
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEarly stages of catalyst aging in the iridium mediated water oxidation reaction†
Dennis G. H.
Hetterscheid
*a,
Cornelis J. M.
van der Ham
a,
Oscar
Diaz-Morales
 a,
M. W. G. M. (Tiny)
Verhoeven
b,
Alessandro
Longo
c,
Dipanjan
Banerjee
c,
J. W. (Hans)
Niemantsverdriet
b,
Joost N. H.
Reek
d and
Martin C.
Feiters
e
a,
M. W. G. M. (Tiny)
Verhoeven
b,
Alessandro
Longo
c,
Dipanjan
Banerjee
c,
J. W. (Hans)
Niemantsverdriet
b,
Joost N. H.
Reek
d and
Martin C.
Feiters
e
aLeiden Institute of Chemistry, Leiden University, PO Box 9502, 2300 RA Leiden, The Netherlands. E-mail: d.g.h.hetterscheid@chem.leidenuniv.nl
bLaboratory for Physical Chemistry of Surfaces, Eindhoven University of Technology, Eindhoven, The Netherlands
cDutch-Belgian Beamline (DUBBLE), ESRF – The European Synchrotron, CS40220, 38043 Grenoble Cedex 9, France
dVan 't Hoff Institute for Molecular Sciences, University of Amsterdam, Amsterdam, The Netherlands
eInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands
First published on 16th March 2016
Abstract
When exposed to a potential exceeding 1.5 V versus RHE for several minutes the molecular iridium bishydroxide complex bearing a pentamethylcyclopentadienyl and a N-dimethylimidazolin-2-ylidene ligand spontaneously adsorbs onto the surface of glassy carbon and gold electrodes. Simultaneously with the adsorption of the material on the electrode, the evolution of dioxygen is detected and modifications of the catalyst structure are observed. XPS and XAS studies reveal that the species present at the electrode interface is best described as a partly oxidized molecular species rather than the formation of large aggregates of iridium oxide. These findings are in line with the unique kinetic profile of the parent complex in the water oxidation reaction.
Introduction
Global climate change and exhaustion of natural resources are among the most important problems that scientists need to solve in the next decades.1 Moving away from fossil fuels would require incorporating alternative energy sources into the energy infrastructure. This must include solar energy as it is the only source of renewable energy that can be harvested in sufficient amounts to power the planet.2 Solar radiation reaching the earth varies considerably over time and location. Hence, the storage of energy derived from solar radiation in the form of a chemical fuel is a key issue. One could achieve such an energy conversion by splitting water, resulting in the formation of O2 and H2. To do this efficiently, water oxidation catalysts that produce dioxygen with high catalytic rates at a minimum overpotential are required.Molecular water oxidation catalysts are in particular important to gain fundamental insight into water oxidation pathways, as such species allow for a high degree of well-defined structural modifications and thereby offer control over the catalyst environment both in terms of electronics and structure. However, molecular systems are susceptible to oxidative degradation,3 and often the resulting reaction products include metal oxo nanoparticles which are the ‘true’ catalytic species.4–7 The ease of formation of nanoparticles is strongly dependent on the reaction conditions applied. Therefore it is important to carefully verify the structure of the catalytically active species, in order to be able to rationalize the observed reaction mechanisms. Several molecular water oxidation catalysts have been described in the literature,8–11 which include catalytic systems based on cheap and abundant metals: manganese,12 iron,13,14 cobalt,15–18 nickel19 and copper.20,21 In terms of mechanistic understanding ruthenium based systems have been extremely valuable22–33 and recent investigations have led to the discovery of very active molecular systems.34
Iridium complexes also have been quite successful in catalytic water oxidation, and especially iridium sites bearing pentamethylcyclopentadienyl (Cp*) ligands have been shown to produce high catalytic rates in the water oxidation reaction.35–46 Longevity of the molecular catalytic site was demonstrated using cyclometallated phenyl-pyridine and phenyl-carbene type ligands.35–37 Nevertheless, the stability of molecular iridium catalysts is under debate as iridium oxo nanoparticles also are excellent catalysts for the water oxidation reaction47–49 and many of the organometallic ligands used are not infinitely stable under the high oxidative conditions employed. Upon treatment with large amounts of sacrificial oxidants the formation of iridium oxo nanoparticles has been observed for several molecular systems under extremely harsh reaction conditions.52 Under milder conditions selective oxygenation of the Cp* ligand was observed