
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The removal of disulfide bonds in amylin oligomers leads to the conformational change of the ‘native’ amylin oligomers†
Vered
Wineman-Fisher
ab,
Lucia
Tudorachi
c,
Einav
Nissim
ab and
Yifat
Miller
*ab
aDepartment of Chemistry, Ben-Gurion University of the Negev, P.O. Box 653, Beér Sheva 84105, Israel. E-mail: ymiller@bgu.ac.il; Fax: +972-86428709; Tel: +972-86428705
bIlse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, Beér-Sheva 84105, Israel
cAl. I. Cuza University of Iasi, 11 Carol I, Iasi-700506, Romania
First published on 13th April 2016
Abstract
The α-helical structure of the N-terminus of the ‘native’ amylin Lys1–Cys7 consists of a disulfide bond between Cys2 and Cys7. The ‘native’ amylin oligomers demonstrate polymorphic states. Removal of the disulfide bonds in the ‘native’ amylin oligomers decreases the polymorphism and induces the formation of longer stable cross-β strands in the N-termini.
Type 2 diabetes (T2D) is a part of a group of amyloidogenic diseases, among them are Alzheimer's disease (AD) and Parkinson's disease (PD). The common hallmarks of these amyloidogenic diseases are the deposition of misfolded proteins, such as Aβ deposits in AD, α-synuclein in PD and amylin or human Islet amyloid polypeptide (hIAPP) in T2D. Amylin is a 37-residue peptide hormone that is normally co-secreted with insulin from the β-cells of the pancreas. In more than 95% of patients with T2D, amylin deposits lead to the loss of β-cell mass and consequently to the reduction in insulin production.1 The soluble amylin under physiological conditions is in an unstructured form.2,3 The N-terminus of amylin consists of a disulfide bond between Cys2 and Cys7, which is required for activity and the C-terminus is amidated.4
Disulfide bonds are presented in more than 50% of misfolded proteins that are involved in amyloidogenic diseases.5 Disulfide bonds are important covalent bonds that are necessary with a wide range of protein properties, such as folding, stability, oligomerization and amyloidogenicity. The role of disulfide bonds in amyloidogenic proteins has been reviewed by Li et al.6 Previous studies have shown that modifications of the disulfide bonds may change the morphology of amyloids7,8 and the chemicals that stabilize or disrupt disulfide bonds may affect the amyloid aggregation.9,10
Native disulfide bonds play crucial roles in protein stability and disruptions of the disulfide bonds usually destabilize the native structure of proteins.11 Consequently, the disruptions of disulfide bonds induce the misfolded proteins to adopt a cross-β structure more easily and thus increase amyloidogenicity.12–14 Furthermore, native disulfide bonds protect hydrophobic residues from being accessed by solvents and thus assist in the folding of the misfolded amyloidogenic proteins,15,16 leading to the formation of oligomers, protofibrils and fibrils.17 The non-native disulfide bonds, especially inter-molecular disulfide cross-linking, play roles in protein aggregation.18,19 The formation of the non-native disulfide bonds usually leads to protein misfolding, aggregation and malfunction.20,21
The disulfide bond in the amylin forms a helix-like structure and therefore does not participate in the cross-β structure of the amylin aggregation.22,23 It has been shown that the disulfide bond in amylin limits the aggregation propensity of amylin.24 The removal of the disulfide bond in amylin eliminates the lag phase of aggregation and accelerates the aggregation.25,26 It was suggested that the disulfide bond in the amylin monomer affects amylin aggregation in two pathways. First, it protects the amylin monomer from adopting a β-sheet structure by stabilizing the N-terminal α-helical structure.27,28 Second, the disulfide loop in the amylin monomer interacts with highly amyloidogenic regions of other amylin monomers and therefore protects these highly amyloidogenic regions from interacting with each other, i.e. prevents the self-assembly of amylin.29 However, the aggregation mechanisms with regard to the role of disulfide bonds in amylin to form oligomers (which are the toxic species) at the molecular level are still elusive. In this paper we examine the effect of the removal of the disulfide bonds in amylin oligomers. We particularly focus on how the removal of the disulfide bonds affects the morphology of amylin oligomers, the conformational change of the various amylin oligomers and the populations.
