 as precatalyst with both Cu(II)-porphyrins at pH 7.0. A considerable O2 yield was only observed for Cu(II)-TClTCPP. Because the reduction potential of Cu(II)-TClTCPP˙+ at +1.22 V is higher than the onset catalytic current at +1.1 V of Co(NO3)2 (Fig. S6, ESI†), Cu(II)-TClTCPP˙+ is thermodynamically capable to induce water oxidation by nanoparticles generated in situ from Co(NO3)2 in pH 7.0 KPi buffer solution.
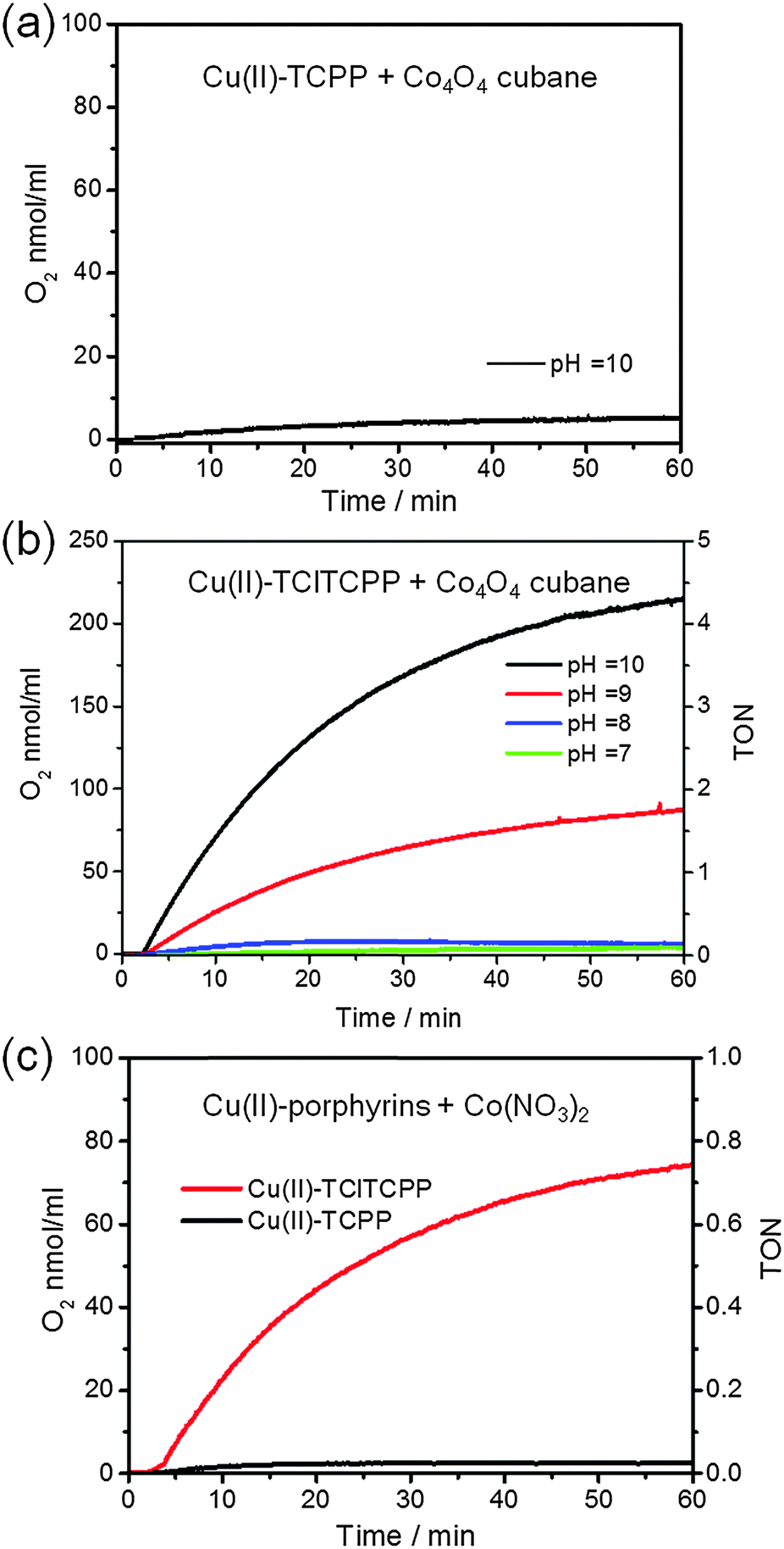 | ||
| Fig. 6 Photochemical oxygen evolution in 1.5 mL of 0.1 M KPi buffer solutions containing K2S2O8 (5.0 × 10−2 M), Cu(II)-porphyrin photosensitizers (6.7 × 10−4 M) and water oxidation catalysts: (a) Co4O4-cubane (5.0 × 10−5 M) at pH 10.0 with Cu(II)-TCPP, (b) Co4O4-cubane (5.0 × 10−5 M) at pH 7.0 to pH 10.0 with Cu(II)-TClTCPP, (c) Co(NO3)2 (1.0 × 10−4 M) at pH 7.0 with both Cu(II) porphyrins. | ||
Fig. 7 shows the time courses of photocatalytic O2 generation by (a) Co4O4-cubane (b) Co(NO3)2 precatalyst with Ni(II)-TCPP and Ni(II)-TClTCPP as photosensitizers at pH 7.0. Both results show that only the Ni(II)-TClTCPP sensitizer leads to O2 production. This conclusion is in agreement with