by the Macchioni group.51,53 The treatment of [IrIIICp*(N^C)NO3] with the sacrificial reagent sodium periodate resulted in the stepwise incorporation of oxygen into the Cp* ligand (N^C = 2-benzoylpyridine) (Fig. 1). Crabtree and coauthors reported the complete loss of the Cp* ligand from their iridium based catalyst and the formation of an iridium(IV) μ-oxo bridged dimer [{IrIV(N^O)(OH2)2}2(μ-O)2], which still carries the complementary N^O ligand (N^O = 2-(2′-pyridyl)-2-propanolate). Despite these findings, a substantial amount of kinetic information has been recorded from which it is concluded that water oxidation in the presence of iridium Cp* complexes is catalyzed by a well-defined molecular species rather than by nanoparticles, as long as a robust complementary ligand is present besides the Cp* ligand.7,35–37,50,54,55
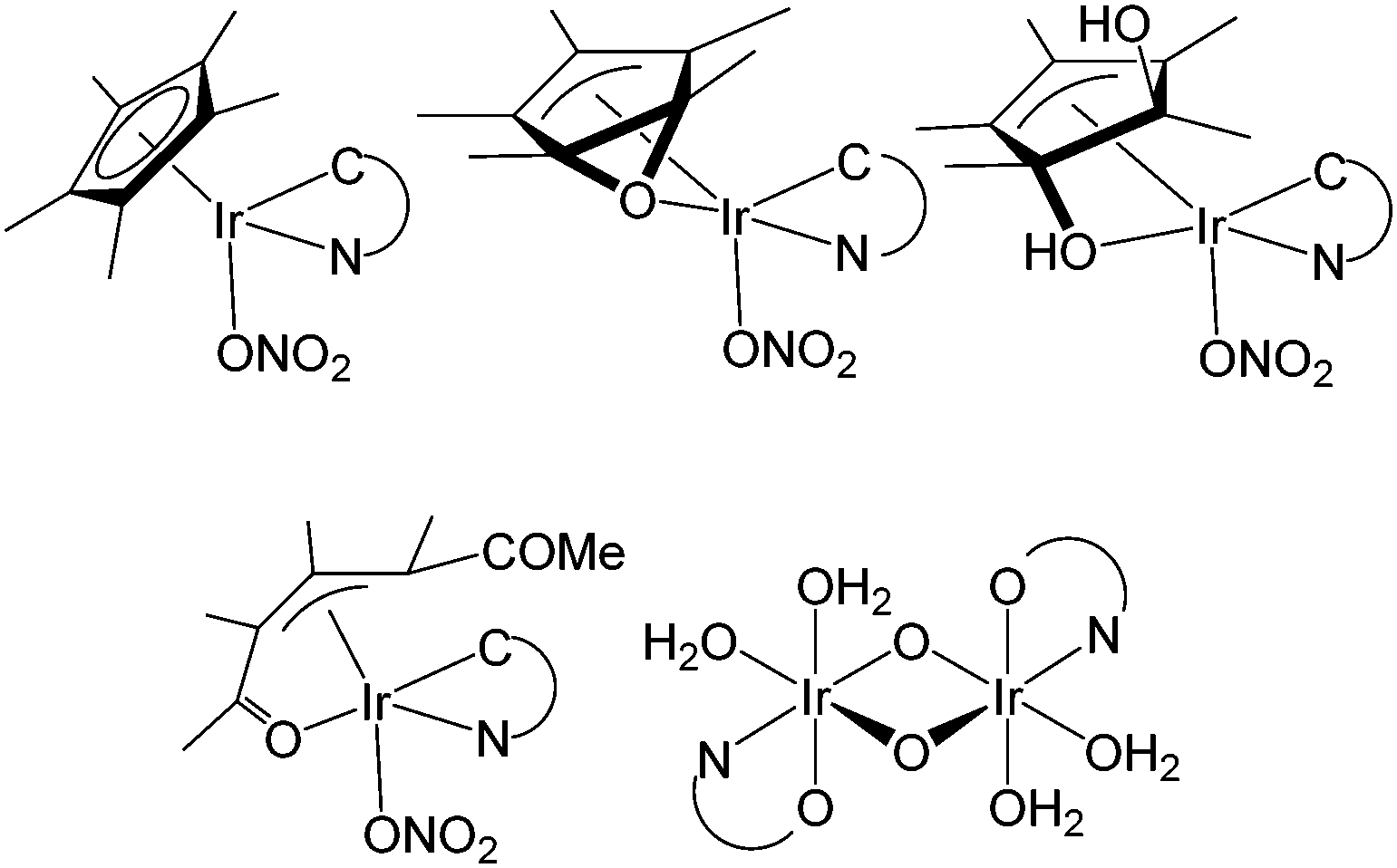 | ||
| Fig. 1 Identified oxidation products of treatment of [IrIIICp*(N^C)NO3]48 and [IrIIICp*(N^O)Cl]50,51 with sodium periodate as a sacrificial oxidant, wherein N^O = 2-(2′-pyridyl)-2-propanolate and N^C = 2-benzoylpyridine. | ||
Under electrochemical conditions it was shown that the iridium based catalyst precursor [IrIIICp*(OH)3]2+ spontaneously decomposed resulting in the formation of a blue iridium oxide deposit (BL) on the electrode. This deposit is believed to be the true active species in catalytic water oxidation in the presence of [IrIIICp*(OH)3]2+.56,57 Besides iridium and oxygen also a considerable amount of carbon (9%) is incorporated into the amorphous deposit and this was proposed to play an important role in catalysis on the basis of computations.58 Electrochemical crystal quartz microbalance (EQCM) experiments showed that the [IrIIICp*(N^O)Cl] complex does not produce any deposit, in contrast to [IrIIICp*(OH)3]2+. Electrochemically driven water oxidation catalysis mediated by [IrIIICp*(N^O)(Cl)] therefore is likely to proceed via a molecular species that still bears the N^O ligand.5 Electrochemical water oxidation mediated by [{IrIV(N^O)(OH2)2}2(μ-O)2] affords a very similar catalytic profile to that of [IrIIICp*(N^O)(Cl)] and points to the loss of the Cp* moiety from the latter species.59 The resulting [{IrIV(N^O)(OH2)2}2(μ-O)2] can also be directly attached to metal oxide surfaces, resulting in a very stable architecture that can undergo more than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 turnovers.60 Such an arrangement shows great potential as it allows for the development of very stable catalysts with a very high surface area while only using a minimum amount of precious metal.
000 turnovers.60 Such an arrangement shows great potential as it allows for the development of very stable catalysts with a very high surface area while only using a minimum amount of precious metal.
In 2011 our group reported that [IrIII(Cp*)(Me2NHC)(OH)2] (1) displays catalytic activity in the oxidation of water in the presence of the sacrificial reagent cerium ammonium nitrate (Me2NHC = N-dimethylimidazolin-2-ylidene).44 Also in the presence of periodate evolution of dioxygen commences, yet in this case it is unclear whether dioxygen originates from water or from periodate instead.61In situ detection of potential catalytic intermediates is in line with a molecular catalytic species. This was supported by DFT calculations, which showed that water oxidation is feasible with such monocationic species.62In situ Raman spectroscopy experiments coupled to electrochemistry showed reversible formation of an iridium(IV) μ-oxo bridged dimer before the onset for catalytic water oxidation (Scheme 1).63 Due to the reversible nature of the process, this species must still bear the Cp* and NHC-ligands right before the onset for catalytic activity. Online mass spectroscopy experiments showed exclusive formation of dioxygen up to 2.0 V versus RHE in cyclic voltammetry, suggesting that neither the Cp* ligand nor the NHC-ligand degenerate to carbon dioxide under harsh electrochemical conditions.
 | ||
| Scheme 1 Reversible formation of 2 under oxidative conditions. | ||
Although the reversible formation of [{IrIV(Cp*)(Me2NHC)}2(μ-O)2]2 (2) was observed right before the onset for catalytic water oxidation, the findings of the Macchioni and Crabtree groups50,51,53 showed that degradation and complete loss of the Cp* ligands may occur to some degree under harsh oxidation conditions. In order to study and develop a new catalyst for the water oxidation reaction, it is crucial to understand the catalyst activation and deactivation pathways. Hence, in this paper we report detailed studies that indicate that structural changes of 1 take place after prolonged electrolysis at potentials exceeding the onset for water oxidation using X-ray techniques.