We have applied our previous amylin oligomeric models:30 M1, M2, M5 and M6 that are based on experimental ssNMR23 and crystal structures.31 In this work we annotated models M1, M2, M5 and M6 as models M1, M2, M3 and M4, respectively. We then removed the disulfide bonds between Cys2 and Cys7 in each amylin monomer for each oligomeric model and constructed eight new amylin oligomeric models: D1–D4 and E1–E4 (Fig. S1 and S2, ESI†). In models D1–D4 the disulfide bonds were removed and the helical structures of the N-termini (Lys1–Cys7) remained. In models E1–E4 the disulfide bonds were removed and the N-termini (Lys1–Cys7) of the amylin monomers were extended to form β-strand structures. In order to study the relative conformational energies and the structures of the various amylin oligomers, we applied all atom explicit molecular dynamics (MD) simulations (ESI†) for each one of these amylin oligomers in the NPT ensemble at 330 K for 50 ns, using the NAMD program32 with the all-atom CHARMM2733,34 force-field. The simulated oligomers E1–E4 are seen in Fig. 1 and the simulated oligomers D1–D4 are seen in Fig. S3 (ESI†).
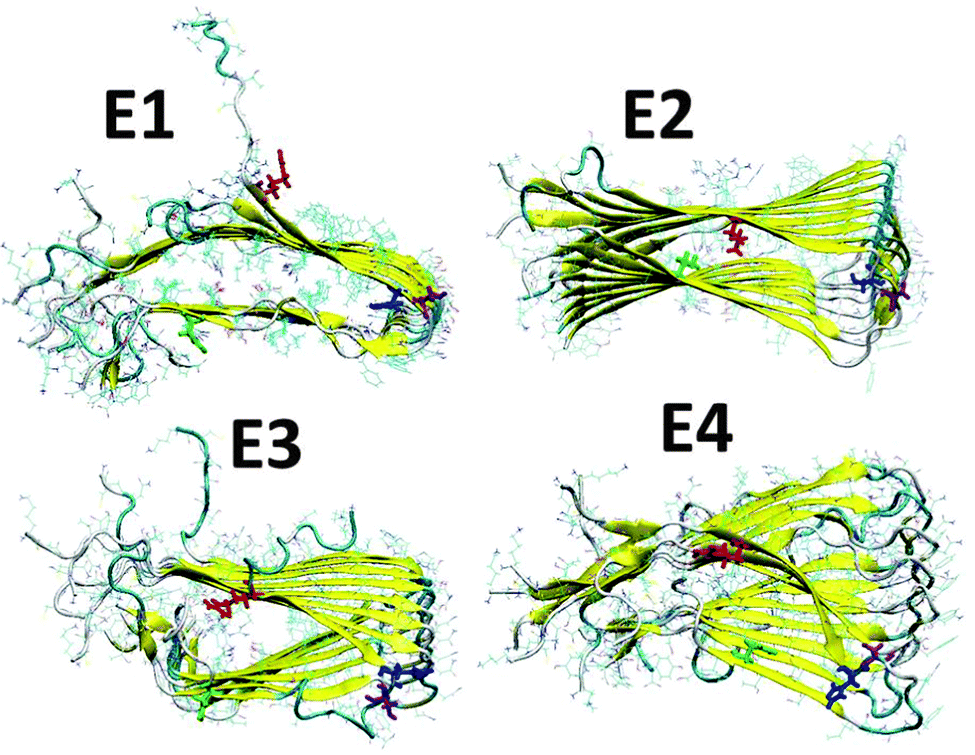 | ||
| Fig. 1 Simulated models of amylin oligomers through which the disulfide bonds between Cys2 and Cys7 were removed. Residue colors: Arg11 (red), His18 (blue), Asn19 (purple) and Asn31 (green). | ||
The preference of each model for a specific arrangement has been obtained by comparing the relative conformational energies of all constructed models, using the generalized Born method with molecular volume (GBMV) (ESI†).35,36 By applying Monte Carlo (MC) simulations, the relative probability of model n was evaluated as Pn = Nn/Ntotal, where Pn is the population of model n, Nn is the total number of conformations visited for model n, and Ntotal is the total steps. The advantages of using Monte Carlo (MC) simulations to estimate conformer probabilities lie on the good numerical stability and on control of the transition probabilities among the conformers, as we have extensively applied in similar systems. We compared the relative conformational energies and the populations between D1–D4 and E1–E4 in order to learn whether starting from helical structures to extended β-strands in the N-termini of amylin oligomers have different conformational energies and different populations after MD simulations. Interestingly, after MD simulations the amylin oligomeric models D1–D4 with the initially helical structure illustrated similar populations to the amylin oligomeric models E1–E4, respectively (Fig. S4, ESI†). These results indicate that different initial structural states of amylin oligomers in which their disulfide bonds are removed lead to similar populations. We also compared the relative conformational