
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of monoglycerides and fatty acids on a ceramide bilayer†
Anna
Akinshina
*a,
Chinmay
Das
b and
Massimo G.
Noro
c
aInstitute of Skin Integrity and Infection Prevention, School of Human and Health Sciences, University of Huddersfield, HD1 3DH, Huddersfield, UK. E-mail: a.akinshina@hud.ac.uk
bSchool of Mathematics, University of Leeds, Leeds, LS2 9JT, UK
cUnilever R&D Port Sunlight, Quarry Road East, Bebington, Wirral, CH63 3JW, UK
First published on 1st June 2016
Abstract
Monoglycerides and unsaturated fatty acids, naturally present in trace amounts in the stratum corneum (top layer of skin) lipid matrix, are commonly used in pharmaceutical, cosmetic and health care formulations. However, a detailed molecular understanding of how the oil additives get incorporated into the skin lipids from topical application and, once incorporated, how they affect the properties and integrity of the lipid matrix remains unexplored. Using ceramide 2 bilayers as skin lipid surrogates, we use a series of molecular dynamics simulations with six different natural oil ingredients at multiple concentrations to investigate the effect of the oils on the properties and stability of the bilayers. The six oils: monoolein, monostearin, monoelaidin, oleic acid, stearic acid and linoleic acid – all having the same length of the alkyl chain, C18, but a varying degree of saturation, allow us to systematically address the effect of unsaturation in the additives. Our results show that at low oil concentration (∼5%) the mixed bilayers containing any of the oils and ceramide 2 (CER2) become more rigid than pure CER2 bilayers due to more efficient lipid packing. Better packing also results in the formation of larger numbers of hydrogen bonds between the lipids, which occurs at the expense of the hydrogen bonds between lipids and water. The mixed bilayers with saturated or trans-unsaturated oils remain stable over the whole range of oil concentration. In contrast, the presence of the oils with at least one cis-double bond leads to bilayer instability and complete loss of bilayer structure at the oil content of about 50–65%. Two cis-double bonds in the lipid tail induce bilayer disruption at even lower concentration (∼30%). The mixed bilayers remain in the gel phase (without melting to a fluid phase) until the phase transition to a non-bilayer phase occurs. We also demonstrate that the stability of the bilayer strongly correlates with the order parameter of the lipid tails.
1. Introduction
The stratum corneum (SC), the outermost layer of skin, plays an important role in protecting our body from the penetration of external pathogens and toxins, and also in preventing internal water loss. The SC is commonly represented by the “brick and mortar” model,1–4 where the “bricks” symbolize corneocytes and the “mortar” is the extracellular lipid matrix. Corneocytes are flat, horny and functionally dead cells filled with proteins (mainly keratins), water and specific low molecular weight hygroscopic compounds, collectively known as Natural Moisturising Factors (NMFs).3–8 Corneocytes are the major water-holding components of the SC. The main components of the SC lipid matrix are ceramides, cholesterol and free fatty acids3,7,9,10 organized in a highly ordered multilamellar structure, interconnected by a strong hydrogen bond network between the head groups.11,12 Contrary to the corneocytes, water content in the SC lipid phase is extremely low12–14 and the lipid “mortar” serves as the main barrier for water loss through the SC.4Ceramides (CERs) form a major component of the skin lipids. There are several types of ceramides found in the SC, among them are “protein-bound” ceramides, covalently attached to the outer surface of the corneocyte envelope, and “free” ceramides, the abundant ingredients of the multi-layered intercellular lipid phase.4,9,10 Together with ceramides, the intercellular lipid matrix consists of cholesterol (CHOL) and free fatty acids (FA) present in roughly equimolar ratios.10 Free fatty acids are mostly long chain (more than 20 carbons) saturated fatty acids; the only unsaturated fatty acids detected in the SC are oleic and linoleic acids, present at low concentrations.10 There are also small amounts of glycerides, glycosphingolipids, cholesterol sulphate, and cholesterol esters present in the SC.10,15–18 We should note that phospholipids, the major components of biological membranes, are not present in the SC. The phospholipids from the keratinocytes of the viable layers are broken down by phospholipases into free fatty acids in the lower SC.4,19–21
Controlled modification of the skin barrier is desired for either enhancing the permeability for topical drug release22–31 or restoring the damaged skin function.10,32,33 Natural oil components such as monoglycerides (MG) and free fatty acids (FA) are natural constituents of the stratum corneum arising from the partial hydrolysis of sebum triglycerides produced by the sebaceous glands.17,18 Unsaturated oils such as monoolein, oleic and linoleic acids are widely used in pharmaceutical and cosmetic industries as drug penetration and solubility enhancers.22–24,26–31 Stearic acid is used as the moisturizing ingredient in the novel body wash products32,33 where it acts as a buffer against lipid extraction by cleanser surfactants. In vitro experiments27,34 show that the effect of a fatty acid as a permeation enhancer depends on chain unsaturation: long saturated fatty acids do not enhance skin permeability, while unsaturated fatty acids markedly enhance skin permeation. However, atomistic understanding of how the oils get incorporated in the skin lipid layer from the topical application or how they affect the structure of the skin lipid matrix is missing. In this study we address these by a series of molecular dynamics simulations of ceramide bilayers as surrogates for the stratum corneum lipid layer along with carefully chosen natural oil components that mostly differ in numbers and positions of unsaturation of the tails.
In spite of the fact that the first studies of the skin structure and properties date back to about 1970 and the essential progress in the understanding of skin structure and function was made over the last 40 years,3,5 the investigations on the roles and effect of the different components of the skin lipid matrix at the molecular and atomistic levels started to develop only during the last decade. Höltje et al.21 performed modelling and experimental studies of the mixed fatty acid bilayers in comparison with the mixed fatty acid–cholesterol bilayers in an equimolar ratio. The authors reported that the well-ordered and rigid structure of pure FA bilayers was strongly affected by the addition of CHOL. Shorter than FA, rigid, planar-shaped CHOL molecules act as lipid fluidisers – they reduce the tail order, bilayer thickness and rigidity as well as increase the area per lipid. The first simulations of the CER bilayer were done by Pandit and Scott,35 who studied symmetric ceramides, CER NS 16:0. The CER bilayer was found to be stable in the liquid-crystalline phase at high temperature (368 K) and the smaller polar headgroups of CER molecules reduce the ordering behaviour of the water molecules.
The first simulations of a hydrated ceramide 2 bilayer with asymmetric lipid tails (the C16 sphingosine chain and the C24 fatty acid chain) at different temperatures were performed by Notman et al.11 It was shown that the designed model of CER2 reproduces the phase behaviour and structural parameters of CER2 bilayers reasonably well. The obtained values of the area per lipid and the bilayer thickness are in good agreement with the experimental values; lipids within the bilayer are well ordered and tightly packed, connected by a strong hydrogen bond network between the headgroups. Pure CER2 bilayers are characterised by the hexagonal gel-phase and the high rigidity compared to phospholipid bilayers. All these findings explain the low water permeability of the stratum corneum lipid phase. This successful model for CER2 was further widely employed in the subsequent modelling studies.12,13,36–40 and it is also used in this work.
Das et al.37 first performed the detailed analysis of the hydrated bilayers composed of mixed skin lipids, CER2, CHOL and FA 24:0 at different compositions and temperatures and compared the properties of the mixed bilayers with the pure bilayer of CER2. The mixed bilayers were generated by transforming the required number of CER2 molecules to either CHOL or FA. The addition of FA into the CER2 bilayer leads to an increase in the bilayer thickness, a slight increase in the chain order and a small reduction of lipid density. Cholesterol, on the contrary, decreases the bilayer thickness and nematic order of the lipids and increases the density at the tail region. Close to the skin composition CER–FA–CHOL (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1), the bilayer becomes softer, with comparatively lower elastic modulus.
:2:1), the bilayer becomes softer, with comparatively lower elastic modulus.
Similar results were also obtained recently by Gupta and Rai41 who studied mixed skin CER:CHOL:FA bilayers at different component molar ratios and a wide temperature range. The authors have reported that the CER bilayers are in a gel phase at normal skin temperature and exhibit a phase transition into a liquid crystalline phase at ∼365 K. Due to the length asymmetry, the CER tails are found to be in a fluid-like state near the bilayer midplane. The presence of FA increases the ordering of CER tails, while the addition of smaller and rigid CHOL decreases the bilayer thickness. The interesting finding is that the cholesterol increases the fluidity of the bilayer below the phase transition temperature of CER and retains the bilayer rigidity above the phase transition temperature.
Notman et al.42 studied the effect of the common penetration enhancer, oleic acid (OA), on the phospholipid (DPPC) bilayer at the temperature of 323 K. The simulations show that OA is able to mix homogeneously with DPPC lipids without significant perturbation of the bilayer at all the concentrations considered (mole fractions 0.05–1.0). In general, OA at all concentrations had very little effect on the overall structure of the bilayer. No phase separation was observed at any of the OA concentrations. The authors suggested that the liquid-crystalline phase DPPC bilayer is sufficiently fluid to accommodate the “kinked” tails of the OA molecules. As the concentration of OA increases a slight decrease in the area compressibility modulus and a minor increase in the diffusion rate of the OA molecules were observed. However, the frequency of water permeation events was found to double, indicating some OA-induced permeability enhancement. Recently Hoopes et al.43 studied the effect of OA on the mixed 1:1:1 CER2–CHOL–FA skin lipid bilayer at the OA concentrations up to 0.1 mol% and T = 300 and 340 K. Similar to the OA-DPPC study discussed above,42 no significant changes in the properties of the skin lipid bilayer upon addition of OA were observed. Addition of a small amount of OA leads to slight enhancement in the diffusion of cholesterol, the fastest species in this gel-like mixture, which in turn increases the diffusion of ceramide and fatty acid. However, no impact on hydrogen bonding between the lipids upon addition of OA was detected.
In this work we are focused on the effect of unsaturation in MG and FA when used as additives in the CER2 bilayer. We present a systematic study of a range of monoglycerides and fatty acids with the same chain length (C18) but with different degrees of saturation of the hydrocarbon tail. We have explored and compared the properties of the mixed bilayers over the whole range of accessible oil concentrations. In the next section we describe the system components and the model parameters used. After that we present and discuss our results, covering bilayer properties such as lipid packing, bilayer rigidity, hydrogen bonding and ordering of the lipid tails. This paper ends with the summary of our findings.