Results
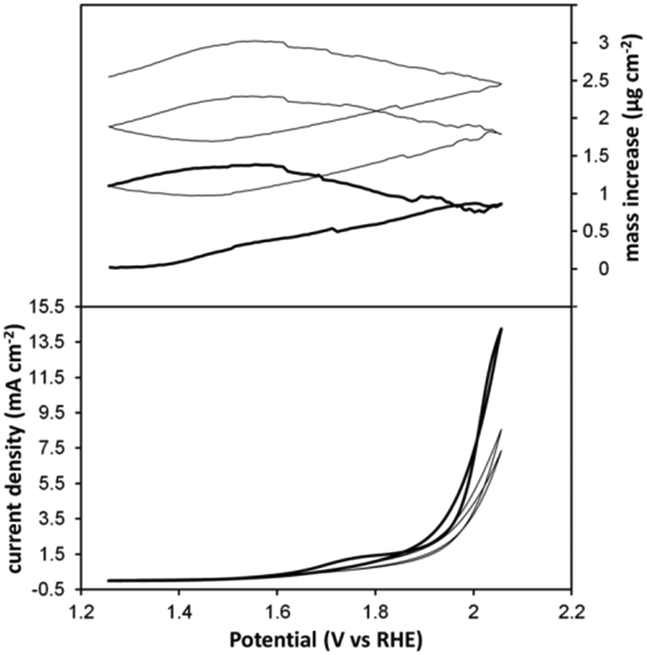
In the presence of 1 the evolution of dioxygen is detected at potentials exceeding 1.5 V versus RHE. The kinetic profile obtained in the presence of 1 is different from that of [IrIIICp*(OH)3]2+ and points to some role of the NHC-ligand in the active species. The iridium oxide blue layer (BL) described by Crabtree and coauthors can be obtained by placing an 2.3 mM solution of [IrIIICp*(OH)3]2+ in 0.1 M KNO3 solution in an electrochemical cell wherein a potential exceeding 1.4 V versus RHE is applied.56 Under these conditions [IrIIICp*(OH)3]2+ apparently loses the Cp* ligand and forms a porous BL of iridium oxide, which is the true active species in this case. This phenomenon is easy to recognize as the surface area of the iridium oxide layer and thereby the catalytic activity of BL increase in time. BL shows a clear reversible iridium(III)/iridium(IV) redox couple at 1.2 V versus RHE in the cyclic voltammogram prior to the catalytic oxygen evolution wave. Repetitive scanning of the potential in the presence of a 1 mM concentration of 1 barely led to an increase of current in the cyclic voltammogram as long as glassy carbon (GC) electrodes were used, and in a typical experiment it was the first scan which showed the maximum current. The observed cyclic voltammogram shows great resemblance to the cyclic voltammetry diagrams of drop cast 1, both in the presence and absence of Nafion.63A first order rate dependence in equivalents of iridium was observed in the case of drop cast 1, pointing to a well-defined molecular catalyst rather than an iridium oxide layer (Fig. S3, ESI†). Nevertheless, when a glassy carbon electrode was removed from a 1 mM solution of 1 after a series of scans, and after rinsing the electrode, some of the catalytic activity was retained upon dipping the electrode into a fresh electrolyte solution in the absence of 1. In other words, some of the catalytic activity comes from some form of iridium deposited onto the electrode. Also in case a gold working electrode was used, the catalytic activity did not increase upon prolonged potential cycling as long as a phosphate buffer was used. When sulfate was used as an electrolyte, a huge current was observed in the first scan, pointing to a non-catalytic oxidative process. In the second cyclic voltammetry scan less current was observed, which from there on increased upon every scan onwards (Fig. 2). Both in phosphate and sulfate media, catalytic activity at the electrode was retained after the removal of 1 from the solution, pointing to some form of the deposited material. From here on, this deposited material (DM) will be referred to as DM|GC in the case of a glassy carbon electrode and DM|Au in the case where a gold electrode was used.
 | ||
| Fig. 2 EQCM experiment wherein the mass balance (top) and current density (bottom) are simultaneously followed as a function of applied potential in the case of a 1 mM solution of 1 in 100 mM Na2SO4 (non-buffered). The first scan is depicted in bold. | ||
Electrochemical quartz crystal microbalance studies
The adsorption kinetics of 1 was studied in detail on a gold working electrode by EQCM techniques (Fig. 2–5, Fig. S2 and S3, ESI†). It was found that the observed catalytic current increases concomitantly with a mass increase of the gold electrode, confirming that adsorption of some form of iridium indeed takes place. Deposition of the material takes place right upon the onset of the catalytic wave and therefore it seems that both processes are intimately connected. Adsorption of the material was observed both in sulfate and phosphate solutions of 1, yet seemed to be limited to roughly 5–100 μg cm−2 (Fig. 2 and 4). This was clearly illustrated by amperometry experiments that after an initiation period eventually showed leveled mass and currents (Fig. 4). | ||
| Fig. 3 EQCM experiment wherein the mass balance (top) and current density (bottom) are simultaneously followed as a function of applied potential in the case of DM|Au in 100 mM Na2SO4 acidified to pH 1. Complex 1 is not present in solution. The first scan is dotted. Note that the mass balance does no longer decrease after the first scan. | ||
 | ||
| Fig. 4 EQCM experiment wherein the mass balance (top) and current density (bottom) are simultaneously followed at an applied potential of 1.8 V versus RHE as a function of time in the case of a 1 mM solution of 1 in 100 mM Na2SO4. | ||
 | ||
| Fig. 5 EQCM experiment wherein the mass balance (top) and current density (bottom) are simultaneously followed as a function of applied potential in the case of a 1 mM solution of 1 in 100 mM Na2SO4 acidified to pH 1. The scan rate is 10 mV s−1 and the first scan is depicted in bold. | ||
After deposition, the DM|Au electrodes were analyzed by EQCM techniques in a fresh sulfate solution in the absence of any 1. Upon positive scanning, desorption of DM from the electrode takes place almost instantaneously (Fig. 3). Since desorption is very fast, it is cumbersome to maintain the resonance of the EQCM machine due to the rapid loss of mass, and to verify whether evolution of dioxygen takes part in these short timeframes. Desorption of DM from the electrode seems to be triggered by the pH as under more acidic conditions a drop of mass is already visible at potentials below 1.5 V (Fig. 5), even in the presence of 1. Desorption of DM appears to be somewhat electrode dependent and more cumbersome in the case of DM|GC compared to DM|Au.
X-ray photoelectron spectroscopy
XPS was carried out to determine the molecular constituents of the deposited material. The XPS iridium 4f signals of benchmark 1, [IrIII(Cp*)(Me2NHC)(Cl)2] (3) and [{IrCp*Cl2}2] powders were found at almost the exact same position (61.4 eV, 61.4 eV and 61.6 eV), and slightly higher than previously recorded values of [{IrCp*Cl2}2].64 The iridium 4f signal of DM|GC at 62.2 eV points to a significantly higher binding energy and fits well with the observed values of the +IV oxidation state of [{IrIV(N^O)(OH2)2}2(μ-O)2] for which a binding energy of 62.4 eV was found.50,60 Identical data sets were obtained for both DM|GC and DM|Au in experiments wherein 1 was quickly adsorbed in a single potential sweep, in multiple potential sweeps and by amperometry. Also DM|GC electrodes from which part of the material had desorbed by treatment in a blank electrolyte solution afford the same XPS characteristics. The sharp signals obtained for the iridium 4f binding energy point to a single and well-defined species, which is in contrast to most examples of electrochemically deposited iridium oxide.65,66 XPS measurements on an iridium oxide foil yields binding energies of 61.8 eV (Ir2O3) and 63.0 eV (IrO2), which are different compared to DM|GC. Also XPS on iridium oxide powders showed binding energies that are markedly different compared to the catalytic active material (Ir2O3: 61.6 eV; IrO2: 62.7 eV).65 The nitrogen 1s signal of 1 was found to be in a 2:1 ratio of nitrogen versus iridium, and was located at 399.7 eV. Also in the case of DM|Au we find a very sharp nitrogen signal at 399.7, although the ratio nitrogen:iridium has shifted to higher nitrogen levels (∼4:1) compared to 1.‡ This is in contrast to XPS measurements on iridium nanoparticles obtained from nitrogen containing [IrCp*(N^N)Cl], for which no nitrogen could be detected by XPS (N^N = 4,4′-bishydroxy-2,2′-bipyridine).55 The XPS measurement of DM|Au illustrates the presence of roughly 18 equivalents of carbon compared to iridium. This is in contrast to BL for which only ∼9% of carbon remained in the catalytic material.56–58 Also a small amount of oxidized carbon was found, suggesting that part of the methyl groups of the Cp* ligand may have been oxidized, in agreement by the studies of Macchioni et al.51,53 Experiments starting from 3 yield the same XPS signals and catalytic activity compared to 1 suggesting that both precursors lead to the same catalytic material. In the case of 3 only traces of chloride were found, confirming that the chloride is not retained in DM|GC.