energies and the populations between all four amylin oligomers D1–D4, all four amylin oligomers E1–E4 and the ‘native’ amylin oligomers M1, M2, M3 and M4 (Fig. 2 and Fig. S5 and Table S1, ESI†). One can see that while in the ‘native’ amylin oligomers there are two preferred polymorphic states (models M1 and M2), but when the disulfide bonds in the native' amylin oligomers are removed only one oligomeric model is preferred: model D2/E2 which is equivalent to model M2. Therefore, we propose that the removal of the disulfide bond leads to a slight decrease of polymorphism in amylin oligomers and induces conformational change of the populations of the ensemble of amylin oligomers.
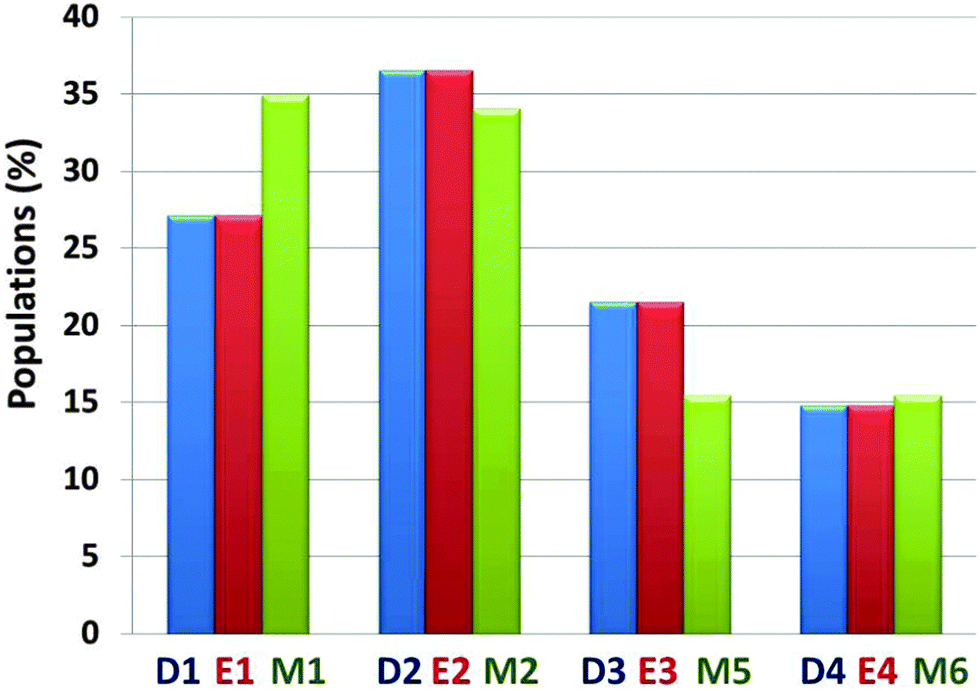 | ||
| Fig. 2 Calculated populations of the constructed models of the ‘native’ amylin oligomers (models M1–M4) and the constructed models after removal of the disulfide bonds (models D1–D4 and E1–E4) using the GBMV method43,44 and Monte Carlo simulations. | ||
There is great interest in investigating the effect of the removal of the disulfide bond in amylin on the structural features of amylin oligomers in order to provide insights into the effect on amylin aggregation. Previous studies have shown that the removal of the disulfide bond in amylin monomers promotes the formation of β-strands along the sequence of amylin monomers.27,28 These studies however were focused on the amylin monomer level and not on the oligomer level, which are known to be the toxic species. Herein, we examine the effect of the removal of the disulfide bond on the structural features of amylin oligomers. After removing the disulfide bond in amylin oligomers the N-termini are free, flexible to move and do not have a helix-like structure. Interestingly, during the MD simulations Asn3 that has shown β-strand properties in the ‘native’ amylin oligomers (with exclusion of model M4) does not exhibit β-strand properties in models D1–D4 and E1–E4 (Fig. 3). However, these models show some β-strand properties in the sequence 4Thr–Ala–Thr–7Lys, which are not shown in the ‘native’ amylin oligomers in this sequence. We, therefore, suggest that the removal of the disulfide bond promotes the elongation of the β-strand that is located in 8Ala–18His and thus may induce the formation of a stable cross-β structure that leads to the promotion of amylin aggregation. Interestingly, the formation of β-strand properties in the N-termini of models D1–D4 and E1–E4 disrupts the C-termini to exhibit β-strand properties (Fig. 3): the sequence of 35Asn–Thr37 Tyr that illustrates β-strand properties in M1–M4 does not exhibit β-strand properties in models D1–D4 and E1–E4.
 | ||