Most of the simulations reviewed so far, and the work presented here, use united atom (UA) representations of the alkyl carbons11–13,21,36–40,43–55 for computational efficiency. Recently Guo et al.39 compared the commonly used Berger lipid force field44 with a modified version of the CHARMM force field developed for ceramides. In this work the lipid tails and OH group parameters were taken from the CHARMM36 topology files and the parameters for the amide group were obtained using ab initio calculations. The force field was applied to the two ceramides, CER NS and CER NP. The authors reported that while both approaches are in good agreement with experiment from a structural perspective under the physiological conditions, the modified CHARMM force field is better able to capture the thermotropic phase transitions observed in the experiment. However, it was noted that together with the accuracy the all atom CHARMM-based force field also brings additional computational cost, compared with the UA GROMOS-based force field, which may limit the simulation timescale and the size of the studied systems.
Tjörnhammar and Edholm56 have developed a new parameterised UA force field based on the Berger44 and Chiu57 parameters to study both the fluid and gel phases of phospholipid bilayers. The force field is characterized by the slightly modified dihedral potential and LJ parameters for the fatty acid chains and new partial charges based on QM calculations. The authors aimed to construct a force field that will be applicable to both fluid and gel phases. The main findings were that the modified force field results in the correct area per lipid, tail tilt, NMR order and the structural factors for both fluid and gel phases. This novel promising UA force field shows potential to be utilized in the future skin lipid simulations.
2. Model systems and simulation aspects
2.1 Lipids
In this study we have simplified the complex multicomponent skin lipid system to a bilayer of pure ceramide 2 (CER2). Ceramides 2 are sphingolipids with a large tail length polydispersity; the most common fatty acid chain length (tail 2 in Fig. 1) in CER2 is C24. This system still captures the key physico–chemical properties of the skin lipid barrier such as the gel phase, the local hexagonal packing of the lipid chains, and the strong and extended hydrogen-bond network across the head groups. CER2 (also known as CER NS) is the predominant ceramide in skin and it was successfully used in the previous simulation studies of the skin lipids.11–13,36–38,45,47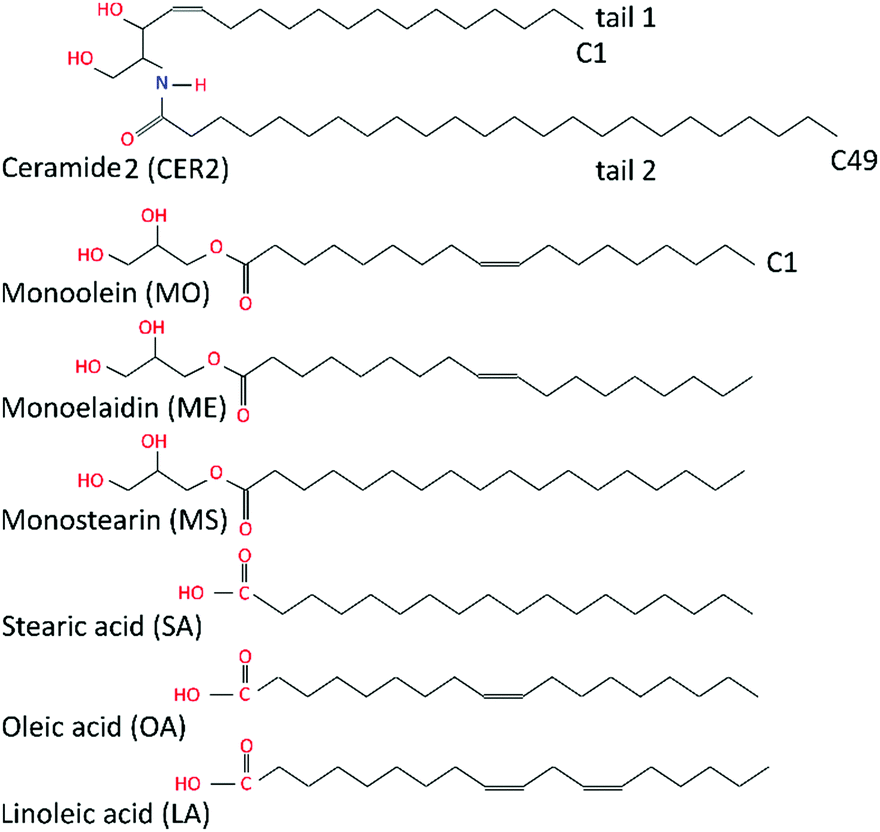 | ||
| Fig. 1 Schematic illustration of ceramide 2, monoglycerides and fatty acids considered in this study. | ||
A series of monoglycerides and fatty acids is considered as additives in this study. While the length of the alkyl chain of the oil molecules was held constant at C18 throughout, the degree and nature of saturation of the hydrocarbon tail were varied. Here we focused on the following oil ingredients: monoolein (2,3-dihydroxypropyl(Z)-octadec-9-enoate), monostearin (2,3-dihydroxypropyl octadecanoate), monoelaidin, (2,3-dihydroxypropyl(E)-octadec-9-enoate), oleic acid, ((9Z)-octadec-9-enoic acid), stearic acid, (octadecanoic acid) and linoleic acid, ((9Z,12Z)-octadeca-9,12-dienoic acid). Monoolein (MO) and oleic acid (OA) differ by the head groups and both featured a cis-9 unsaturated double bond that makes the hydrocarbon tail “kinked”. Monostearin (MS) and stearic acid (SA) both have a saturated hydrocarbon tail; monoelaidin (ME) is the trans isomer of monoolein; and linoleic acid (LA) is a poly-unsaturated fatty acid (C18:2) and it contains two cis-double bonds at C9 and C12 atoms. The chemical structures of the molecules studied are shown in Fig. 1.
2.2 Computational details
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond parameters for unsaturated acyl tails were taken from POPC simulations,51–53 where the double bond was described via improper dihedral. For monoelaidin (ME), with the trans-double bond, the dihedral angle was changed from 0 to 180° to keep the trans-double bond planar. Such parameters were previously used in simulations of oleic acid43 and sphingomyelin.48,54 Partial charges for the glycerol head group of monoglycerides were taken from 1-lauroyl-glycerol (1mlg.itp file available in Gromacs package), and the values for bonds, angles and dihedrals were extracted from topologies of POPC51–53 and CER211,13,37 lipids and the gromos force field “ffgmx”, derived from GROMOS87.59 The water was modelled using the SPC potential. The examples of the topology files for monoolein and stearic acid are provided in the ESI.†
C double bond parameters for unsaturated acyl tails were taken from POPC simulations,51–53 where the double bond was described via improper dihedral. For monoelaidin (ME), with the trans-double bond, the dihedral angle was changed from 0 to 180° to keep the trans-double bond planar. Such parameters were previously used in simulations of oleic acid43 and sphingomyelin.48,54 Partial charges for the glycerol head group of monoglycerides were taken from 1-lauroyl-glycerol (1mlg.itp file available in Gromacs package), and the values for bonds, angles and dihedrals were extracted from topologies of POPC51–53 and CER211,13,37 lipids and the gromos force field “ffgmx”, derived from GROMOS87.59 The water was modelled using the SPC potential. The examples of the topology files for monoolein and stearic acid are provided in the ESI.†
The simulations were carried out at constant temperature (T = 350 K) and pressure (1 bar) with a time step of 2 fs. We have used the Nosé–Hoover thermostat62,63 with a time constant of 0.1 ps and the anisotropic Parrinello–Rahman barostat64,65 with a time constant of 5 ps and a compressibility of 4.5 × 10−5 bar−1. Water and lipids were coupled separately. The cut-off of 1.2 nm was used for both the van der Waals and electrostatic interactions. Electrostatics was treated using the Particle Mesh Ewald (PME)66,67 algorithm. Standard periodic boundaries were employed in all three directions and all the bonds were constrained using the P-LINCS58 algorithm. Water molecules were kept rigid using the SETTLE68 algorithm. The temperature (T = 350 K) in our simulations is significantly higher than the physiological temperature (300–305 K). Previous studies37,39 show that ceramide bilayers, represented by the UA force field used here, do not have any structural transition in the temperature range 300–360 K. All structural quantities vary smoothly with temperature in this range. Thus we expect that the only effect of the elevated simulation temperature is to accelerate all dynamics without changing the qualitative features.