the cyclic voltammetry (CV) of Co4O4-cubane or the Co(NO3)2 precatalyst and Ni(II)-TClTCPP at pH 7.0. The potential of oxidation of Ni(II)-TClTCPP (1.33 V vs. NHE) is higher than the onset of the catalytic wave due to water oxidation of Co4O4-cubane (∼1.3 V vs. NHE) or the Co(NO3)2 precatalyst (1.1 V vs. NHE). Therefore, Ni(II)-TClTCPP˙+ is thermodynamically capable of promoting water oxidation with these two Co-based catalysts in neutral phosphate buffer solution. The potential of Ni(II)-TCPP˙+ is too low to efficiently oxidize the catalysts. It is interesting to note that the Co4O4-cubane, which has a larger overpotential than the Co(NO3)2 derived catalyst, gives a substantially higher TOF with the Ni(II)-TClTCPP photosensitizer.
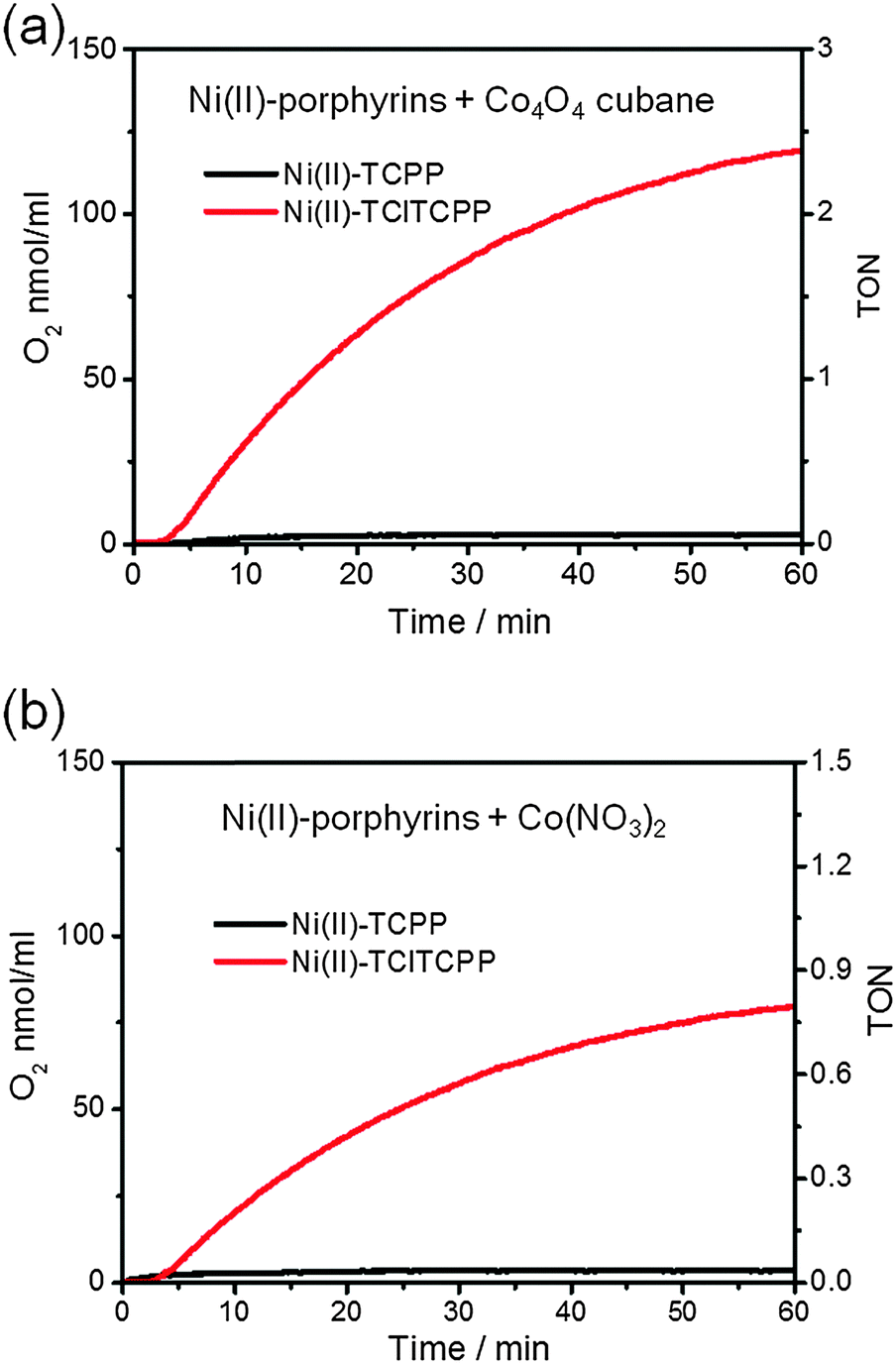 | ||
| Fig. 7 Photochemical oxygen evolution in 1.5 mL of pH 7.0, 0.1 M KPi buffer solutions containing K2S2O8 (5.0 × 10−2 M), Ni(II)-porphyrin photosensitizers (6.7 × 10−4 M) and water oxidation catalysts: (a) Co4O4-cubane (5.0 × 10−5 M), (b) Co(NO3)2 (1.0 × 10−4 M). | ||
Light control experiment of the photochemical water oxidation