| Fig. 3 The secondary structure of the simulated models of the ‘native’ amylin oligomers (models M1–M4) and the constructed models after removal of the disulfide bonds (models D1–D4 and E1–E4) along the sequence of amylin. The arrows illustrate the β-strand structure. | ||
We further compared the stability of the ‘native’ amylin oligomers M1–M4 with models D1–D4 and E1–E4 by investigating their backbone solvation. It is expected that the N-termini (residues Lys1–Cys7) of models D1–D4 and E1–E4 will be highly exposed to the solute in comparison with the helix-like structure of the N-termini of models M1–M4, and thus these models will be more solvated in this domain. Interestingly, all the ‘native’ amylin oligomers and the models studied here show a similar solvation backbone (Fig. S6, ESI†). The N-terminal domain (Lys1–Ala8), Leu12, Ser29, and the C-terminal domain (Gly33–Tyr37) are relatively highly solvated. The turn domain (Ser19–Gly24) is also highly solvated in all models.
To examine the structural stability of the ‘native’ amylin oligomers in comparison to the models studied here, it is important to investigate whether the fibril-like structure is well packed by following the percentage of the hydrogen bonds along the cross-β structure during the MD simulations, and by measuring the inter-sheet distance between two β-strands within each monomer in each model. Furthermore, one can monitor the root-mean-square deviations (RMSDs) and the root-mean-square fluctuations (RMSFs) of all models in order to investigate their stability. All the ‘native’ amylin oligomers and the models studied here demonstrate a well-packed fibril-like structure (Fig. S7 and S8, ESI†) with a relatively large percentage of hydrogen bonds along the cross-β structure, which are retained through the MD simulations (Fig. S9 and S10, ESI†). Interestingly, one can see from Fig. S11 (ESI†) that the inter-sheet distances of the ‘native’ amylin oligomer models M1 and M2 are similar to those obtained in models D1 and D2 and E1 and E2 (Fig. S7 and S8, ESI†). However, the inter-sheet distances of models D3 and D4 and E3 and E4 are dramatically smaller than those of models M3 and M4. Interestingly, models M1, M2, D1, D2, E1 and E2 have similar orientations of the residues along the β-strands, and the orientations of residues along the β-strands are similar for models M3, M4, D3, D4, E3 and E4. Therefore, one can divide these models into two groups, according to the orientations of the residues along the β-strands: group one includes models M1, M2, D1, D2, E1 and E2, and the second group includes models M3, M4, D3, D4, E3 and E4. We therefore suggest that the removal of disulfide bonds in amylin oligomers does not affect the packing of the cross-β structure of amylin oligomers in group one, while the disulfide bonds stabilize the packing of the cross-β structure of amylin oligomers in the second group.