| N CER2 | N OIL | X OIL |
|---|---|---|
| 128 | 0 | 0 |
| 126 | 2 | 0.016 |
| 124 | 4 | 0.031 |
| 122 | 6 | 0.047 |
| 120 | 8 | 0.063 |
| 118 | 10 | 0.078 |
| 116 | 12 | 0.094 |
| 102 | 26 | 0.203 |
| 90 | 38 | 0.297 |
| 78 | 50 | 0.391 |
| 64 | 64 | 0.500 |
| 52 | 76 | 0.594 |
| 38 | 90 | 0.703 |
| 32 | 96 | 0.750 |
| 28 | 100 | 0.781 |
| 20 | 108 | 0.844 |
| 12 | 116 | 0.906 |
| 0 | 128 | 1 |
3. Results, analysis and discussion
3.1 Spontaneous permeation
Prior to investigating the influence of the specific oils at a certain concentration on the properties of the CER2 bilayer we have examined the spontaneous penetration of the oil molecules into the bilayer. This was done for the example of monoolein (MO). Even though it is logical to expect that amphiphilic oil molecules would prefer the lipid phase to water, the dynamics of embedding MO from the water to lipid phase is far from obvious. The result of our reasonably long (1.2 μs) simulation is quite interesting and is presented in Fig. 2. We have started by placing six MO molecules randomly in the water region of the well equilibrated CER2 bilayer. Prior to NPT simulation, the system was subjected to energy minimization and a short NVT equilibration.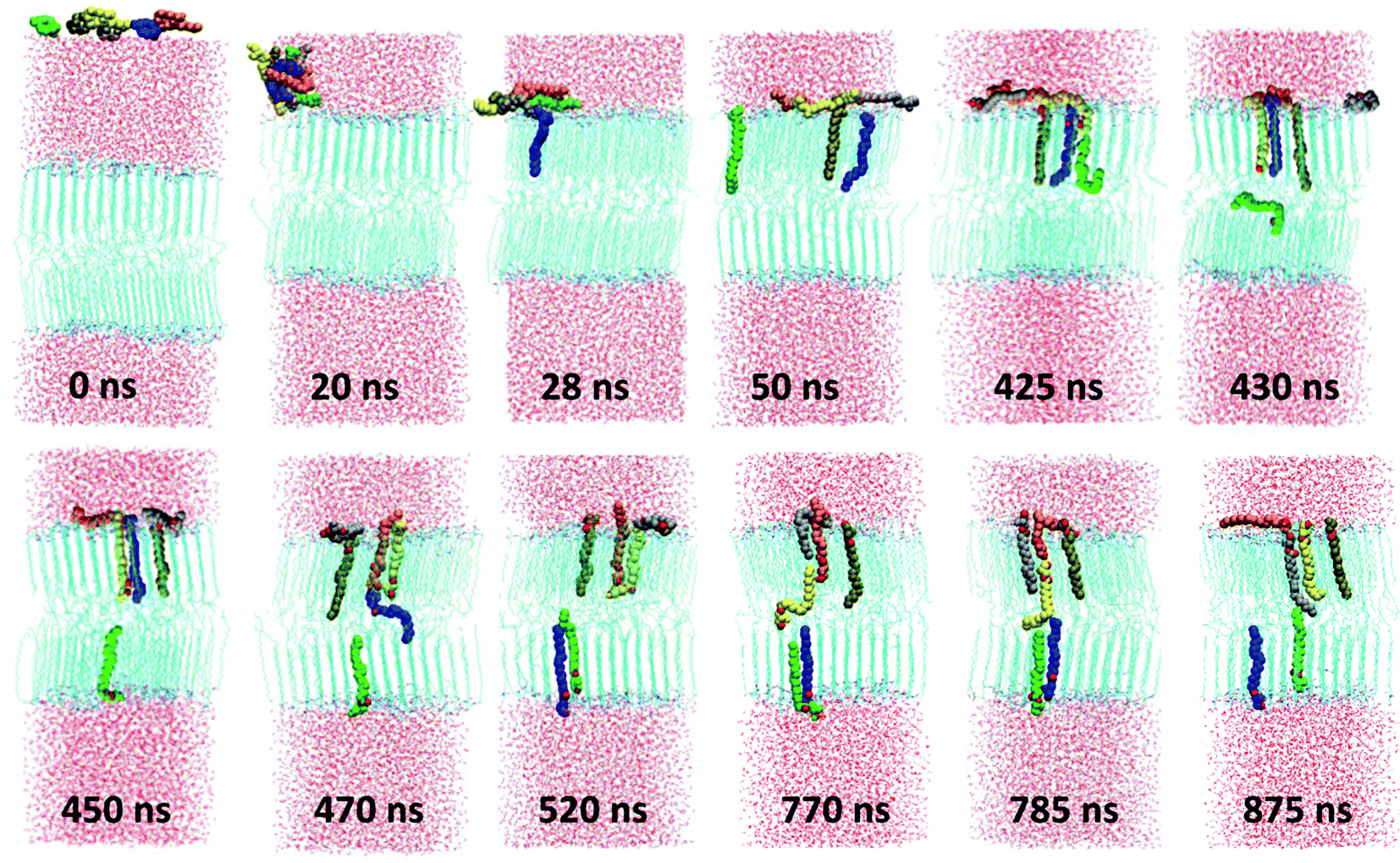 | ||
| Fig. 2 Spontaneous permeation of monoolein molecules. The MO molecules are coloured differently in order to visualize the dynamics and location of the individual molecules. | ||
After about 3 ns of simulation the molecules self-assemble into a relatively spherical cluster to minimize contacts of the hydrophobic tails with water. The cluster diffuses towards the lipid phase and then flattens at the lipid head group surface after 22 ns of simulation time. Then MO molecules start to move inside the bilayer tail first straight away one by one and the first three molecules integrate into the bilayer very quickly. The first one (blue) penetrates after 28 ns of simulation, the second one (green) after 39 ns, and the third one (brown) joined them after 41 ns of simulation time. We expected a similar penetration rate for the other three molecules, but during the next ∼385 ns the system “was trapped” under that condition (three molecules stay inside the bilayer and the other three remain on the head group surface). That can be explained by the fact that the leaflet in which the three MO get in has higher density than the opposing leaflet. Thus, further inclusion of another MO has lower probability. After 425 ns we observe that the fourth molecule (yellow one) moves inside the bilayer, now head group first, pushing another MO molecule (green one) further down to the bilayer. That (green) molecule then jumps to the bilayer midplane and shortly after that moves to another leaflet. The same procedure occurs again shortly, after 460 ns of simulation time: the orange molecule inserted into the bilayer head group first, pushing the blue one to the bottom leaflet. At skin pH, and reflected in our choice of partial charges, the small polar head group of MO does not pay much penalty for being present in the hydrocarbon region. The excess density in the leaflet is relieved by transporting MO in the inter-leaflet liquid region. The penetration by the head groups also involves formation of a hydrogen bond with the fellow MO lipid, which then is pushed to the bilayer midplane. After penetration of these two molecules, the system again stays under that condition (five molecules are inside the bilayer and the last one outside) for the following ∼250 ns. After about 770 ns of simulation the last grey molecule penetrates into the bilayer by the tail first, pushing the yellow one to the midplane. However, the yellow molecule does not go to the bottom leaflet, as expected and as the previous two molecules did, it forms an H-bond with the orange molecule and pushes it out of the bilayer, back to the water phase. When the molecule happens to be between the leaflets, it has the possibility to pass to any leaflet of the bilayer. A similar effect was observed in the study of CHOL flip-flop,12 where the frequent exchange of CHOL molecules between the ordered tail phase and the tail–tail inter-leaflet region was observed. Interestingly, after visiting the midplane, the yellow molecule, which penetrates the bilayer by the head group, inserts back to the leaflet in a correct head group position. The last molecular exchange occurs after ∼875 ns of simulation and then the system again remains under this condition for another ∼300 ns.
During our 1.2 μs long MD simulation we have observed that 5 out of 6 molecules integrated into the bilayer. Among them 2 molecules are in one leaflet and 3 in another one. All the inserted molecules are oriented correctly within the bilayer with their head groups in the head group region. We have observed the events of molecular penetration by either the tail first or by the head group first, the molecular flip-flop to another leaflet, the reversal of the orientation of the molecule within the bilayer by moving to the midplane area and inserting again to the ordered region, and the exchange between the already inserted molecule and that which was outside the bilayer in the water phase.
The penetration process appears to be spontaneous and irregular and we do not see that the insertion of some of the molecules into the bilayer facilitates faster penetration of the remaining ones. However, we have observed that after the first 3 molecules have penetrated, the insertion of the next ones involves interaction with these already inserted molecules by pushing them down to the bilayer. We have also registered that all the penetrated molecules are located near each other within each leaflet and during our 1.2 μs long simulation they do not distribute uniformly within the bilayer.
3.2 Phase behaviour of mixed bilayers
:1) a first order phase transition occurs from the stable bilayer to a structure with different symmetry.
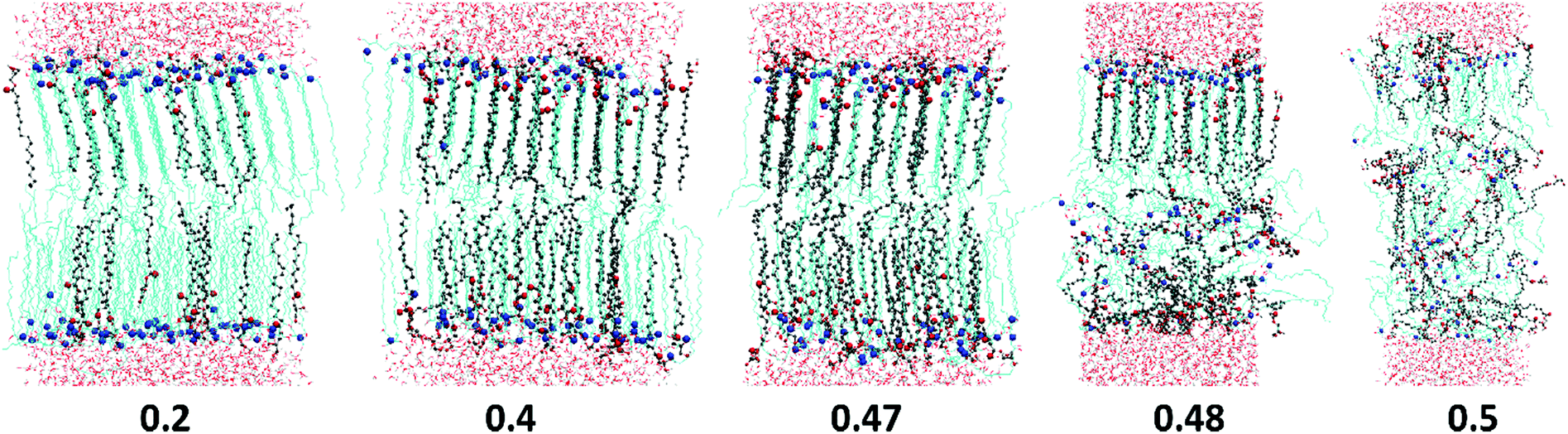 | ||
| Fig. 3 Snapshots of CER2–MO bilayers at different MO mole fractions, XMO, indicated on the image. Ceramide 2 tails are shown as cyan lines and MO tails as grey balls and sticks. Red balls are ester oxygen (O) atoms from MO head groups and blue balls are nitrogen (N) atoms from the CER2 head groups. Water molecules are shown as light grey and red lines, above and below the bilayers. | ||
 | ||
| Fig. 4 Snapshots of CER2–LA bilayers at different LA mole fractions, XLA, indicated on the image. Ceramide 2 tails are shown as cyan lines and LA tails as grey balls and sticks. Red balls are ester oxygen (O) atoms from LA head groups and blue balls are nitrogen (N) atoms from the CER2 head groups. Water molecules are shown as light grey and red lines, above and below the bilayers. | ||
 | ||
| Fig. 5 Snapshots of CER2–SA bilayers at different SA mole fractions, XSA, indicated on the image. Ceramide 2 tails are shown as cyan lines and SA tails as grey balls and sticks. Red balls are ester oxygen (O) atoms from SA head groups and blue balls are nitrogen (N) atoms from the CER2 head groups. Water molecules are shown as light grey and red lines, above and below the bilayers. | ||
Cis-unsaturated lipids such as monoolein,28–31 oleic acid22–28,71 and linoleic acid26,72 are commonly used as skin penetration enhancers for topical (to the skin) and transdermal (across the skin) drug delivery. There are two proposed mechanisms of action of lipidic penetration enhancers: lipid fluidisation and lipid phase separation. The first one involves disruption of the highly ordered SC lipid matrix, thereby increasing the diffusion of drugs22,25,73,74 and the second one proposes that the enhancer forms a separate domain within the lipid phase which acts as permeable defects of the skin barrier.22,24,73,75 In our simulations we have not observed any lipid phase separation; in all the studies oils are found to be completely miscible with the ceramides. However, the limited system size in our simulations may artificially disfavour phase separation. Also, lipid domain formation is usually associated with the mismatch of the lipid tail length and in the considered model CER2 lipids with the C16 and C24 tails are found to be well miscible with C18 FA and MG. Our simulations confirm the suggestion of the lipid tail order disruption by the oils containing cis-double bonds (MO, OA and LA). However, in our simple ceramide model for SC lipids these oils behave not as a lipid phase fluidizer but rather as a “bilayer destroyer” at high concentration. Such an effect probably occurs due to strong rigidity and high tail order of a pure ceramide bilayer. It is worth investigating the subject further using a more realistic mixed lipid model membrane, which is shown to be softer and slightly less ordered than pure CER bilayers.