A light control experiment shows that the catalytic water oxidation in our systems is truly driven by light (Fig. 8). These results clearly confirm the light-driven water oxidation by the Co-based catalysts and halogenated metalloporphyrins (Cu(II) and Ni(II)). A slight [O2] decrease during the “off” period was observed which results from equilibration of dissolved oxygen with the headspace. Importantly, the production of oxygen in Fig. 8 can be observed for several hours. This indicates that the halogenated metalloporphyrins are photostable throughout the whole experiment. Fig. S10 (ESI†) shows the color pictures and absorption spectra of the photocatalytic solutions before and after the time course of illumination. Both nonhalogenated metalloporphyrins (Cu(II)-TCPP and Ni(II)-TCPP) changed from red to dark brown, respectively. This indicates that decomposition of Cu(II)-TCPP and Ni(II)-TCPP occurs during the illumination. In contrast, it can be seen by eye that the halogenated metalloporphyrins (Cu(II)-TClTCPP and Ni(II)-TClTCPP) in Fig. S9(b) and (d) (ESI†), are stable as the color of solution before and after illumination is identical. This visual impression is more quantitatively supported by a comparison of the UV-Vis absorption spectra.13 The absorption spectra (Fig. S10(b) and (d), ESI†) of both halogenated metalloporphyrins were still identical with the initial spectra.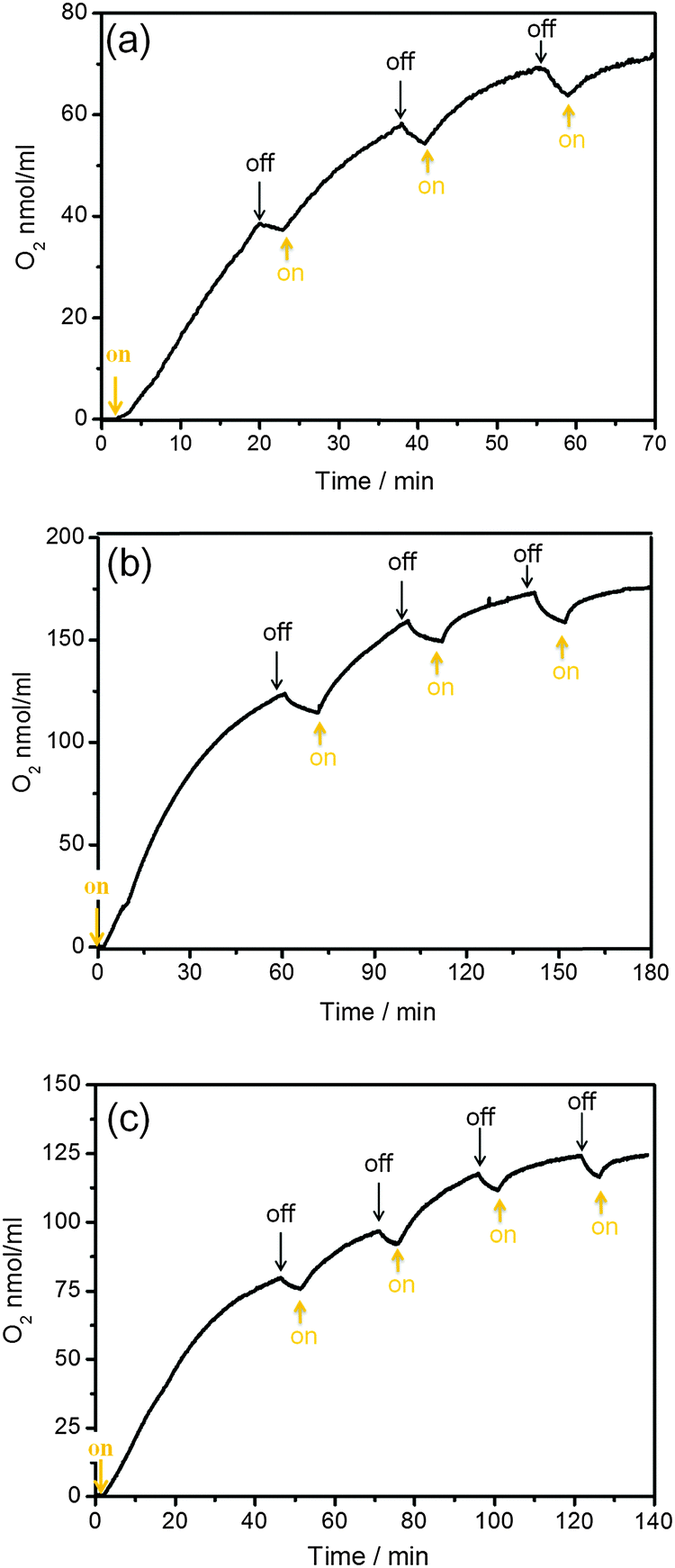 | ||
| Fig. 8 Photochemical oxygen evolution with switching on/off excitation light in 1.5 mL of pH 7.0, 0.1 M KPi buffer solutions containing K2S2O8 (5.0 × 10−2 M) with (a) Cu(II)-TClTCPP (6.7 × 10−4 M) and Co(NO3)2 precatalyst (1.0 × 10−4 M); (b) Ni(II)-TClTCPP (6.7 × 10−4 M) and Co4O4-cubane (5.0 × 10−5 M); (c) Ni(II)-TClTCPP (6.7 × 10−4 M) and Co(NO3)2 precatalyst (1.0 × 10−4 M). | ||
We conclude that tetrachlorination at the β-pyrrole positions of tetraarylporphyrins dramatically improves the photo-stability of the porphyrin photosensitizers. In addition, two metalloporphyrins (Cu(II)-TClTCPP and Ni(II)-TClTCPP) with the same tetrachloro-porphyrin macrocycle show long term photostability in photocatalytic water oxidation. This indicates that photostability is a property of the tetrachloro-porphyrin macrocycle, not related to the central metal in tetrachloro-metalloporphyrins.