X-ray absorption spectroscopy
For the determination of the structure of the catalytic material as a function of applied potential above 1.5 V versus RHE we rely on ex situ X-ray absorption spectroscopy. In initial experiments we tried to record the Ir EXAFS of the catalyst on gold electrodes but this turned out to be a problem due to the proximity of the Ir and Au L edges, so we changed to the catalyst on carbon electrodes instead. Samples were prepared by dipping a carbon sheet into a 1 mM stock solution of 1. The iridium catalyst was deposited onto these electrodes by amperometry at potentials between 0.8 and 2.0 V versus RHE. The lower potential samples contained relatively little iridium in line with what one would expect from the earlier described EQCM experiments, yet by simply folding the bendable DM|GC electrode so that a stack of layered material is formed, reliable XAS data could be obtained. Since there is an apparent fluxional behavior between 1, DM|Au or DM|GC and the desorption product of DM, the lifetime of iridium at the electrode interface is difficult to estimate. Hence we refrained from the analysis of long term experiments.§X-ray absorption near edge spectra
The DM|GC and DM|Au electrodes were analyzed by X-ray absorption techniques and their spectra were carefully compared to those of reference samples of 1, 3, [{IrCp*Cl2}2], K2IrCl6 and IrO2.67–69 The XANES of a number of reference compounds are shown in Fig. 6, top. Going from 1 to IrO2, the intensity of the white line increases significantly, and there is an apparent shift of the edge to lower energy, which is however not significant when the energies at half-height (11213.5 eV for both) are considered. The Cl-coordinated compounds 3 and [{IrCp*Cl2}2] have white-line intensities comparable to that of 1 and edge energies at half height that are subtly higher (11214.0) respectively lower (11213.0 eV) and moreover have an increased intensity in the region on the high-energy side of the white line (11225 eV). In Fig. 6, bottom, the XANES of the carbon electrode exposed to various potentials are compared with those of 1 and IrO2. It can be seen that the edge position starts to move to lower energy at the lower potentials (1.1, 1.3 V) and that the intensity of the white line increases with a large step at 1.5 V to values even exceeding that of IrO2 at 1.9 and 2.0 V. It is of interest to compare our results to related experiments reported in the literature, starting from another material than 1 (i.e. IrCl3 and H2O2), while our work was in progress.70 Although our experiments were extended to higher potentials (2.0 V vs. 1.6 V), we did not observe any shift of the edge to energies higher than that of IrO2. Inspection of the derivative spectra of the edge region of the electrode spectra in the range 1.3–2.0 V (see Fig. S7, ESI†) revealed the presence of a shoulder at approx. 11220 eV on the high-energy side of the white line (11217 eV), which showed its highest relative intensity in the 1.5 V sample. The maxima did not correspond to any of the white lines of any of the benchmark compounds. Following the interpretation in the literature,70–72 in which they correspond to electronic transitions of the Ir 2p to respectively the t2g and eg orbitals of the Ir 5d level, we conclude from the estimated separation (approx. 3 eV) that no oxidation state higher than iridium(IV) is reached in our system. | ||
| Fig. 6 Normalized iridium L3 edges of reference compounds (top): dotted blue, 1; orange, [{IrCp*Cl2}2]; purple, 3; dotted red, IrO2, and (bottom) electrode samples, dotted blue, 1; dotted red, IrO2; DM|GC deposited at 1.1 V, 1.3 V, 1.5 V, 1.7 V, 1.9 V and 2.0 V are depicted in grey tones on going from dark to light. | ||
Extended X-ray absorption fine structure
Fig. 7 compares the EXAFS of the Ir catalyst on the carbon electrodes exposed to increasing potentials (top to bottom) to that of the starting material 1 (top) and a possible end product, IrO2 (bottom). The intensity of the EXAFS, as reflected in the amplitude of the major peak in the Fourier transform (Fig. 7, right), reaches a minimum at 1.5 V. Moreover the position of the major peak in the Fourier transform shifts from a position corresponding to that of the main shell in the starting material to a shorter distance of the main shell without quite reaching that of the IrO2 reference material. The signal-to-noise ratio of the electrode data is low compared to that of the reference benchmarks 1 (Fig. 7, top trace), IrO2 (Fig. 7, bottom trace), 3, [{IrCp*Cl2}2] and K2IrCl6.73 This is due to the small amount of catalyst deposited on the carbon electrode, despite putting a number of layers on top of each other in the EXAFS transmission experiment. In spite of this it is clear that none of the electrode spectra contain contributions at high (8–12 Å−1) k in the EXAFS and high R in the Fourier transform, which are characteristic of pure solid IrO2 (see Fig. 7). | ||
| Fig. 7 k 3-weighted iridium L3 edge EXAFS (left) and the modulus of the corresponding phase-corrected Fourier transforms (right) of 2 reference compounds and carbon electrodes exposed to various potentials. Top to bottom: 1, DM|GC deposited at 1.1 V, 1.3 V, 1.5 V, 1.7 V, 1.9 V and 2.0 V, and benchmark IrO2. Grey traces, experimental, black and red traces; best single shell fits with carbon and oxygen as atom types, respectively. Parameters for the best and other fits in Table 1. | ||
In contrast to pyridine and triazole ligands74 and in line with parallel studies,73,75 the contribution of N-heterocyclic carbene in the Fourier transform is very weak in 1 and 3. Hence, it is difficult to exclude its presence or even that of cyclopentadienyl in the Ir coordination sphere of Ir in the electrode material. In view of a possible change of a coordination sphere dominated by C (from cyclopentadienyl and carbene) to O we analyzed the EXAFS by single-shell simulations with either carbon or oxygen as the atom type, for which not only the threshold energy, the Debye–Waller factors, and the distance to iridium were refined, but also the occupancy (coordination number); the results are given in Table 1. It turned out that the best fit indices were obtained with C as the atom type for the starting material 1, and the carbon electrodes till 1.5 V and that O was the better atom type at higher potentials.