3.3 Area per lipid and area per chain
The analysis of the lipid packing can be expressed either in terms of the area per lipid, ALIP, defined as the bilayer area projected onto the xy-plane divided by the number of lipids in one leaflet, ALIP = X·Y/NLIP or in terms of the area per chain, or lipid tail, ACHAIN, calculated as the ratio of the same cross-sectional area and the number of lipid tails, ACHAIN = X·Y/NCHAIN. It is reasonable to analyse both quantities because by construction of the mixed system the number of lipids in each leaflet remains constant, NLIP = 64, while the total number of acyl chains, NCHAIN, decreases as the oil mole fraction increases. The higher the oil concentration, the more double-tail CER2 molecules are substituted by single tail MG or FA molecules. For a pure CER2 bilayer NCHAIN = 128, but when all the CER2 are replaced by the oil molecules, the number of hydrocarbon tails in each leaflet becomes equal to the number of lipids, NCHAIN = NLIP = 64. The results for ALIP (XOIL) and ACHAIN (XOIL) are given in Fig. 6.For all the mixed bilayers ALIP gradually decreases with increasing concentration of the oil molecules. This is an expected result because with the increase of oil concentration the total number of acyl chains decreases. The values of ALIP are about 0.4 nm2 for the pure CER2 bilayer and decrease practically linearly down to about 0.2 nm2. Monotonous behaviour of the ALIP (XOIL) is slightly disrupted at low MG or FA content (XOIL < 0.1), where the sharp decrease of the ALIP (XOIL) line for XOIL ≤ 0.05 is followed by the plateau for XOIL = 0.05–0.1. In contrast, the ACHAIN (XOIL) seems to display a small minimum at low concentrations of MG or FA (XOIL = 0.05–0.1) and then it slightly increases with the increase of the oil concentration. We attribute the appearance of this minimum to a more efficient packing of the lipid chains upon replacement of the small amount of double-tail CER2 lipids with a single tail monoglyceride or fatty acid molecules. The errors for ALIP and ACHAIN were calculated as the standard deviation from the mean values obtained from at least three independent simulations and do not exceed 0.007 nm2 or 3.5% for both quantities.
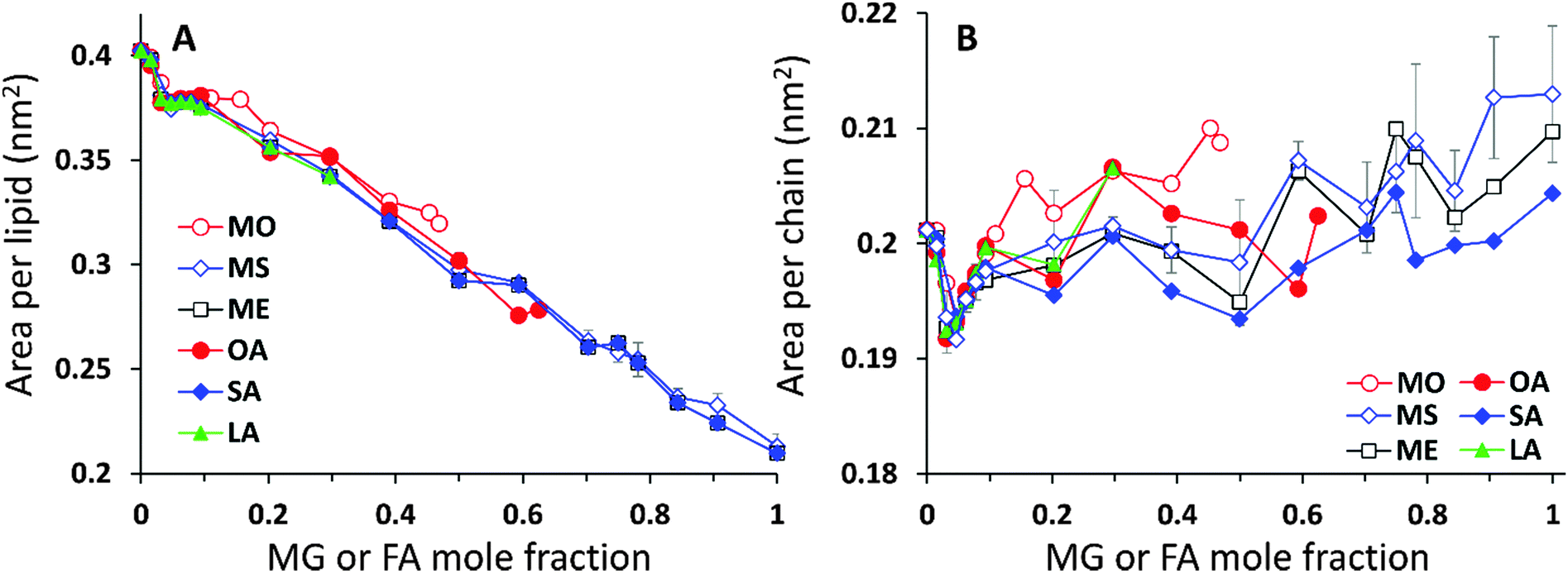 | ||
| Fig. 6 (A) Area per lipid and (B) area per chain for CER2–MG and CER2–FA bilayers vs. mole fraction of MG or FA. The error bars are shown for CER2–MS only; the other error bars are omitted for clarity and are comparable to the error bars of CER2–MS. | ||
3.4 Area compressibility modulus
The area compressibility modulus κA is defined as the energy needed to stretch the bilayer per unit surface area; it is a measure of bilayer stiffness and it is easily calculated from the fluctuations of the bilayer projected area as: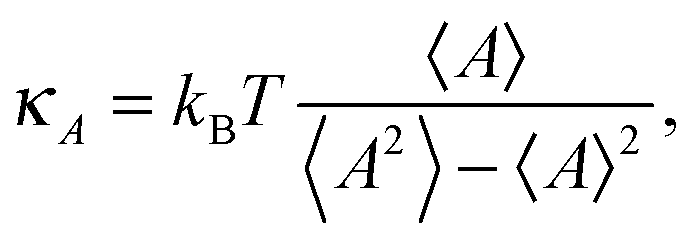 | (1) |
The κA values for mixed bilayers as a function of MG or FA concentration in the CER2 bilayer are presented in Fig. 7A and B, respectively. We found that for both types of added oils at low oil concentration the values for the area compressibility modulus increase by about 50%, up to ∼6000 dyne cm−1, compared against the pure CER2 bilayer. The maximum in κA occurs at XOIL ≈ 0.05 and then the κA values gradually decrease with increasing oil content. The increased membrane rigidity at low oil content correlates very well with the small decrease in the area per chain values (Fig. 6B). A small amount of a single-chain MG or FA promotes tighter local packing of the lipid chains (area per chain becomes slightly smaller), and therefore makes the bilayer more difficult to stretch. It is interesting to note that this effect is observed not only for the saturated or trans-unsaturated oils, but also for those carrying one or two cis-double bonds. Another observation is that we do not see any clear dependence of the bilayer rigidity and lipid packing on the oil head group size. It sounds reasonable that the addition of fatty acids with a smaller head group would encourage tighter packing leading to a more rigid bilayer, but we observe this effect also for monoglycerides, whose head group is not much smaller than that of ceramides. Therefore, we suggest that the tighter lipid packing and the resulting increase in the bilayer rigidity could be explained by the absence of the second hydrocarbon tail in FA and MG and also by the increased amount of the hydrogen bonds between the lipids30 (see the next section).
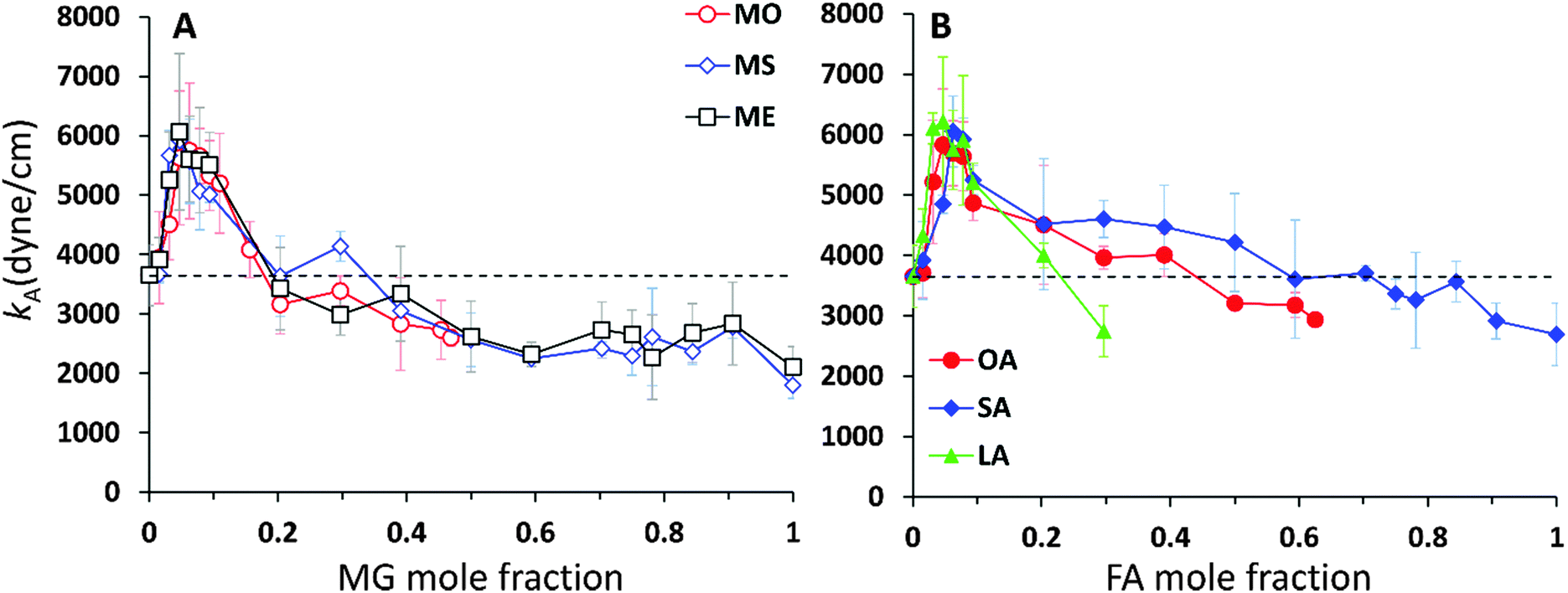 | ||
| Fig. 7 Area compressibility modulus, κA, vs. mole fraction of the monoglyceride or fatty acid for (A) CER2–MG and (B) CER2–FA bilayers indicated on the graphs. The dashed horizontal line on the graph indicates κA for the pure CER2 bilayer. | ||
With increase of oil content, κA values gradually decrease and eventually the mixed bilayers become softer than the pure CER2 bilayer. For mixed CER2–MG bilayers κA values becomes smaller than those for the CER2 bilayer at ∼40% of MG concentration. For LA the transition occurs at ∼30%, for OA – at ∼50%, and the mixed CER2–SA bilayers remain highly rigid until the content of SA reaches 90%. The lowest values of the compressibility modulus for the mixed bilayers are found to be of the order of 2000 dyne cm−1. This value is practically half than that for the pure CER2 bilayer, but it is still much higher than that for liquid crystalline phospholipid bilayers. The mixed bilayers remain in the gel phase (without melting to a fluid phase) over the whole range of concentrations or until the phase transition boundary. A similar effect was also observed upon addition of ethanol to the CER2 bilayer.38 The bilayer retains the ordered gel phase even when the ethanol-induced water pores are formed.