The advantages of a tetrachloro metallo-tetraarylporphyrins for sunlight driven water oxidation
One of the essential properties of photoactive chromophores applied to molecule-based artificial photosynthetic devices is their ability to capture photons over a large part of the solar spectrum. Fig. S11(a) (ESI†) shows the UV-Vis spectra of the four chromophores: Cu(II)-TClTCMePP, Ni(II)-TClTCMePP, Ru(bpy)32+, and Chl a (related to the monomer of P680 in natural oxygenic photosynthesis). In addition, Fig. S11(b) (ESI†) shows the photon absorption rates by 2 μM solutions of chromophores under AM1.5G sunlight in the range 300 to 730 nm. The integrated molar absorptivities and percentages of photons absorbed by 2 μM chromophore solutions are shown in Table 2. Both the values for Cu(II)-TClTCMePP and Ni(II)-TClTCMePP are at least five times larger than those of Ru(bpy)32+ because they show much more intense absorption in the visible light range. Another parameter representing the photon-absorption ability of a chromophore is the 50% photon capture threshold (PCT50),55 which is the concentration of a chromophore needed to absorb 50% of incident solar photons in the given solar spectrum range. The PCT50 of Cu(II)-TClTCMePP and Ni(II)-TClTCMePP are about one order smaller than that of Ru(bpy)32+. Moreover, compared to our previously reported photoactive chromophore, Pt(II)-TCMePP,22 the broadening and red-shift of absorption by tetrachlorination for Cu(II)-TClTCMePP and Ni(II)-TClTCMePP make the PCT50 a factor four smaller than that of Pt(II)-TCMePP. The photon absorption rates of 2 μM for both tetrachloro-metalloporphyrins are also higher than that of chlorophyll a. Even though chlorophyll a has broader bands that span a large portion of the sunlight spectrum, the PCT50 values of both of our tetrachloro-metalloporphyrins are similar to that of chlorophyll a.| Molecule | Integrated molar absorptivityb (M−1) | AM1.5G photon capturec (2 μM) | 50% photon capture thresholdd (PCT50) | Redox potential V vs. NHE |
|---|---|---|---|---|
| a 300–730 nm. b The absorption spectra of Ru(bpy)32+ and chlorophyll a are from ref. 56. c Percentage of incident solar photons absorbed for a solution of a given concentration (1 cm path length). d Concentration required to absorb 50% of the incident solar photons (1 cm path length). e Ref. 22. f Ref. 54. g Ref. 57. h Ref. 58 herein, NHE = SHE + 6 mV. i Ref. 59. j Ref. 60. | ||||
| Cu(II)-TClTCMePP | 6.5 × 108 | 9.6% | 32 μM | 1.44 |
| Ni(II)-TClTCMePP | 7.0 × 108 | 10.3% | 31 μM | 1.51 |
| Pt(II)-TCMePP | 4.5 × 108 | 6.0% | 122 μM | 1.50e |
| Ru(bpy)32+ | 1.2 × 108 | 1.7% | 360 μM | 1.26f |
| Chlorophyll a | 4.8 × 108 | 7.2% | 38 μM | 0.81g,h |
| P680 | 1.26i | |||
| Ru(bpy)2(4,4′-(PO3H2)2bpy)2+ | 1.30j | |||
The second essential property of a photosensitizer for light-driven water oxidation is the oxidation potential.19 Particularly, the E1/2(PS˙+/PS) of Ni(II)-TClTCMePP is 250 mV higher than E1/2(PS˙+/PS) of Ru(bpy)32+. Thus, choosing tetrachloro-metalloporphyrins as photosensitizers for light-driven water oxidation not only affords a better light harvesting function under solar excitation but also provides a larger driving force for electron transfer from the WOC to the radical cation of the photosensitizer. We previously studied the noble-metal based photo-active porphyrin, Pt(II)-TCPP, which was shown to be much more photostable in phosphate buffer solution, and to have a better photon capture ability than Ru(bpy)32+.22 The halogenated metalloporphyrins introduced in the present work provide a further improvement of both properties.