| C | O | |||||
|---|---|---|---|---|---|---|
| Threshold energy (eV) | Number of atoms@distance (Å) and Debye–Waller factor (2σ2, Å2) | Fit indexb with k3-weighting (×103) | Threshold energy (eV) | Number of atoms@distance (Å) and Debye–Waller factor (2σ2, Å2) | Fit indexb with k3-weighting (×103) | |
| a Fitting errors in parentheses; except for the distances, where the fitting error was always lower than the accepted systematic error (0.02 Å). b The best fit index (C or O) is highlighted in bold. | ||||||
| 1 | −10.5607 | 5.7@2.147 (0.007) | 0.4903 | −14.6284 | 4.1@2.110 (0.009) | 0.5555 |
| 1.1 V | −16.8836 | 6.8@2.139 (0.016) | 1.9858 | −19.6258 | 5.0@2.090 (0.019) | 2.1008 |
| 1.3 V | −9.1949 | 5.9@2.146 (0.011) | 1.0433 | −12.1444 | 4.5@2.101 (0.014) | 1.0929 |
| 1.5 V | −7.99290 | 5.0@2.100 (0.010) | 1.3133 | −10.5973 | 4.0@2.053 (0.015) | 1.3161 |
| 1.7 V | −7.3754 | 5.7@2.068 (0.004) | 1.3189 | −11.095 | 4.4@2.031 (0.015) | 1.3000 |
| 1.9 V | −11.1800 | 6.1@2.075 (0.003) | 1.2304 | −15.147 | 4.6@2.039 (0.005) | 1.1833 |
| 2.0 V | −9.08911 | 6.1@2.087 (0.005) | 0.5581 | −12.6264 | 4.5@2.049 (0.007) | 0.4625 |
| IrO2 | −7.26908 | 6.6@2.010 (0.002) | 3.2154 | −12.1225 | 4.5@1.976 (0.014) | 3.3055 |
The selected simulation for each spectrum is highlighted in bold in Table 1 and included in Fig. 7. It should be noted that the real coordination numbers for the benchmark compounds ((5 + 1)C + 2O = 8 for 1; 4C + 2O = 6 for IrO2) are underestimated by the occupancies obtained in this way (respectively 5.7(3) and 4.5(1)). This is due to (i) the static variation in Ir–ligand distances as discussed for both cases elsewhere,73 and (ii) the inevitable underestimation when the O contributions in the former compound are simulated by C, which has a lower backscattering amplitude. It is a warning not to rely too much on the coordination numbers and atom types derived from these EXAFS simulations; the latter problem is underlined by the observation that for solid IrO2 the simulation with C (not included in Fig. 7) has a slightly more favourable fit index than with O (Fig. 7, bottom trace). Nonetheless the EXAFS clearly shows that the coordination sphere of Ir in the catalyst on the carbon electrodes changes from the one dominated by carbon atoms (Cp*, NHC) at a relatively long average distance (2.15(2) Å) to one dominated by O (iridium oxides) at a shorter distance (2.05(2) Å). For the electrode at 1.5 V the small difference in the quality of the fit between C and O and low amplitude indicate that each iridium has a mixed C/O environment, and/or that there is a mixture of compounds. This is probably the case to varying extents for all the electrode materials.
Discussion
In a previous study the pH dependence of water oxidation mediated by 1 was discussed.63 The maximum catalytic activity was pinpointed near pH 4. This is in sharp contrast to iridium oxides for which the overpotential (measured at 0.5 mA cm−2) is independent of the RHE reference scale76 and molecular iridium catalysts including [IrIIICp*(N^O)(Cl)] that typically operate best under alkaline conditions.39 Moreover, the activity in the presence of 1 appeared to be inhibited by phosphate,63 while the presence of buffers including phosphate is essential in the activity of BL.77 This suggests that the predominant active species in the catalytic water oxidation reaction in the presence of 1 is unique with respect to that of [IrIIICp*(OH)3]2+ and [IrIIICp*(N^O)(Cl)] and points to some role of the N-heterocyclic carbene in the catalyst structure, while the Cp* ligand is a common factor in the structure of these catalysts.The structure of the iridium species changes with the applied redox potential. At potentials below 1.0 V versus RHE the iridium complex remains in the +III oxidation state. Although no clear oxidation wave was observed under catalytic conditions, a quasi-irreversible oxidation wave was visible in the case of 1 in acetonitrile at 1.0 V versus RHE.44 Since the formation of 2 takes place prior to the onset for water oxidation (estimated at ∼1.0 V vs. RHE), further oxidation steps are compulsory before water oxidation can commence.
When a potential above 1.55 V versus RHE is applied a catalytic current is recorded and the formation of dioxygen is detected instantaneously by in situ mass spectrometry experiments. Several phenomena are taking place under these catalytic conditions which is best visible in sulfate media at neutral pH in the presence of high concentrations of 1 in solution. Under these conditions the huge current in the first cyclic voltammetry scan points to a non-catalytic oxidative process, which probably relates to a catalyst modification reaction. Simultaneously upon oxygen evolution the catalytic material deposits on the electrode, leading to a slow increase in catalytic activity upon every cycle. In phosphate media and when the catalyst is drop cast onto the electrode these phenomena are much less evident although XPS measurements illustrate that eventually the same material is formed. The deposition of material is limited to a maximum value, with a huge variation which most likely relates to the roughness of the electrode used. Apparently, there is a maximum loading capacity of the electrode surface, unlike deposition of iridium oxide layers such as BL. Also the very high rates of DM desorption is in contrast to deposited iridium oxide and direct binding of iridium clusters to the electrode surface.60 The onset of the current does not change upon repetitive scanning and coincides with the in situ detection of dioxygen.63 This suggests that catalyst modification and catalytic water oxidation are somehow linked to each other and are most likely triggered by the initial oxidation of 2. It is difficult to judge whether structural modifications are obligatory to obtain catalytic activity. Since one-electron-oxidation of 2 is most likely not connected to a H+ transfer, its formation in the rate determining step of the water oxidation reaction would be in line with the observed first order rate dependence in iridium (see ESI†) and the pH dependence we found earlier (Fig. 8).63
 | ||
| Fig. 8 Schematic summary of events taking place at initial stages of water oxidation in the presence of 1. | ||
The XPS spectra of DM|GC and DM|Au prepared above 1.5 V versus RHE point to a different electronic environment of iridium in DM|GC and DM|Au compared to 1. Only one set of iridium signals was observed by XPS analysis, suggesting that the material on the electrodes is highly uniform. This contrasts with the electrodeposition of iridium oxide, which typically gives rise to a wide distribution of iridium binding energies.65,66,78 Since the binding energy of DM|GC and DM|Au is similar to that of the iridium(IV) complex [{IrIV(N^O)(OH2)2}2(μ-O)2]50,59 and relatively close to that of iridium(IV) oxide, the oxidation state of iridium in DM|GC and DM|Au under ex situ conditions is best assigned as +IV. Also the XANES data show a closer (but not an exact) resemblance between DM|GC and iridium(IV) oxide spectra compared to the iridium(III) precursors 1, 3 and [{IrCp*Cl2}2]. According to the XPS analysis, the elemental constitution of DM|GC and DM|Au prepared above 1.6 V versus RHE contains roughly 18 equivalents of carbon per iridium center, while the amount of nitrogen appears to be overestimated. In line with possible modification reactions under oxidative conditions large oxygen loadings were detected. Yet these oxygen loadings must at least in part be ascribed to the oxidation of the carbon and gold electrode surface rather than to the organometallic species absorbed.