3.5 Hydrogen bonds
CER2 bilayers are characterised by a strong network of hydrogen bonds between the lipid head groups which is holding the bilayer in an ordered gel phase and is responsible for the bilayer rigidity and its very low permeability.11,13,37,78 We have examined the hydrogen bond network of the mixed bilayers as a function of oil concentration. In Fig. 8 we present the number of hydrogen bonds per lipid for mixed CER2–MS and CER2–SA bilayers. We have chosen to present the results for those mixed bilayers, which are stable over the whole range of concentrations. For the bilayers containing other MG and FA the results are very similar till the point the bilayer structure becomes unstable. We have divided all the hydrogen bonds in the system into the following groups: (i) intra-lipid bonds (or self-bonds, formed within one lipid molecule), lipid–lipid bonds (include inter-molecular bonds, formed between the lipids of the same or different types), mixed-lipid bonds (formed between CER2 and oil molecules) and lipid–water bonds. The total number of hydrogen bonds is also calculated. The H-bonds were calculated using the Gromacs tool g_hbond with default parameters for the definition of hydrogen bonds.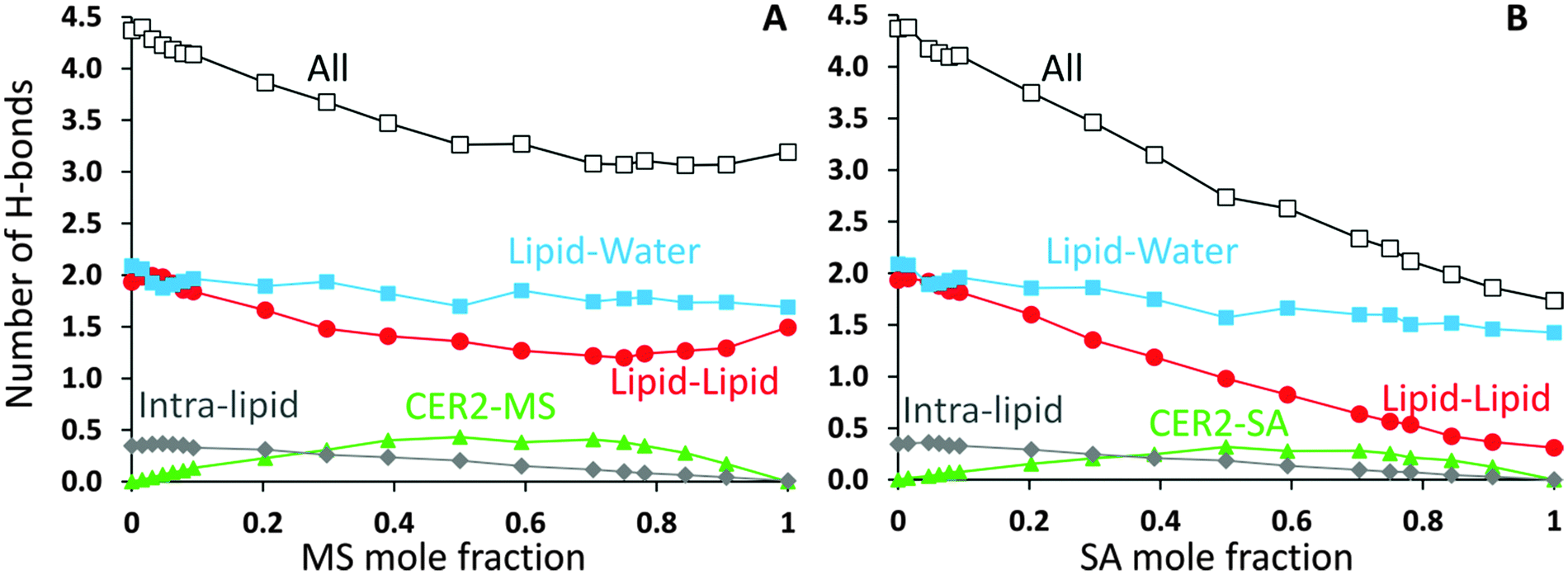 | ||
| Fig. 8 Number of H-bonds per one lipid for (A) CER2–MS and (B) CER2–SA bilayers. The total number of H-bonds (black, open squares) is divided into intra-lipids bonds, mixed bonds, inter-lipid bonds and lipid–water bonds. See text for details. | ||
The average number of intra-molecular bonds is presented by grey diamond lines. It was reported that both ceramides and monoglycerides are capable of forming intra-molecular hydrogen bonds,35,78–81 however we have found that the number of intra-molecular H-bonds in monoglycerides is very low and practically zero (on average 0.01 bonds per one MG). The average number of intra-molecular H-bonds formed within a CER2 lipid was found to be approximately 0.36 and this number is more or less constant throughout all the oil concentrations and also independent of the oil type. Fatty acids do not form intramolecular hydrogen bonds. Therefore, the number of intra-lipid H-bonds per one lipid has a maximum at the highest concentrations of CER2 and then decreasing with oil concentration (as the number of CER2 molecules, contributing mostly into this type of bonds, decreases).
The number of H-bonds formed between different types of lipids is given by green triangle lines. For all systems considered, the number of mixed-lipid bonds is equal to zero at the oil concentration of 0 or 1 as there is only one type of lipid present. The graphs have a broad maximum at the oil mole fractions of about 0.5–0.7 where the amount of both types of lipids is high.
The average number of lipid–lipid hydrogen bonds is given by red circle lines. The bonds formed between lipids have three contributions: inter-lipid bonds from each type of lipids and the mixed-lipid bonds. The number of H-bonds formed between the lipids slightly increases (from 1.9 to 2.0) at low oil concentration (∼5%) and then gradually decreases. The small maximum is more pronounced in the CER2–MS system, but it is also present in other CER2–MG and CER2–FA systems. In the case of MS the number of lipid–lipid H-bonds starts to slightly increase again at an oil content of ∼0.8, reaching the value of 1.5 bonds per lipid. We believe that this occurs due to the increasing number of inter-lipid bonds between monoglycerides, even though the number of inter-lipid bonds involving ceramides strongly reduced. In the case of SA, the number of inter-lipid H-bonds continues to decrease down to 0.3 bonds per lipid.
The number of H-bonds formed between lipids and water molecules is presented by light blue squares. The number of bonds between lipids and water has a small minimum at low oil content and this minimum correlates well with the maximum in lipid–lipid bonds. With increasing oil concentration the number of bonds slowly decreases from 2.1 to 1.7 for CER2–MS bilayers and to 1.4 for CER2–SA bilayers.
The maximum in the lipid–lipid H-bonds and the minimum in lipid–water H-bonds at about 5% of oil concentration are more clearly pronounced in the enlarged graphs, which include the data for all studied mixed bilayers and are presented in Fig. 10C and D in the Summary section. Taking into account the tighter lipid packing and higher values of the area compressibility modulus at these concentrations, we propose that the more efficient lipid packing leads to a higher amount of H-bonds formed between the lipid head groups what contributes to the increased bilayer rigidity.11,46 In the pure ceramide bilayers the excluded volume of the alkyl tails prevents the formation of the 2D percolating H-bond network; instead, the CER head groups arrange in small clusters which are not connected by H-bonds.46 Thus, the inclusion of a small amount of single-tail molecules with smaller head groups may help to fill the gaps between the CER head groups and, therefore, generate the increase in the number of H-bonds formed between lipids. The increase in the number of lipid–lipid H-bonds also results in the reduction of the H-bonds formed between the lipids and water.
The total number of H-bonds per lipid for both types of mixed bilayers is presented by black lines with open squares. Replacing CER2 lipids, which are highly susceptible to H-bonding interactions, by MG or FA, which are less capable to form H-bonds, leads to the reduction of the total number of H-bonds per lipid with increasing oil concentration.
Albeit the total number of H-bonds formed by the lipids decreases with the oil concentration (and this decrease is quite significant in the case of SA, from 4.4 to 1.7), the integrity of the bilayers remains intact for all the oils types considered, with a saturated or trans-unsaturated tail. The reduction of the number of H-bonds, however, occurs gradually, without any sharp transitions, opposite to what was observed in the case of disrupted CER2 bilayers by addition of DMSO molecules.11 It could be expected that strong reduction in hydrogen bonding would promote faster disruption of the bilayer in the case of CER2–FA systems. However, taking into account that the CER2–MO bilayers break at lower oil concentration, compared to the CER2–OA bilayers, we conclude that the reduction in the number of H-bonds formed by lipids does not significantly affect the bilayer stability in our case.