Finally, the RMSDs (Fig. S12 and S13, ESI†) show that all the ‘native’ amylin oligomers and all the models studied here exhibit similar values of RMSDs. Therefore, all models have relatively stable structures thus indicating that all models converged during the simulations of 50 ns. The RMSFs (Fig. 4) for all the models of the ‘native’ amylin oligomers and the models studied here show a similar scenario. However, one can see that the N-termini (residues Lys1–Asn3) in models D1–D4 and E1–E4 relatively show more fluctuations in comparison to these residues in models M1–M4. But these fluctuations compensate the relatively low fluctuations in the turn domain of models D1–D4 and E1–E4. While in the ‘native’ amylin oligomers (particularly model M4) the turn domain fluctuates relatively more, in models D1–D4 and E1–E4 the fluctuations in the turn domain are smaller. We therefore suggest that the removal of the disulfide bond in the amylin oligomers stabilizes the turn domain and therefore may induce amylin aggregation.
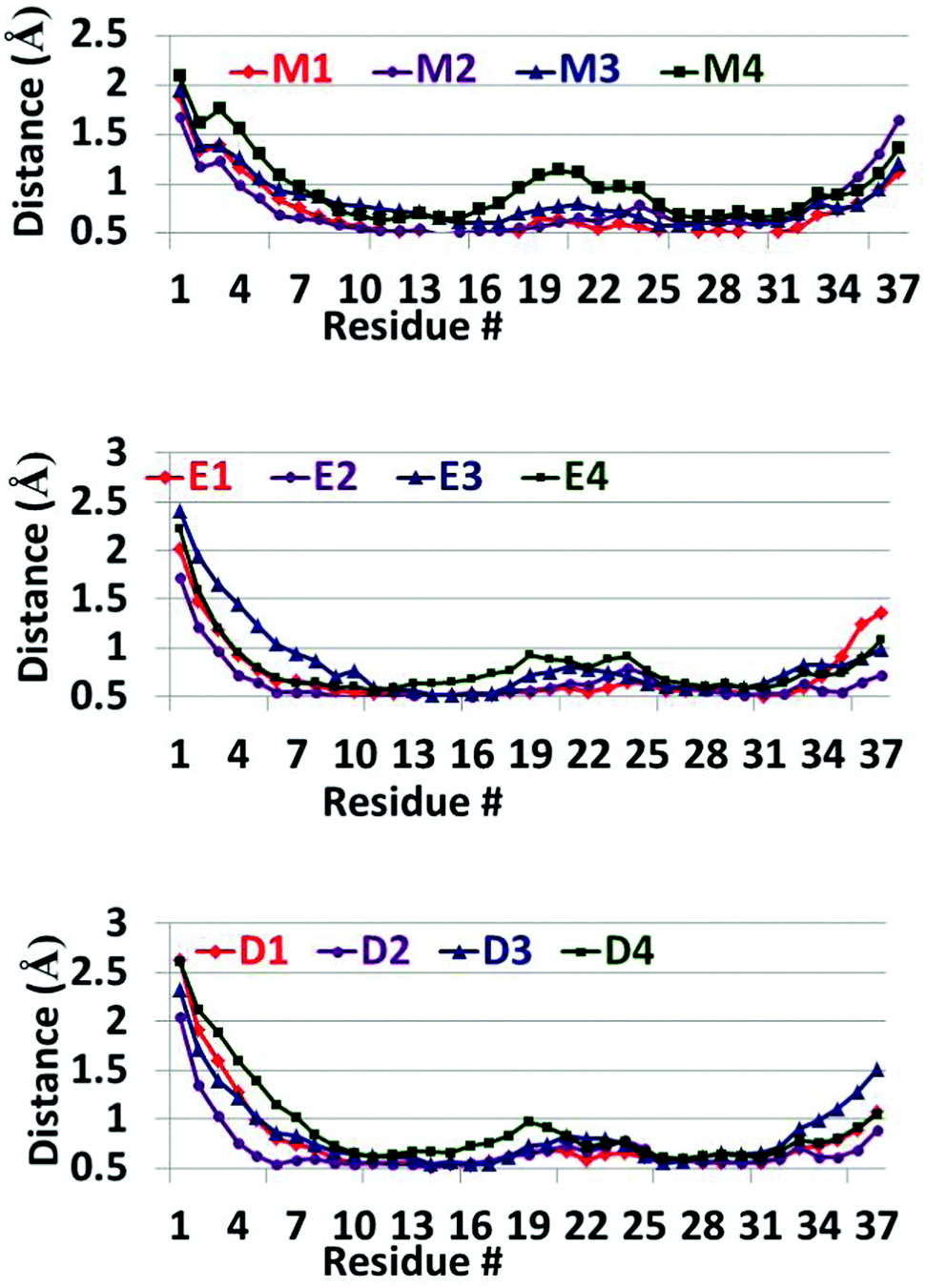 | ||
| Fig. 4 RMSFs of the simulated models of the ‘native’ amylin oligomers (models M1–M4) and the constructed models after removal of the disulfide bonds (models D1–D4 and E1–E4). | ||
We also estimated the helicity pitch values of the ‘native’ amylin oligomers (models M1–M4) and models D1–D4 and E1–E4 (Table S2, ESI†). We compared the computed helicity pitch values with the experimental value that has been obtained by Eisenberg and coworkers.31 While models M1–M4 show similar values of helicity pitch and agree with the experimental value, models D1–D4 and E1–E4 do not agree with the experimental value and exhibit relatively large values. The experimental helicity pitch value has been obtained for the ‘native’ amylin oligomers. Therefore, it is expected that removal of the disulfide bond may change the helicity pitch values. We thus propose that removal of disulfide bonds in amylin oligomers allows the N-termini to be flexible and therefore induces the formation of a more twisted helical cross-β structure and larger helicity pitch values.