The identification of iridium–iridium distances by EXAFS requires data acquisition over a large k range. Powder samples of [{IrCp*Cl2}2] and iridium(IV) oxide showed oscillations up to a k value of 18 Å−1 and for both benchmark materials we were able to find reliable iridium distances that match literature values.73 For example [{IrCp*Cl2}2] produced a characteristic additional maximum in the EXAFS envelope at k = 13–18 Å−1.73 Sampling at a very large k in the case of DM|GC required very long irradiation times leading to unsatisfactory results which point to the decay of the material. We refrained from the in-depth analysis of such samples, and analyzed electrode data up to k = 12.5 Å−1 instead. At lower k, it is considerably more difficult to find evidence for an iridium–iridium bond,69 although the Ir–Ir contacts in the range 3–4 Å already contribute to the EXAFS of IrO2 from k = 8 Å−1 (Fig. 7, bottom traces). Therefore, the comparison of the EXAFS of the electrode and IrO2 and their Fourier transforms taken over the shorter data range (Fig. 7) clearly indicate that large aggregates of iridium are not formed, in particular for the data at 2.0 V which have a relatively good signal-to-noise ratio.
As one would expect upon oxidation the metal–ligand bond distances become significantly shorter in the case of DM|GC compared to 1. Moreover, the fit-index of the EXAFS data increases when ‘carbon’ is (partly) replaced by ‘oxygen’ in the direct coordination sphere of iridium. Apparently, upon prolonged electrolysis above 1.5 V versus RHE, the Cp* ligand does not remain intact and undergoes at least some oxidative degradation. The iridium–oxygen distance in DM|GC was found at 2.03–2.05 Å by the EXAFS which is significantly (estimated error ± 0.02 Å) longer than that of IrO2 (1.976 Å). BL, the reaction product of [IrIIICp*(OH)3]2+ deposition, has only a slightly shorter iridium oxygen distance than DM|GC (BL: 2.01 Å with XAFS,58 2.02 Å with X-ray pair distribution function analysis57); however, for this material, clear Ir–Ir contributions are visible in the EXAFS in the k range 9–12 Å−158 which are absent in DM|GC. Apparently the N-heterocyclic carbene has a considerable effect of the structure of DM|GC and is most likely (in some form) retained. The full oxidative decomposition of organometallic iridium complexes typically involves the formation of carbon dioxide,55 although the Cp* ligand in principle can be oxidized to five equivalents of acetic acid. Online mass spectrometry experiments of 1 drop cast on gold did not indicate any formation of CO2 or NO. Since we have been able to detect these gasses for several positive controls, this suggests that full oxidative decomposition of 1 has not yet occurred. Most likely, the structures of DM|GC and DM|Au after ten minutes of electrolysis are still molecular architectures that lie somewhere on the path of ligand oxidation similar to the complexes isolated by Crabtree and Macchioni and their coauthors, which are depicted in Fig. 1.
Conclusions
The structure of the iridium complexes bearing cyclopentadienyl and the N-heterocyclic carbene ligand changes over time when a strong oxidative potential is applied. Simultaneously with structural modifications of the complex catalytic water oxidation activity is displayed. Although water oxidation catalysis by an intact complex cannot be ruled out, it is evident that structural modifications of the ligand framework play an important role in the observed catalytic activity over prolonged reaction times. The catalytic activity, pH dependence and reaction kinetics vary greatly with the ligands employed in the organometallic iridium precursors, showing that one can control the catalytic activity by the ligand(s). Such structure reactivity relationships will be very important for the development of very active water oxidation electrocatalysts that contain very low precious metal content, yet show very high catalytic rates and stabilities.Experimental section
Synthetic procedures
:3 mixture of concentrated HNO3/concentrated H2SO4 to remove organic contaminants after which it was boiled five times in water. The water used for cleaning glassware and preparing solutions was demineralized and ultra-filtrated using a Milipore MiliQ system (resistivity > 18.2 MΩ cm and TOC < 5 ppb). Electrolyte solutions were prepared using high quality chemicals. Dissolved oxygen in solutions was removed prior to measurements by purging with argon (purity grade 5.0) for at least 30 min, and argon was kept flowing above the solution during experiments. Electrochemical measurements were carried out in a homemade three-electrode and a two-compartment cell with the reference electrode separated by a Luggin capillary. A gold wire was used as the counter electrode, and a reversible hydrogen electrode (RHE) was used as a reference; a platinum wire was connected to the reference electrode through a capacitor of 10 μF, acting as a low-pass filter to reduce the noise in the low current measurements.
EQCM
The EQCM experiments were recorded on an Autolab PGSTAT310N equipped with an EQCM module. Commercial gold–EQCM electrodes were treated electrochemically above 1.0 V versus RHE and kept above 0.8 V versus RHE in order to avoid mechanical destruction of the thin gold electrode by repetitive scanning over the gold oxidation and reduction potentials.81 Once a steady mass balance signal was obtained upon cycling the potential between 0.9 and 2.0 V versus RHE, the blank electrolyte solution was displaced for a solution of 1. A gold wire was used as a counter electrode and a commercial Metro-Ohm Ag/AgCl electrode (3.0 M KCl) was used as a reference electrode. Potentials were recalculated and reported versus RHE.XPS
The XPS measurements are carried out using a Thermo Scientific K-Alpha, equipped with a monochromatic small-spot X-ray source and a 180° double focusing hemispherical analyzer with a 128-channel detector. Spectra were obtained using an aluminium anode (Al Kα = 1486.6 eV) operating at 72 W and a spot size of 400 μm. Survey scans were measured at a constant pass energy of 200 eV and region scans at 50 eV. The background pressure was 2 × 10−8 mbar and during measurement 4 × 10−7 mbar argon because of the charge compensation dual beam source. Resolution: the full width at half-maximum is 0.50 eV in the case of a Ag 3d5/2 peak.XAS
X-ray absorption spectra were collected at the iridium L3-edge (11215 eV) on the EXAFS station (BM26A) of the Dutch-Belgian beamline (DUBBLE)82 at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France. The solid benchmark materials were diluted with boron nitride and measured as pressed pellets in the transmission mode; 2 scans were averaged together to improve the signal-to-noise ratio. The DM|GC and DM|Au electrode samples were also recorded in the transmission mode and 6–8 scans were averaged. The EXAFS spectra were processed using Viper83 and simulations were performed in EXCURVE.84,85Acknowledgements
The authors thank Dutch Research Council (NWO) for the provision of beam time at the DUBBLE beam line at the ESRF and M. Tromp for useful discussions. D. G. H. H. thanks NWO (VENI research grant 700.59.410) and Leiden University for funding.Notes and references
- IPCC Fifth Assessment Report (AR5), www.ipcc.ch/report/ar5/index.shtml.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20142 CAS.
- B. Limburg, E. Bouwman and S. Bonnet, Coord. Chem. Rev., 2012, 256, 1451 CrossRef CAS.
- J. W. Vickers, H. J. Lv, J. M. Sumliner, G. B. Zhu, Z. Luo, D. G. Musaev, Y. V. Geletii and C. L. Hill, J. Am. Chem. Soc., 2013, 135, 14110 CrossRef CAS PubMed.
- N. D. Schley, J. D. Blakemore, N. K. Subbaiyan, C. D. Incarvito, F. D'Souza, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2011, 133, 10473 CrossRef CAS PubMed.
- S. Fukuzumi and D. C. Hong, Eur. J. Inorg. Chem., 2014, 645 CrossRef CAS.