Conclusions
Tetrachloro-metallo-porphyrins with earth abundant metal ions Cu(II) and Ni(II) are very photostable sensitizers for visible-light-driven water oxidation coupled to cobalt based water oxidation catalysts. The relatively high electrochemical potential of the tetrachloro-metalloporphyrins allows these chromophores to be used to study WOCs with moderate overpotentials (∼400 mV) in neutral phosphate buffer solution. Especially Ni(II)-TClTCPP performs well with the Co4O4-cubane WOC. More importantly, the tetrachloro-metalloporphyrins show exceptionally high photostability in concentrated phosphate buffer solution during light-driven water oxidation. They also show at least five times more solar photon capture ability and 170–250 mV more oxidizing power than the extensively used Ru(bpy)32+. Moreover, they also show similar oxidation potentials and better solar photon capture abilities relative to the photoactive noble-metal porphyrin, Pt(II)-TCMePP.22 Therefore, the introduction of halogenated metalloporphyrins with earth-abundant metal ions is a step forward in the development of robust photosensitizers in molecule based artificial photosynthesis.Acknowledgements
This work is part of the research program of the Foundation for Fundamental Research on Matter (FOM), which is part of the Netherlands Organisation for Scientific Research (NWO). This research is financed in part by the BioSolar Cells open innovation consortium, supported by the Dutch Ministry of Economic Affairs, Agriculture and Innovation.References
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- S. Bensaid, G. Centi, E. Garrone, S. Perathoner and G. Saracco, ChemSusChem, 2012, 5, 500–521 CrossRef CAS PubMed.
- L. Duan, L. Tong, Y. Xu and L. Sun, Energy Environ. Sci., 2011, 4, 3296–3313 CAS.
- S. Romain, L. Vigara and A. Llobet, Acc. Chem. Res., 2009, 42, 1944–1953 CrossRef CAS PubMed.
- D. G. H. Hetterscheid and J. N. H. Reek, Chem. Commun., 2011, 47, 2712–2714 RSC.
- G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia and G. F. Swiegers, Acc. Chem. Res., 2009, 42, 1935–1943 CrossRef CAS PubMed.
- N. S. McCool, D. M. Robinson, J. E. Sheats and G. C. Dismukes, J. Am. Chem. Soc., 2011, 133, 11446 CrossRef CAS PubMed.
- J. L. Fillol, Z. Codola, I. Garcia-Bosch, L. Gomez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef CAS PubMed.
- M.-T. Zhang, Z. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048–2051 CrossRef CAS PubMed.
- D. G. H. Hetterscheid and J. N. H. Reek, Angew. Chem., Int. Ed., 2012, 51, 9740–9747 CrossRef CAS PubMed.
- A. Sartorel, M. Carraro, F. M. Toma, M. Prato and M. Bonchio, Energy Environ. Sci., 2012, 5, 5592–5603 CAS.
- X. Deng and H. Tüysüz, ACS Catal., 2014, 4, 3701–3714 CrossRef CAS.