In summary, the ‘native’ amylin oligomers demonstrate polymorphism, i.e. there are several structural models of amylin oligomers that are produced. The polymorphism is a common phenomenon in amyloids, among them are Aβ,37,38 prions39 and tau and mutated repeats.40–42 Herein, by applying all-atom explicit MD simulations we examined the effect of the removal of the disulfide bonds in the ‘native’ amylin self-assembled oligomers considering the amylin oligomers that have been observed by the experiment.23,31 Our study leads to several conclusions: first, the removal of the disulfide bonds decreases the polymorphism of the ‘native’ amylin oligomers. While in the ‘native’ amylin oligomers there are two preferred structural models of amylin oligomers, after the removal of the disulfide bonds there is one preferred structural model of amylin oligomers. Second, the removal of the disulfide bonds in amylin oligomers induces the formation of β-strand properties in the N-termini and thus promotes this domain to adopt a cross-β structure. Therefore, our study suggests that the removal of the disulfide bonds in amylin oligomers induces longer cross-β strands in the N-termini of amylin oligomers and consequently increases the amyloidogenicity. In some amylin oligomers the removal of the disulfide bonds does not affect the packing of the cross-β structure, while in others it stabilizes the cross-β structure and leads to more packed structures. Third, these longer cross-β strands in the N-termini of amylin oligomers fluctuate more, but their turn domains fluctuate less also in comparison with the turn domain of the ‘native’ amylin oligomers. We therefore propose that the turn domains stabilize the cross-β structures and thus induce amylin aggregation.
Because amylin aggregation is a complex process a full understanding of the pathology caused by the formation of amylin aggregates requires establishing both the identity of the morphology of the intermediates along the amylin aggregation pathway and the kinetics of their formation. The propensity for native and non-native amylin oligomers to aggregate may also be linked to kinetic aspects and future work is needed to provide insights into these aspects.