3.6 Tail order
The chemical structure of the lipid tails is clearly reflected in the lipid tail order parameter. The lipid tail order parameter, S, describes the average orientation of the hydrocarbon tails with respect to the bilayer normal. The z-component of the order parameter tensor S for an atom Cn is defined as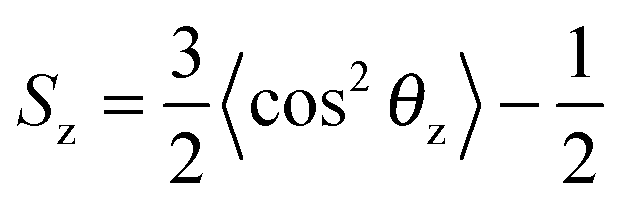 , where θz is the angle between the z-axis of the simulation box (normal to the bilayer) and the molecular axis. The average here uses all molecules of the same type and different time frames from the simulation. For atom n, the molecular axis is defined as the vector from the atom Cn−1 to the atom Cn+1. As defined, the value of Sz depends both on the tilt of the molecular axis about the z-axis and on the variation of the tilt angle either between different molecules or with time. Values of Sz equal to 1 indicate full alignment of the hydrocarbon tails with the bilayer normal, and −1/2 full order perpendicular to the normal.49,82,83 A small Sz value can indicate either an isotropic distribution of the tilt angle, or the tilt angle being close to the “magic angle” θz ≈ cos−1(1/√3). For the cases when the motion of the lipids around the bilayer normal is isotropic, Sz is related to the deuterium order parameter SCD measured in NMR experiments of lipids as Sz = −2SCD.50,83 The order parameter calculations were performed using the Gromacs tool g_order.
, where θz is the angle between the z-axis of the simulation box (normal to the bilayer) and the molecular axis. The average here uses all molecules of the same type and different time frames from the simulation. For atom n, the molecular axis is defined as the vector from the atom Cn−1 to the atom Cn+1. As defined, the value of Sz depends both on the tilt of the molecular axis about the z-axis and on the variation of the tilt angle either between different molecules or with time. Values of Sz equal to 1 indicate full alignment of the hydrocarbon tails with the bilayer normal, and −1/2 full order perpendicular to the normal.49,82,83 A small Sz value can indicate either an isotropic distribution of the tilt angle, or the tilt angle being close to the “magic angle” θz ≈ cos−1(1/√3). For the cases when the motion of the lipids around the bilayer normal is isotropic, Sz is related to the deuterium order parameter SCD measured in NMR experiments of lipids as Sz = −2SCD.50,83 The order parameter calculations were performed using the Gromacs tool g_order.
In Fig. 9A we present the lipid tail order parameter Sz as a function of C atom index n for the long (C24) tail of CER2 lipids at different concentrations of MO. The ordering of the short tail of CER2 (C16) shows a similar behaviour to the long one11,37,38 and therefore the data for that are not presented here. The middle part of the CER2 tail shows strong nematic order with the chains aligning along the z-axis, Sz(n) > 0.8, for all the MO concentration where the mixed bilayer is stable. The high degree of alignment is lost for the terminal bonds (n > 42), as the terminal atoms in the ceramide long tail are in a liquid-like disordered environment and are free to explore configurations not constrained by the local hexagonal packing. A similar loss of alignment can be observed near the head group (n = 27), because the lipid tail needs to respond to the local tilting of the headgroups, as they try to maximise the number of intra-bilayer hydrogen bonds. The high degree of alignment is retained till close to the phase transition concentration: for bilayers containing MO, the tail ordering Sz(n) remains close to 0.8 at XMO = 0.47, close to the bilayer disintegration concentration. However, for the 1:1 CER2–MO composition (where the bilayer structure is no longer stable), the Sz(n) profile drastically drops down to Sz(n) < 0.25. The probability distribution of the tilt angle θz (as shown in Fig. 9C) changes from being peaked around 10–15 degrees for XMO = 0.3 to a broad distribution for XMO = 0.5, indicating an almost complete loss the of tail order. (The extended data for the tilt angle distributions are available in the ESI.†)
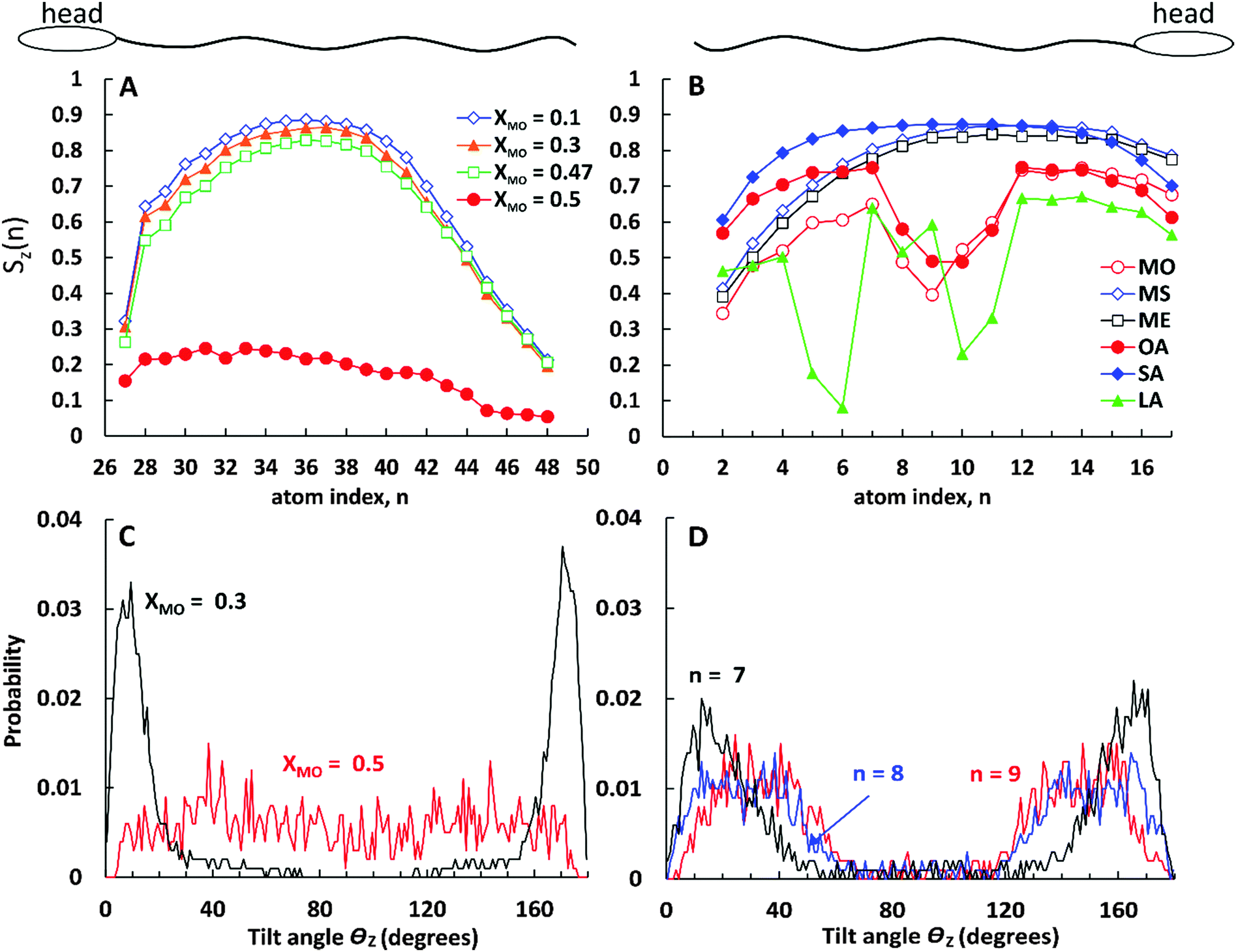 | ||
| Fig. 9 (A and B) Tail order parameter Sz(n) for (A) the C24 (fatty acid) tail of CER2 at different MO concentrations indicated on the graph and (B) for monoglycerides and fatty acids indicated on the graph at the fixed mole fraction of the oils, XOIL = 0.2. Relative locations of the head groups compared to the tail position are illustrated on top of the graphs. (C and D) Tilt angle distribution for (C) atom n = 34 in the long tail of CER2 for MO mole fractions XMO = 0.3 and 0.5 (indicated on the graph) and (D) for atoms n = 7–9 in the oleic acid tail at XOA = 0.2. | ||
Fig. 9B shows the order parameter Sz(n) for monoglycerides and fatty acids at a fixed oil concentration of XOIL = 0.2. Similar to the case of mixed CER2–MO bilayers, the ordering of the lipid tails does not depend much on the oil concentration (except for the cases when the bilayer breaks) and therefore the results are presented for a single oil concentration only.
As can be seen from the plot, all the ingredients with the “straight” tails, i.e. with the saturated tails and the tail with only trans-double bond, show high values of the tail orders (Sz(n) > 0.8 at the middle of the chains) similar to the CER2 tails. The most ordered are the stearic acid (fully saturated) tails. The Sz(n) parameter for SA is near or above 0.8 for all the C(n) atoms except for the one near the head group (n = 16) and the two at the end of the chain (n = 1 and 2). Such a strong order along the chain for SA molecules correlates well with the highest values of the compressibility modulus, κA, reflecting the high rigidity of the mixed CER2–SA bilayers at a high SA content. As for the “straight-chain” monoglycerides, ME (containing one trans-double bond) and MS (fully saturated), their tail order parameter decreases towards the end of the chain (n < 6), which agrees with the decrease of the bilayer rigidity at high MG concentrations. The decrease in Sz here is associated with increasing disorder of the monoglyceride tails because the distribution of the tilt angles for these terminal atoms became broader (see the ESI†).