- Y. Xu, L. Duan, L. Tong, B. Åkermark and L. Sun, Chem. Commun., 2010, 46, 6506–6508 RSC.
- W. J. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef CAS PubMed.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760–775 CAS.
- S. Berardi, G. La Ganga, M. Natali, I. Bazzan, F. Puntoriero, A. Sartorel, F. Scandola, S. Campagna and M. Bonchio, J. Am. Chem. Soc., 2012, 134, 11104–11107 CrossRef CAS PubMed.
- G. S. Nahor, P. Neta, P. Hambright, A. N. Thompson and A. Harriman, J. Phys. Chem., 1989, 93, 6181–6187 CrossRef CAS.
- A. Lewandowska-Andralojc and D. E. Polyansky, J. Phys. Chem. A, 2013, 117, 10311–10319 CrossRef CAS PubMed.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357–2387 RSC.
- A. Harriman and M. C. Richoux, J. Phys. Chem., 1983, 87, 4957–4965 CrossRef CAS.
- G. F. Moore, J. D. Blakemore, R. L. Milot, J. F. Hull, H. Song, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Energy Environ. Sci., 2011, 4, 2389–2392 CAS.
- H.-C. Chen, D. G. H. Hetterscheid, R. M. Williams, J. I. van der Vlugt, J. N. H. Reek and A. M. Brouwer, Energy Environ. Sci., 2015, 8, 975–982 CAS.
- J. R. Swierk, D. D. Méndez-Hernández, N. S. McCool, P. Liddell, Y. Terazono, I. Pahk, J. J. Tomlin, N. V Oster, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1681–1686 CrossRef CAS PubMed.
- The potential of 0.209 V vs. NHE corresponds to 0.0 V vs. Ag/AgCl, NaCl.
- C. J. Gagliardi, A. K. Vannucci, J. J. Concepcion, Z. Chen and T. J. Meyer, Energy Environ. Sci., 2012, 5, 7704–7717 CAS.
- D. Wang and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579–15584 CrossRef CAS PubMed.
- M. Hara, C. C. Waraksa, J. T. Lean, B. A. Lewis and T. E. Mallouk, J. Phys. Chem. A, 2000, 104, 5275–5280 CrossRef CAS.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109–114 RSC.
- D. K. Dogutan, R. McGuire and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178–9180 CrossRef CAS PubMed.
- B. Mondal, K. Sengupta, A. Rana, A. Mahammed, M. Botoshansky, S. G. Dey, Z. Gross and A. Dey, Inorg. Chem., 2013, 52, 3381–3387 CrossRef CAS PubMed.
- D. Dolphin, T. G. Traylor and L. Y. Xie, Acc. Chem. Res., 1997, 30, 251–259 CrossRef CAS.
- T. G. Traylor and S. Tsuchiya, Inorg. Chem., 1987, 26, 1338–1339 CrossRef CAS.
- H. Y. Liu, T. S. Lai, L. L. Yeung and C. K. Chang, Org. Lett., 2003, 5, 617–620 CrossRef CAS PubMed.
- P. Ochsenbein, K. Ayougou, J. Fischer, R. Weiss, N. Rachel, K. Jayaraj, A. Gold, J. Terner and J. Fajer, Angew. Chem., Int. Ed., 1994, 33, 348–350 CrossRef.
- D. Hong, J. Jung, J. Park, Y. Yamada, T. Suenobu, Y.-M. Lee, W. Nam and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7606–7616 CAS.
- G. F. Swiegers, J. K. Clegg and R. Stranger, Chem. Sci., 2011, 2, 2254 RSC.
- H. Shahroosvand, S. Zakavi, A. Sousaraei and M. Eskandari, Phys. Chem. Chem. Phys., 2015, 17, 6347–6358 RSC.
- T. Takeuchi, H. B. Gray and W. A. Goddard, J. Am. Chem. Soc., 1994, 116, 9730–9732 CrossRef CAS.