Acknowledgements
This research was supported by the Israel Science Foundation (grant No. 532/15) and partly by the FP7-PEOPLE-2011-CIG, research grant No. 303741. All simulations were performed using the high-performance computational facilities of the Miller Lab in the BGU HPC computational center. The support of the BGU HPC computational center staff is greatly appreciated.References
- J. W. Hoppener and C. J. Lips, Int. J. Biochem. Cell Biol., 2006, 38, 726 CrossRef PubMed.
- C. E. Higham, E. T. Jaikaran, P. E. Fraser, M. Gross and A. Clark, FEBS Lett., 2000, 470, 55 CrossRef CAS PubMed.
- R. Kayed, J. Bernhagen, N. Greenfield, K. Sweimeh, H. Brunner, W. Voelter and A. Kapurniotu, J. Mol. Biol., 1999, 287, 781 CrossRef CAS PubMed.
- A. Clark, G. J. Cooper, C. E. Lewis, J. F. Morris, A. C. Willis, K. B. Reid and R. C. Turner, Lancet, 1987, 2, 231 CrossRef CAS.
- M. F. Mossuto, B. Bolognesi, B. Guixer, A. Dhulesia, F. Agostini, J. R. Kumita, G. G. Tartaglia, M. Dumoulin, C. M. Dobson and X. Salvatella, Angew. Chem., 2011, 50, 7048 CrossRef CAS PubMed.
- Y. Li, J. Yan, X. Zhang and K. Huang, Proteins, 2013, 81, 1862 CrossRef CAS PubMed.
- G. L. Devlin, T. P. Knowles, A. Squires, M. G. McCammon, S. L. Gras, M. R. Nilsson, C. V. Robinson, C. M. Dobson and C. E. MacPhee, J. Mol. Biol., 2006, 360, 497 CrossRef CAS PubMed.
- S. S. Wang, K. N. Liu and B. W. Wang, Eur. Biophys. J., 2010, 39, 1229 CrossRef CAS PubMed.
- R. Maeda, K. Ado, N. Takeda and Y. Taniguchi, Biochim. Biophys. Acta, 2007, 1774, 1619 CrossRef CAS PubMed.
- N. Sarkar, M. Kumar and V. K. Dubey, Biochimie, 2011, 93, 962 CrossRef CAS PubMed.
- M. V. Trivedi, J. S. Laurence and T. J. Siahaan, Curr. Protein Pept. Sci., 2009, 10, 614 CrossRef CAS PubMed.
- C. M. Dobson, Trends Biochem. Sci., 1999, 24, 329 CrossRef CAS PubMed.
- J. W. Kelly, Curr. Opin. Struct. Biol., 1998, 8, 101 CrossRef CAS PubMed.
- J. C. Rochet and P. T. Lansbury, Jr., Curr. Opin. Struct. Biol., 2000, 10, 60 CrossRef CAS PubMed.
- Y. Harpaz, M. Gerstein and C. Chothia, Structure, 1994, 2, 641 CrossRef CAS PubMed.
- W. J. Wedemeyer, E. Welker, M. Narayan and H. A. Scheraga, Biochemistry, 2000, 39, 4207 CrossRef CAS PubMed.
- J. C. Rochet, Expert Rev. Mol. Med., 2007, 9, 1 Search PubMed.
- L. Banci, I. Bertini, A. Durazo, S. Girotto, E. B. Gralla, M. Martinelli, J. S. Valentine, M. Vieru and J. P. Whitelegge, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11263 CrossRef CAS PubMed.
- K. Huang, N. C. Maiti, N. B. Phillips, P. R. Carey and M. A. Weiss, Biochemistry, 2006, 45, 10278 CrossRef CAS PubMed.
- N. S. Dangoria, M. L. DeLay, D. J. Kingsbury, J. P. Mear, B. Uchanska-Ziegler, A. Ziegler and R. A. Colbert, J. Biol. Chem., 2002, 277, 23459 CrossRef CAS PubMed.