All the ingredients containing at least one cis-double bond have a pronounced minimum in Sz(n) around the location of the bond. The tilt angle distributions (Fig. 9D) show that this lowering of Sz is associated with the molecular tilt being closer to the magic angle instead of increased disorder. For OA the Sz(n) graph is symmetrical with a minimum in the middle where Sz(n) drops below 0.5 reflecting the symmetrical position of the cis-double bond. The minimum in the Sz(n) for OA has the smallest depth compared to the other two cis-bonded oils and also the values of the order parameter near the head group (n = 11–16) and the tail end (n = 1–6) are reasonably high, Sz(n) > 0.6 for the most of them. These observations are consistent with the fact that the CER2–OA bilayer remains stable at the highest level of added ingredients (until XOA = 0.62) among the other ones with the cis-bond. The MO tail has deeper minimum, which is slightly skewed to the end of the tail, probably because of the larger size of the glycerol head group and therefore more space available for the tail to deviate from the bilayer normal direction. The order of tail atoms near the head group is similar to that of OA but the order of the tail-end is noticeably decreased. These changes in the tail ordering leads to disruption of the mixed bilayer at lower oil content, at XMO = 0.5. Linoleic acid (LA) with two cis-double bonds at C9 and C12 atoms has the weakest tail ordering among all the considered molecules. The two kinks at the tail are well illustrated by the two deep minima at n = 9 and n = 5, where Sz(n) ≈ 0.2 for the first double bond and Sz(n) < 0.1 for the second. It is interesting to note that the location of the minima for LA is not centered around the double bonds as it occurs in the case of OA; the first minimum is slightly shifted to the head of the molecule and the second one to the end of the tail. This could be explained by the fact that the LA chains adjust their conformation to accommodate into the CER2 bilayer and also tend to either go deeper into the bilayer towards the midplane or jump out of it with the large part of the molecule being near the head groups, as can be seen in Fig. 4 illustrating mixed CER2–LA bilayers. The strongly reduced ordering in the LA tail results in the poor stability of the mixed CER2–LA bilayer, which falls apart at XLA ≥ 0.31.
4. Summary
We have carried out atomistic molecular dynamic simulations of fully hydrated mixed bilayers containing the major component of the stratum corneum lipid matrix, ceramide 2, and single-chain natural oil ingredients (monoglycerides and fatty acids) at different concentrations. We studied the dynamics involved in the inclusion of the oil components from long simulations, and have addressed the structural effects induced in a CER2 bilayer by replacing some amount of CER2 lipids by either monoglycerides or fatty acid molecules. The concentration of natural oils in the CER2 bilayer was varied from zero (implying a pure CER2 bilayer) to either one (implying a pure oil bilayer) or up to the concentration at which the mixed bilayer remains stable. We have considered six oil ingredients, all with the C18 tail length: monoolein, monostearin, monoelaidin, oleic acid, stearic acid and linoleic acid. The range of oils studied includes two types of the head groups, glycerol and carboxyl ones, and saturated and mono- and polyunsaturated alkyl tails. Our main findings are summarized as follows.When the lipids are placed randomly in the water phase, they insert spontaneously into the CER2 bilayer. The molecules penetrate into the bilayer interior one by one in an irregular manner. The molecular penetration events into the bilayer occur either by the tail first or head first. We also observed incidents of molecular flip-flop across the leaflets, the reversal of the molecular orientation within the bilayer and the exchange of the molecules between the bilayer and water regions.
The analysis of the area per lipid and area per lipid chain, bilayer rigidity and hydrogen bonding between the different components of the system reveals that at low oil concentrations (∼5%) the substitution of the double-tail CER2 lipids by a single-tail monoglycerides or fatty acids promotes better lipid packing. Better lipid packing leads to the formation of a higher amount of hydrogen bonds between lipids, which also coincides with the reduction of the hydrogen bonds formed between lipids and water. This leads to an increase in the rigidity of the mixed bilayers compared to the pure CER2 bilayer. Probably, the increased membrane rigidity at those oil concentrations arises from extra hydrogen bonds between lipids.46 These conclusions are illustrated in Fig. 10.
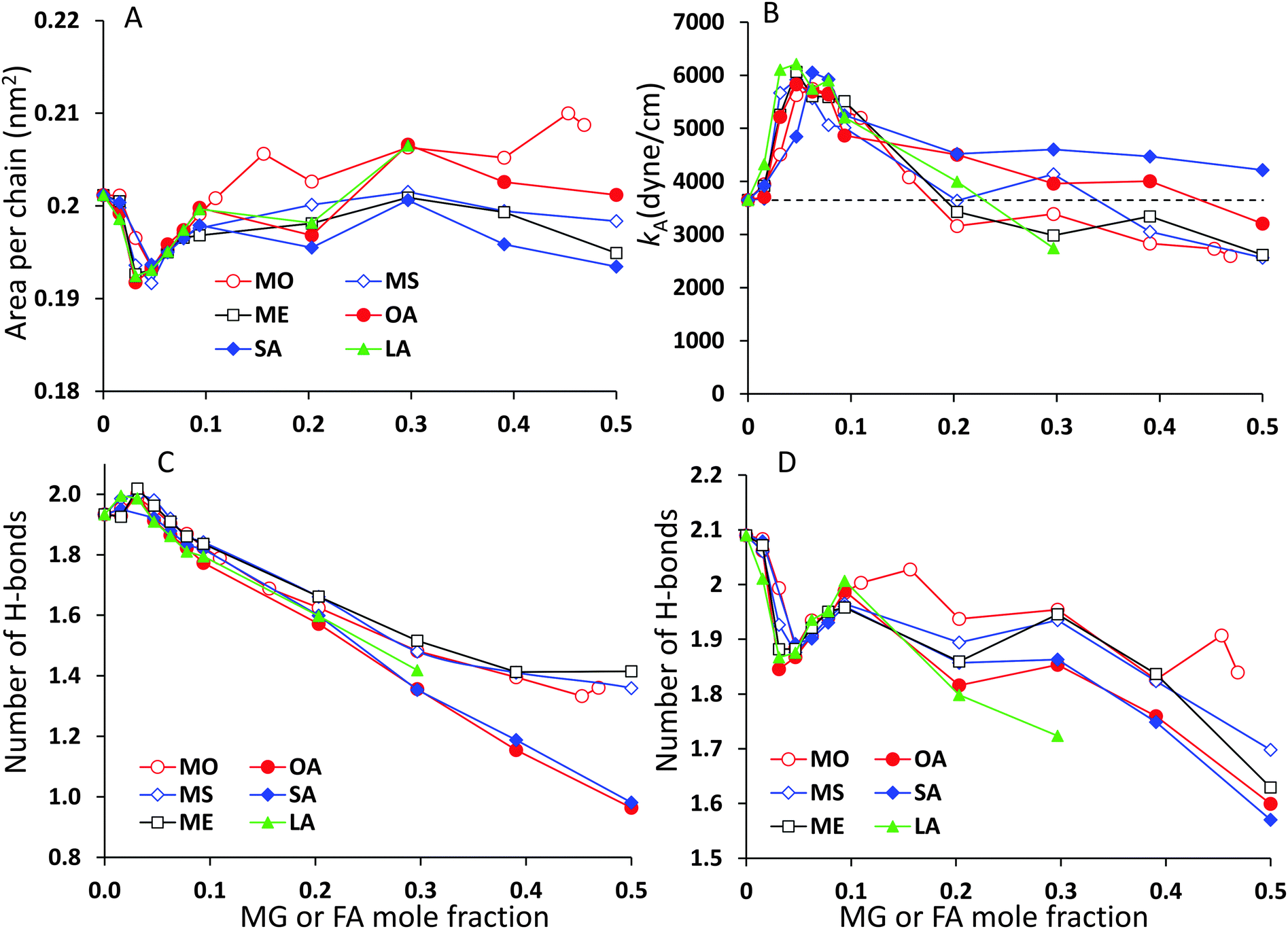 | ||
| Fig. 10 Comparison of (A) compressibility modulus, κA, (B) area per chain, (C) number of hydrogen bonds between lipids per lipid, and (D) number of hydrogen bonds between lipids and water per lipid for different oil ingredients. | ||
All the oil ingredients with the “straight” hydrocarbon tail, saturated one (stearic acid, monostearin) or containing only a trans-double bond (monoelaidin), accommodate well into the CER2 bilayer, so that the mixed CER2–oil bilayer is stable over the whole range of oil concentrations. Alternatively, the unsaturated oils containing a cis-double bond which causes a “kink” in the acyl chain, such as monoolein and oleic acid, induce bilayer instability followed by a further disintegration with increasing oil concentration. When the composition of lipids in the bilayer approaches some critical value (50–65%), the bilayer loses its integrity and undergoes a phase transition to a phase with different mesostructure. Apparently, poly-unsaturated linoleic acid, containing two cis-double bonds on its acyl chain, induces bilayer disruption at even lower content (about 30%). However, the bilayers remain in the gel phase until the phase transition to the non-lamellar phase occurs.
Acknowledgements
The authors thank Prof. Peter Olmsted (University of Georgetown, USA) and Dr Rebecca Notman (University of Warwick, UK) for fruitful discussions and help. The authors thank Unilever R&D Port Sunlight, Huddersfield University and STFC (SCARF) HPC facilities for computational resources.References
- A. S. Michaels, S. K. Chandrasekaran and J. E. Shaw, AIChE J., 1975, 21, 985–996 CrossRef CAS.
- P. M. Elias, J. Controlled Release, 1991, 15, 199–208 CrossRef CAS.
- C. R. Harding, Dermatol. Ther., 2004, 17, 6–15 CrossRef PubMed.
- R. R. Wickett and M. O. Visscher, Am. J. Infect. Control, 2006, 34, S98–S110 CrossRef.
- A. V. Rawlings, I. R. Scott, C. R. Harding and P. A. Bowser, J. Invest. Dermatol., 1994, 103, 731–740 CrossRef CAS PubMed.
- Y. Jokura, S. Ishikawa, H. Tokuda and G. Imokawa, J. Invest. Dermatol., 1995, 104, 806–812 CrossRef CAS PubMed.
- A. V. Rawlings and C. R. Harding, Dermatol. Ther., 2004, 17, 43–48 CrossRef PubMed.
- A. Akinshina, E. Jambon-Puillet, P. B. Warren and M. G. Noro, BMC Biophys., 2013, 6, 12 CrossRef PubMed.
- M. Ponec, A. Weerheim, P. Lankhorst and P. Wertz, J. Invest. Dermatol., 2003, 120, 581–588 CrossRef CAS PubMed.
- J. J. Leyden and A. V. Rawlings, Skin Moisturization, Marcel Dekker, New York, 2002 Search PubMed.
- R. Notman, W. K. den Otter, M. G. Noro, W. J. Briels and J. Anwar, Biophys. J., 2007, 93, 2056–2068 CrossRef CAS PubMed.
- C. Das, M. G. Noro and P. D. Olmsted, Soft Matter, 2014, 10, 7346–7352 RSC.
- C. Das, P. D. Olmsted and M. G. Noro, Soft Matter, 2009, 5, 4549–4555 RSC.
- M. A. Kiselev, N. Y. Ryabova, A. M. Balagurov, S. Dante, T. Hauss, J. Zbytovska, S. Wartewig and R. H. H. Neubert, Eur. Biophys. J., 2005, 34, 1030–1040 CrossRef CAS PubMed.