- W. Jentzen, M. C. Simpson, J. D. Hobbs, X. Song, T. Ema, N. Y. Nelson, C. J. Medforth, K. M. Smith and M. Veyrat, J. Am. Chem. Soc., 1995, 117, 11085–11097 CrossRef CAS.
- K. L. Cunningham, K. M. McNett, R. A. Pierce, K. A. Davis, H. H. Harris, D. M. Falck and D. R. McMillin, Inorg. Chem., 1997, 36, 608–613 CrossRef CAS.
- R. L. Milot, G. F. Moore, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, J. Phys. Chem. C, 2013, 117, 21662–21670 CAS.
- P. Brodard and E. Vauthey, Chem. Phys. Lett., 1999, 309, 198–208 CrossRef CAS.
- J. A. S. Cavaleiro, H. Görner, P. S. S. Lacerda, J. G. MacDonald, G. Mark, M. G. P. M. S. Neves, R. S. Nohr, H.-P. Schuchmann, C. von Sonntag and A. C. Tomé, J. Photochem. Photobiol., A, 2001, 144, 131–140 CrossRef CAS.
- T. Kumpulainen, B. H. Bakker, M. Hilbers and A. M. Brouwer, J. Phys. Chem. B, 2015, 119, 2515–2524 CrossRef CAS PubMed.
- J. Rodriguez, C. Kirmaier and D. Holten, J. Am. Chem. Soc., 1989, 111, 6500–6506 CrossRef CAS.
- M. Asano, Y. Kaizu and H. Kobayashi, J. Chem. Phys., 1988, 89, 6567–6576 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- G. F. Moore, S. J. Konezny, H. Song, R. L. Milot, J. D. Blakemore, M. L. Lee, V. S. Batista, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, J. Phys. Chem. C, 2012, 116, 4892–4902 CAS.
- G. A. Spyroulias, A. P. Despotopoulos, C. P. Raptopoulou, A. Terzis, D. de Montauzon, R. Poilblanc and A. G. Coutsolelos, Inorg. Chem., 2002, 41, 2648–2659 CrossRef CAS PubMed.
- Y. Zhao, N. M. Vargas-Barbosa, E. A. Hernandez-Pagan and T. E. Mallouk, Small, 2011, 7, 2087–2093 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- K. Henbest, P. Douglas, M. S. Garley and A. Mills, J. Photochem. Photobiol., A, 1994, 80, 299–305 CrossRef CAS.
- G. La Ganga, F. Puntoriero, S. Campagna, I. Bazzan, S. Berardi, M. Bonchio, A. Sartorel, M. Natali and F. Scandola, Faraday Discuss., 2012, 155, 177–190 RSC.
- L. Duan, Y. Xu, P. Zhang, M. Wang and L. Sun, Inorg. Chem., 2010, 49, 209–215 CrossRef CAS PubMed.
- M. T. Whited, P. I. Djurovich, S. T. Roberts, A. C. Durrell, C. W. Schlenker, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2011, 133, 88–96 CrossRef CAS PubMed.
- J. M. Dixon, M. Taniguchi and J. S. Lindsey, Photochem. Photobiol., 2005, 81, 212–213 CrossRef CAS PubMed.
- M. Kobayashi, S. Ohashi, K. Iwamoto, Y. Shiraiwa, Y. Kato and T. Watanabe, Biochim. Biophys. Acta, 2007, 1767, 596–602 CrossRef CAS PubMed.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- H. Dau and I. Zaharieva, Acc. Chem. Res., 2009, 42, 1861–1870 CrossRef CAS PubMed.
- Y. Gao, X. Ding, J. Liu, L. Wang, Z. Lu, L. Li and L. Sun, J. Am. Chem. Soc., 2013, 135, 4219–4222 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental procedures for the synthesis of all the porphyrins, the electrochemical characterization, spectroscopy and photocatalytic setup are described. See DOI: 10.1039/c6cp01352j |
| This journal is © the Owner Societies 2016 |