- H. Junge, N. Marquet, A. Kammer, S. Denurra, M. Bauer, S. Wohlrab, F. Gartner, M. M. Pohl, A. Spannenberg, S. Gladiali and M. Beller, Chem. – Eur. J., 2012, 18, 12749 CrossRef CAS PubMed.
- D. J. Wasylenko, R. D. Palmer and C. P. Berlinguette, Chem. Commun., 2013, 49, 218 RSC.
- D. G. H. Hetterscheid and J. N. H. Reek, Angew. Chem., Int. Ed., 2012, 51, 9740 CrossRef CAS PubMed.
- M. D. Karkas, O. Verho, E. V. Johnston and B. Akermark, Chem. Rev., 2014, 114, 11863 CrossRef PubMed.
- X. Sala, I. Romero, M. Rodriguez, L. Escriche and A. Llobet, Angew. Chem., Int. Ed., 2009, 48, 2842 CrossRef CAS PubMed.
- J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524 CrossRef CAS PubMed.
- J. L. Fillol, Z. Codola, I. Garcia-Bosch, L. Gomez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807 CrossRef CAS PubMed.
- W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, J. Borau-Garcia and C. P. Berlinguette, Chem. Commun., 2011, 47, 4249 RSC.
- H. J. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kogerler, P. F. Zhuk, J. Bacsa, G. B. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268 CrossRef CAS PubMed.
- Q. S. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342 CrossRef CAS PubMed.
- E. Pizzolato, M. Natali, B. Posocco, A. M. Lopez, I. Bazzan, M. Di Valentin, P. Galloni, V. Conte, M. Bonchio, F. Scandola and A. Sartorel, Chem. Commun., 2013, 49, 9941 RSC.
- M. Zhang, M. T. Zhang, C. Hou, Z. F. Ke and T. B. Lu, Angew. Chem., Int. Ed., 2014, 53, 13042 CrossRef CAS PubMed.
- M. K. Coggins, M. T. Zhang, Z. F. Chen, N. Song and T. J. Meyer, Angew. Chem., Int. Ed., 2014, 53, 12226 CrossRef CAS PubMed.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498 CrossRef CAS PubMed.
- A. M. Angeles-Boza, M. Z. Ertem, R. Sarma, C. H. Ibanez, S. Maji, A. Llobet, C. J. Cramer and J. P. Roth, Chem. Sci., 2014, 5, 1141 RSC.
- S. Maji, I. Lopez, F. Bozoglian, J. Benet-Buchholz and A. Llobet, Inorg. Chem., 2013, 52, 3591 CrossRef CAS PubMed.
- S. Maji, L. Vigara, F. Cottone, F. Bozoglian, J. Benet-Buchholz and A. Llobet, Angew. Chem., Int. Ed., 2012, 51, 5967 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, M. A. Henderson, B. D. Koivisto, H. D. Osthoff and C. P. Berlinguette, J. Am. Chem. Soc., 2010, 132, 16094 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, B. D. Koivisto, M. A. Henderson and C. P. Berlinguette, Inorg. Chem., 2010, 49, 2202 CrossRef CAS PubMed.
- Z. F. Chen, J. J. Concepcion, X. Q. Hu, W. T. Yang, P. G. Hoertz and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7225 CrossRef CAS PubMed.
- J. J. Concepcion, J. W. Jurss, M. R. Norris, Z. F. Chen, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2010, 49, 1277 CrossRef CAS PubMed.
- J. J. Concepcion, M. K. Tsai, J. T. Muckerman and T. J. Meyer, J. Am. Chem. Soc., 2010, 132, 1545 CrossRef CAS PubMed.
- L. L. Duan, A. Fischer, Y. H. Xu and L. C. Sun, J. Am. Chem. Soc., 2009, 131, 10397 CrossRef CAS PubMed.
- J. Nyhlen, L. L. Duan, B. Akermark, L. C. Sun and T. Privalov, Angew. Chem., Int. Ed., 2010, 49, 1773 CrossRef CAS PubMed.
- T. Privalov, B. Akermark and L. C. Sun, Chem. – Eur. J., 2011, 17, 8313 CrossRef CAS PubMed.
- X. Sala, M. Z. Ertem, L. Vigara, T. K. Todorova, W. Z. Chen, R. C. Rocha, F. Aquilante, C. J. Cramer, L. Gagliardi and A. Llobet, Angew. Chem., Int. Ed., 2010, 49, 7745 CrossRef CAS PubMed.
- L. L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. C. Sun, Nat. Chem., 2012, 4, 418 CrossRef CAS PubMed.
- R. Lalrempuia, N. D. McDaniel, H. Muller-Bunz, S. Bernhard and M. Albrecht, Angew. Chem., Int. Ed., 2010, 49, 9765 CrossRef CAS PubMed.
- A. Petronilho, J. A. Woods, H. Mueller-Bunz, S. Bernhard and M. Albrecht, Chem. – Eur. J., 2014, 20, 15775 CrossRef CAS PubMed.
- J. A. Woods, R. Lalrempuia, A. Petronilho, N. D. McDaniel, H. Muller-Bunz, M. Albrecht and S. Bernhard, Energy Environ. Sci., 2014, 7, 2316 CAS.
- J. F. Hull, D. Balcells, J. D. Blakemore, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2009, 131, 8730 CrossRef CAS PubMed.
- J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017 CrossRef CAS PubMed.
- A. Savini, G. Bellachioma, G. Ciancaleoni, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Chem. Commun., 2010, 46, 9218 RSC.
- A. Bucci, A. Savini, L. Rocchigiani, C. Zuccaccia, S. Rizzato, A. Albinati, A. Llobet and A. Macchioni, Organometallics, 2012, 31, 8071 CrossRef CAS.
- A. Savini, G. Bellachioma, S. Bolano, L. Rocchigiani, C. Zuccaccia, D. Zuccaccia and A. Macchioni, ChemSusChem, 2012, 5, 1415 CrossRef CAS PubMed.
- W. I. Dzik, S. E. Calvo, J. N. H. Reek, M. Lutz, M. A. Ciriano, C. Tejel, D. G. H. Hetterscheid and B. de Bruin, Organometallics, 2011, 30, 372 CrossRef CAS.
- D. G. H. Hetterscheid and J. N. H. Reek, Chem. Commun., 2011, 47, 2712 RSC.
- A. Lewandowska-Andralojc, D. E. Polyansky, C. H. Wang, W. H. Wang, Y. Himeda and E. Fujita, Phys. Chem. Chem. Phys., 2014, 16, 11976 RSC.
- K. E. deKrafft, C. Wang, Z. G. Xie, X. Su, B. J. Hinds and W. B. Lin, ACS Appl. Mater. Interfaces, 2012, 4, 608 CAS.
- N. Marquet, F. Gartner, S. Losse, M. M. Pohl, H. Junge and M. Beller, ChemSusChem, 2011, 4, 1598 CrossRef CAS PubMed.
- A. A. Gambardella, N. S. Bjorge, V. K. Alspaugh and R. W. Murray, J. Phys. Chem. C, 2011, 115, 21659 CAS.
- C. C. L. McCrory, S. H. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed.
- U. Hintermair, S. W. Sheehan, A. R. Parent, D. H. Ess, D. T. Richens, P. H. Vaccaro, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2013, 135, 10837 CrossRef CAS PubMed.