- J. A. Bouwstra, F. E. R. Dubbelaar, G. S. Gooris and M. Ponec, Acta Derm.-Venereol. Suppl., 2000, 208, 23–30 CAS.
- P. W. Wertz, Acta Derm.-Venereol. Suppl., 2000, 208, 7–11 CrossRef CAS.
- L. Blasco, L. Duracher, J. P. Forestier, L. Vian and G. Marti-Mestres, J. Dispersion Sci. Technol., 2006, 27, 799–810 CrossRef.
- M. Picardo, M. Ottaviani, E. Camera and A. Mastrofrancesco, Dermatoendocrinol, 2009, 1, 68–71 CrossRef CAS PubMed.
- P. W. Wertz and B. Van Den Bergh, Chem. Phys. Lipids, 1998, 91, 85–96 CrossRef CAS PubMed.
- J. W. Fluhr, J. Kao, M. Jain, S. K. Ahn, K. R. Feingold and P. M. Elias, J. Invest. Dermatol., 2001, 117, 44–51 CrossRef CAS PubMed.
- M. Höltje, T. Förster, B. Brandt, T. Engels, W. Von Rybinski and H. D. Höltje, Biochim. Biophys. Acta, Biomembr., 2001, 1511, 156–167 CrossRef.
- M. E. Lane, Int. J. Pharm., 2013, 447, 12–21 CrossRef CAS PubMed.
- B. W. Barry, J. Controlled Release, 1987, 6, 85–97 CrossRef CAS.
- A. C. Williams and B. W. Barry, Adv. Drug Delivery Rev., 2012, 64, 128–137 CrossRef.
- S. Songkro, Songklanakarin J. Sci. Technol., 2009, 31, 299–321 Search PubMed.
- S. Santoyo and P. Ygartua, Eur. J. Pharm. Biopharm., 2000, 50, 245–250 CrossRef CAS PubMed.
- H. Tanojo, J. A. Bouwstra, H. E. Junginger and H. E. Bodde, Pharm. Res., 1997, 14, 42–49 CrossRef CAS.
- L. B. Lopes, C. M. Brophy, E. Furnish, C. R. Flynn, O. Sparks, P. Komalavilas, L. Joshi, A. Panitch and M. V. L. B. Bentley, Pharm. Res., 2005, 22, 750–757 CrossRef CAS PubMed.
- L. B. Lopes, J. H. Collett and M. V. Bentley, Eur. J. Pharm. Biopharm., 2005, 60, 25–30 CrossRef CAS PubMed.
- L. B. Lopes, J. L. Lopes, D. C. Oliveira, J. A. Thomazini, M. T. Garcia, M. C. Fantini, J. H. Collett and M. V. Bentley, Eur. J. Pharm. Biopharm., 2006, 63, 146–155 CrossRef CAS PubMed.
- L. B. Lopes, F. F. Speretta and M. V. Bentley, Eur. J. Pharm. Sci., 2007, 32, 209–215 CrossRef CAS PubMed.
- K. P. Ananthapadmanabhan, L. Yang, C. Vincent, L. Tsaur, K. Vetro, V. Foy, S. Zhang, A. Ashkenazi, E. Pashkovski and V. Subramanian, Cosmet. Dermatol., 2009, 22, 307–316 Search PubMed.
- K. P. Ananthapadmanabhan, S. Mukherjee and P. Chandar, Int. J. Cosmet. Sci., 2013, 35, 337–345 CrossRef CAS PubMed.
- K. Morimoto, H. Tojima, T. Haruta, M. Suzuki and M. Kakemi, J. Pharm. Pharmacol., 1996, 48, 1133–1137 CrossRef CAS PubMed.
- S. A. Pandit and H. L. Scott, J. Chem. Phys., 2006, 124, 014708 CrossRef PubMed.
- R. Notman, J. Anwar, W. J. Briels, M. G. Noro and W. K. Den Otter, Biophys. J., 2008, 95, 4763–4771 CrossRef CAS PubMed.
- C. Das, M. G. Noro and P. D. Olmsted, Biophys. J., 2009, 97, 1941–1951 CrossRef CAS PubMed.
- R. Thind, D. W. O'Neill, A. Del Regno and R. Notman, Chem. Commun., 2015, 51, 5406–5409 RSC.
- S. Guo, T. C. Moore, C. R. Iacovella, L. A. Strickland and C. McCabe, J. Chem. Theory Comput., 2013, 9, 5116–5126 CrossRef CAS PubMed.
- M. Paloncyova, R. H. DeVane, B. P. Murch, K. Berka and M. Otyepka, Langmuir, 2014, 30, 13942–13948 CrossRef CAS PubMed.
- R. Gupta and B. Rai, J. Phys. Chem. B, 2015, 119, 11643–11655 CrossRef CAS PubMed.
- R. Notman, M. G. Noro and J. Anwar, J. Phys. Chem. B, 2007, 111, 12748–12755 CrossRef CAS PubMed.
- M. I. Hoopes, M. G. Noro, M. L. Longo and R. Faller, J. Phys. Chem. B, 2011, 115, 3164–3171 CrossRef CAS PubMed.
- O. Berger, O. Edholm and F. Jahnig, Biophys. J., 1997, 72, 2002–2013 CrossRef CAS PubMed.
- C. Das, M. G. Noro and P. D. Olmsted, Phys. Rev. Lett., 2013, 111, 148101 CrossRef PubMed.
- C. Das and P. D. Olmsted, Phil. Trans. R. Soc. A 20150126, 2016, arXiv:1510.08939.
- R. Notman and J. Anwar, Adv. Drug Delivery Rev., 2013, 65, 237–250 CrossRef CAS PubMed.
- P. Niemela, M. T. Hyvonen and I. Vattulainen, Biophys. J., 2004, 87, 2976–2989 CrossRef PubMed.
- D. Van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- M. Patra, E. Salonen, E. Terama, I. Vattulainen, R. Faller, B. W. Lee, J. Holopainen and M. Karttunen, Biophys. J., 2006, 90, 1121–1135 CrossRef CAS PubMed.
- D. P. Tieleman, H. J. C. Berendsen and M. S. P. Sansom, Biophys. J., 1999, 76, 3186–3191 CrossRef CAS PubMed.
- D. P. Tieleman, H. J. C. Berendsen and M. S. P. Sansom, Biophys. J., 1999, 76, 1757–1769 CrossRef CAS PubMed.
- D. P. Tieleman, M. S. P. Sansom and H. J. C. Berendsen, Biophys. J., 1999, 76, 40–49 CrossRef CAS PubMed.
- E. Mombelli, R. Morris, W. Taylor and F. Fraternali, Biophys. J., 2003, 84, 1507–1517 CrossRef CAS PubMed.
- M. Laner, B. A. C. Horta and P. H. Hünenberger, J. Mol. Graphics Modell., 2015, 55, 48–64 CrossRef CAS PubMed.
- R. Tjörnhammar and O. Edholm, J. Chem. Theory Comput., 2014, 10, 5706–5715 CrossRef PubMed.
- S. W. Chiu, S. A. Pandit, H. L. Scott and E. Jakobsson, J. Phys. Chem. B, 2009, 113, 2748–2763 CrossRef CAS PubMed.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed.
- W. F. van Gunsteren, P. Kruger, S. R. Billeter, A. E. Mark, A. A. Eising, W. R. P. Scott, P. H. Huneberg and I. G. Tironi, Biomolecular Simulation: The GROMOS96 Manual and User Guide., Vdf Hochschulverlag AG an der ETH Zürich, Zürich, Switzerland, 1996 Search PubMed.
- W. L. Jorgensen and J. Tiradorives, J. Am. Chem. Soc., 1988, 110, 1657–1666 CrossRef CAS.
- J. P. Ryckaert and A. Bellemans, Chem. Phys. Lett., 1975, 30, 123–125 CrossRef CAS.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef.
- S. Nose, Mol. Phys., 1984, 52, 255–268 CrossRef CAS.
- S. Nose and M. L. Klein, Mol. Phys., 1983, 50, 1055–1076 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- K. Larsson, Lipids – Molecular Organization, Physical Functions and Technical Applications, The Oily Press Ltd, Dundee, UK, 1994 Search PubMed.
- M. T. Hyvönen, K. Öörni, P. T. Kovanen and M. Ala-Korpela, Biophys. J., 2001, 80, 565–578 CrossRef.
- M. J. Kim, H. J. Doh, M. K. Choi, S. J. Chung, C. K. Shim, D. D. Kim, J. S. Kim, C. S. Yong and H. G. Choi, Drug Delivery, 2008, 15, 373–379 CrossRef CAS PubMed.
- S. A. Ibrahim and S. K. Li, Pharm. Res., 2010, 27, 115–125 CrossRef CAS PubMed.
- A. Naik, L. A. R. M. Pechtold, R. O. Potts and R. H. Guy, J. Controlled Release, 1995, 37, 299–306 CrossRef CAS.
- E. H. Mojumdar, R. W. Helder, G. S. Gooris and J. A. Bouwstra, Langmuir, 2014, 30, 6534–6543 CrossRef CAS PubMed.
- B. Ongpipattanakul, R. R. Burnette, R. O. Potts and M. L. Francoeur, Pharm. Res., 1991, 8, 350–354 CrossRef CAS.
- S. L. Duncan and R. G. Larson, Biophys. J., 2008, 94, 2965–2986 CrossRef CAS PubMed.
- J. J. Lopez Cascales, A. Garro, R. D. Porasso and R. D. Enriz, Phys. Chem. Chem. Phys., 2014, 16, 21694–21705 RSC.
- I. Pascher, Biochim. Biophys. Acta, Biomembr., 1976, 455, 433–451 CrossRef CAS.
- A. Holmgren, G. Lindblom and L. B. Å. Johansson, J. Phys. Chem., 1988, 92, 5639–5642 CrossRef CAS.
- H. L. Brockman, M. M. Momsen, R. E. Brown, L. He, J. Chun, H. S. Byun and R. Bittman, Biophys. J., 2004, 87, 1722–1731 CrossRef CAS PubMed.
- M. Öğütcü, N. Arifoğlu and E. Yilmaz, J. Am. Oil Chem. Soc., 2015, 92, 459–471 CrossRef.
- E. Egberts, S. J. Marrink and H. J. C. Berendsen, Eur. Biophys. J., 1994, 22, 423–436 CrossRef CAS PubMed.
- D. P. Tieleman, D. Van Der Spoel and H. J. C. Berendsen, J. Phys. Chem. B, 2000, 104, 6380–6388 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp01238h |
| This journal is © the Owner Societies 2016 |