- J. Niwa, S. Yamada, S. Ishigaki, J. Sone, M. Takahashi, M. Katsuno, F. Tanaka, M. Doyu and G. Sobue, J. Biol. Chem., 2007, 282, 28087 CrossRef CAS PubMed.
- A. V. Kajava, U. Aebi and A. C. Steven, J. Mol. Biol., 2005, 348, 247 CrossRef CAS PubMed.
- S. Luca, W. M. Yau, R. Leapman and R. Tycko, Biochemistry, 2007, 46, 13505 CrossRef CAS PubMed.
- G. G. Tartaglia, A. P. Pawar, S. Campioni, C. M. Dobson, F. Chiti and M. Vendruscolo, J. Mol. Biol., 2008, 380, 425 CrossRef CAS PubMed.
- C. Goldsbury, K. Goldie, J. Pellaud, J. Seelig, P. Frey, S. A. Muller, J. Kistler, G. J. Cooper and U. Aebi, J. Struct. Biol., 2000, 130, 352 CrossRef CAS PubMed.
- B. W. Koo and A. D. Miranker, Protein Sci., 2005, 14, 231 CrossRef CAS PubMed.
- R. Laghaei, N. Mousseau and G. Wei, J. Phys. Chem. B, 2010, 114, 7071 CrossRef CAS PubMed.
- I. T. Yonemoto, G. J. Kroon, H. J. Dyson, W. E. Balch and J. W. Kelly, Biochemistry, 2008, 47, 9900 CrossRef CAS PubMed.
- S. M. Vaiana, R. B. Best, W. M. Yau, W. A. Eaton and J. Hofrichter, Biophys. J., 2009, 97, 2948 CrossRef CAS PubMed.
- V. Wineman-Fisher, Y. Atsmon-Raz and Y. Miller, Biomacromolecules, 2015, 16, 156 CrossRef CAS PubMed.
- J. J. Wiltzius, S. A. Sievers, M. R. Sawaya, D. Cascio, D. Popov, C. Riekel and D. Eisenberg, Protein Sci., 2008, 17, 1467 CrossRef CAS PubMed.
- L. Kale, R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan and K. Schulten, J. Comput. Phys., 1999, 151, 283 CrossRef CAS.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187 CrossRef CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586 CrossRef CAS PubMed.
- M. S. Lee, M. Feig, F. R. Salsbury and C. L. Brooks, J. Comput. Chem., 2003, 24, 1821 CrossRef CAS.
- M. S. Lee, F. R. Salsbury and C. L. Brooks, J. Chem. Phys., 2002, 116, 10606 CrossRef CAS.
- D. Thirumalai, G. Reddy and J. E. Straub, Acc. Chem. Res., 2012, 45, 83 CrossRef CAS PubMed.
- M. G. Krone, L. Hua, P. Soto, R. Zhou, B. J. Berne and J. E. Shea, J. Am. Chem. Soc., 2008, 130, 11066 CrossRef PubMed.
- G. Reddy, J. E. Straub and D. Thirumalai, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21459 CrossRef CAS PubMed.
- Y. Miller, B. Ma and R. Nussinov, Biochemistry, 2011, 50, 5172 CrossRef CAS PubMed.
- Y. Raz, J. Adler, A. Vogel, H. A. Scheidt, T. Haupl, B. Abel, D. Huster and Y. Miller, Phys. Chem. Chem. Phys., 2014, 16, 7710 RSC.
- Y. Raz and Y. Miller, PLoS One, 2013, 8, e73303 CAS.
- M. S. Lee, M. Feig, F. R. Salsbury, Jr. and C. L. Brooks, 3rd, J. Comput. Chem., 2003, 24, 1348 CrossRef CAS PubMed.
- M. S. Lee, J. F. R. Salsbury and C. L. Brooks III, J. Chem. Phys., 2002, 116, 10606 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Constructed models, MD protocol, analysis details, values for conformational energies, populations, and helicity pitch values provided in tables and additional structural features demonstrated in Fig. S1–S12. See DOI: 10.1039/c6cp01196a |
| This journal is © the Owner Societies 2016 |