- C. Zuccaccia, G. Bellachioma, O. Bortolini, A. Bucci, A. Savini and A. Macchioni, Chem. – Eur. J., 2014, 20, 3446 CrossRef CAS PubMed.
- D. B. Grotjahn, D. B. Brown, J. K. Martin, D. C. Marelius, M. C. Abadjian, H. N. Tran, G. Kalyuzhny, K. S. Vecchio, Z. G. Specht, S. A. Cortes-Llamas, V. Miranda-Soto, C. van Niekerk, C. E. Moore and A. L. Rheingold, J. Am. Chem. Soc., 2011, 133, 19024 CrossRef CAS PubMed.
- A. Savini, P. Belanzoni, G. Bellachioma, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Green Chem., 2011, 13, 3360 RSC.
- M. Zhou, U. Hintermair, B. G. Hashiguchi, A. R. Parent, S. M. Hashmi, M. Elimelech, R. A. Periana, G. W. Brudvig and R. H. Crabtree, Organometallics, 2013, 32, 957 CrossRef CAS.
- D. C. Hong, M. Murakami, Y. Yamada and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 5708 CAS.
- J. D. Blakemore, N. D. Schley, G. W. Olack, C. D. Incarvito, G. W. Brudvig and R. H. Crabtree, Chem. Sci., 2011, 2, 94 RSC.
- J. Huang, J. D. Blakemore, D. Fazi, O. Kokhan, N. D. Schley, R. H. Crabtree, G. W. Brudvig and D. M. Tiede, Phys. Chem. Chem. Phys., 2014, 16, 1814 RSC.
- J. D. Blakemore, M. W. Mara, M. N. Kushner-Lenhoff, N. D. Schley, S. J. Konezny, I. Rivalta, C. F. A. Negre, R. C. Snoeberger, O. Kokhan, J. Huang, A. Stickrath, L. A. Tran, M. L. Parr, L. X. Chen, D. M. Tiede, V. S. Batista, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2013, 52, 1860 CrossRef CAS PubMed.
- J. M. Thomsen, S. W. Sheehan, S. M. Hashmi, J. Campos, U. Hintermair, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2014, 136, 13826 CrossRef CAS PubMed.
- S. W. Sheehan, J. M. Thomsen, U. Hintermair, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, Nat. Commun, 2015, 6, 6469 CrossRef CAS PubMed.
- D. G. H. Hetterscheid and J. N. H. Reek, Eur. J. Inorg. Chem., 2014, 742 CrossRef CAS.
- A. Venturini, A. Barbieri, J. N. H. Reek and D. G. H. Hetterscheid, Chem. – Eur. J., 2014, 20, 5358 CrossRef CAS PubMed.
- O. Diaz-Morales, T. J. P. Hersbach, D. G. H. Hetterscheid, J. N. H. Reek and M. T. M. Koper, J. Am. Chem. Soc., 2014, 136, 10432 CrossRef CAS PubMed.
- Y. Hayashi, K. Toriumi and K. Isobe, J. Am. Chem. Soc., 1988, 110, 3666 CrossRef CAS.
- J. Augustynski, M. Koudelka, J. Sanchez and B. E. Conway, J. Electroanal. Chem., 1984, 160, 233 CrossRef CAS.
- S. Le Vot, L. Roue and D. Belanger, Electrochim. Acta, 2012, 59, 49 CrossRef CAS.
- G. Pannetier and D. Macarovici, J. Therm. Anal., 1972, 4, 187 CrossRef CAS.
- A. R. Hillman, M. A. Skopek and S. J. Gurman, Phys. Chem. Chem. Phys., 2011, 13, 5252 RSC.
- E. Prouzet, J. Phys.: Condens. Matter, 1995, 7, 8027 CrossRef CAS.
- A. Minguzzi, O. Lugaresi, E. Achilli, C. Locatelli, A. Vertova, P. Ghigna and S. Rondinini, Chem. Sci., 2014, 5, 3591 RSC.
- J. H. Choy, D. K. Kim, S. H. Hwang, G. Demazeau and D. Y. Jung, J. Am. Chem. Soc., 1995, 117, 8557 CrossRef CAS.
- J. H. Choy, D. K. Kim, G. Demazeau and D. Y. Jung, J. Phys. Chem., 1994, 98, 6258 CrossRef CAS.
- M. C. Feiters, A. Longo, D. Banerjee, C. J. M. Van der Ham and D. G. H. Hetterscheid, J. Phys. Conf. Series 2005, XAFS 16 Proceedings.
- V. S. I. Sprakel, M. C. Feiters, W. M. Klaucke, M. Klopstra, J. Brinksma, B. L. Feringa, K. D. Karlin and R. J. M. Nolte, Dalton Trans., 2005, 3522 RSC.
- B. J. A. v. Weerdenburg, A. H. J. Engwerda, A. L. N. Eshuis, D. Banerjee, M. Tessari, C. F. Guerra, F. P. J. T. Rutjes, F. M. Bickelhaupt and M. C. Feiters, Chem. – Eur. J., 2015, 21, 10482 CrossRef PubMed.
- T. Nakagawa, C. A. Beasley and R. W. Murray, J. Phys. Chem. C, 2009, 113, 12958 CAS.
- M. N. Kushner-Lenhoff, J. D. Blakemore, N. D. Schley, R. H. Crabtree and G. W. Brudvig, Dalton Trans., 2013, 42, 3617 RSC.
- J. Y. Xu, M. Wang, G. Y. Liu, J. L. Li and X. D. Wang, Electrochim. Acta, 2011, 56, 10223 CrossRef.
- X. Q. Xiao and G. X. Jin, J. Organomet. Chem., 2008, 693, 3363 CrossRef CAS.
- A. Yates, C. White, P. M. Maitlis and D. M. Heinekey, Inorg. Synth, 1992, 29, 228 CrossRef.
- O. Diaz-Morales, F. Calle-Vallejo, C. de Munck and M. T. M. Koper, Chem. Sci., 2013, 4, 2334 RSC.
- S. Nikitenko, A. M. Beale, A. M. J. van der Eerden, S. D. M. Jacques, O. Leynaud, M. G. O'Brien, D. Detollenaere, R. Kaptein, B. M. Weckhuysen and W. Bras, J. Synchrotron Radiat., 2008, 15, 632 CrossRef CAS PubMed.
- K. V. Klementev, J. Phys. D: Appl. Phys., 2001, 34, 209 CrossRef CAS.
- S. J. Gurman, N. Binsted and I. Ross, J. Phys. C: Solid State Phys., 1984, 17, 143 CrossRef CAS.
- S. J. Gurman, N. Binsted and I. Ross, J. Phys. C: Solid State Phys., 1986, 19, 1845 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp00751a |
| ‡ The GC electrodes contained significant amounts of N and hence an accurate N:Ir ratio could not be obtained. |
| § Long term electrolysis has occasionally been reported as a measure to indicate the stability of molecular water oxidation catalysts. As long as there is diffusion of fresh catalyst from the bulk to the electrode surface, such measurements are meaningless. |
| This journal is © the Owner Societies 2016 |