
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling an electrostatic repulsion by oppositely charged surfactants towards positively charged fluorescent gold nanoclusters†
Ryan D.
Corpuz
,
Yohei
Ishida
and
Tetsu
Yonezawa
*
Division of Material Science and Engineering, Faculty of Engineering, Hokkaido University, Kita 13, Nishi 8, Kita-ku, Sapporo, Hokkaido 060-8628, Japan. E-mail: tetsu@eng.hokudai.ac.jp
First published on 9th February 2016
Abstract
A novel positively charged fluorescent gold nanocluster was successfully synthesized using the shortest cationic thiol, thiocholine. Effective control of electrostatic repulsion by the introduction of an anionic surfactant afforded a nanocluster that showed blue fluorescence emission.
Nanocluster synthesis is an interesting field of study, and various researchers are aiming at manipulating individual atoms, molecules, or groups of molecules to produce novel hybrid materials with unprecedented structures and properties.1–8 A typical example is the synthesis of gold nanoclusters with a non-metallic structure, whose distinctive optical properties are derived from the constituent gold nanoparticles, which generally show surface plasmon resonance at relatively larger diameters (>3 nm). The optical properties of gold nanoclusters are hypothesized to originate from the metallic cluster core, and they can be altered depending on the attached ligands. Unlike the collective oscillation of conduction electrons in the gold nanoparticle, a single electron transition results in molecule-like absorption and emission from the UV to NIR region in a gold nanocluster.9–11 Experimentally, it has been determined that the sharp contrast between the optical properties of the gold nanocluster and gold nanoparticles become observable when the particle size is below 2.4 nm and the gold particles no longer exhibit metallic properties.12
Among the tunable properties of gold nanoclusters, we focused on their optical properties, particularly photoluminescence. Photoluminescence is an important property because of its potential application in biomedical fields, especially for imaging, sensing, and therapy.13 The nanomaterials commonly used for these applications are quantum dots,14 which contain toxic components such as Cd, Pd, Se, and Te, and are highly discouraged. Hence, many researchers have proposed that gold could be one of the most viable alternative because of its chemical inertness, photostability, and biocompatibility. Unfortunately, gold in its nanoparticle form shows size-related toxicity,15 and thus, a synthetic scheme for fluorescent gold nanoclusters is an urgent demand.
There are several reported synthetic routes to fluorescent gold nanoclusters using polymers,16 dendrimers,17 DNA,18 or phosphine19 as the stabilizing compounds. We focus on gold nanoclusters with thiol stabilizers,20 pioneered by Murray and Whetten. In this regard, there are two commonly exploited methods involving the use of neutral and anionic thiol ligands including alkylthiols,21 tiopronin,22 phenylethylthiolate,23 and thiolate cyclodextrin.24 However, there is no established method for the synthesis of fluorescent gold nanoclusters by means of conventional chemical reduction using cationic thiols. Such nanoclusters are expected to play a significant role in bioimaging25 and sensing as cellular proteins show high affinity for positively charged nanocomposites than for neutral and anionic nanocomposites. In particular, quaternary ammonium-terminated thiols have rarely been used for the preparation of gold nanoparticles.26,27 Quaternary ammonium groups are always positively charged under any pH conditions. Thus, we hypothesized that a positively charged gold nanocluster could easily be absorbed inside the cell and potentially be used to study intercellular activities that are imperative in cancer research.13 The main limitation of cationic thiolate-protected gold nanoclusters is that the electrostatic repulsion between the cationic ligands on the surface of the nanoparticles hinders the formation of small clusters (ca. <2 nm) during nucleation in solvents. Our previous research showed the formation of a red dispersion of thiocholine bromide-stabilized gold nanoparticles even at a high molar ratio of the thiocholine ligand as compared with the gold ions.26 Thiocholine is the smallest quaternary ammonium terminated mercapto ligand. Moreover, silver,28 palladium,29 and platinum30 nanoparticles can be obtained by chemical reduction using thiocholine as the stabilizing reagent, but these nanoparticles, too, are large. Our objective is the utilization of the smallest cationic thiolate ligand, thiocholine (HS–(CH2)2–N(CH3)3+), to synthesize fluorescent gold nanoclusters by chemical reduction. Cationic thiolate-protected fluorescent gold nanoparticles or nanoclusters have been obtained only by a physical synthesis method by our group.31 This paper reports the first successful synthesis of positively charged nanoclusters by the conventional chemical reduction method.
In this study, the shortest cationic thiolate, thiocholine chloride (TC), was synthesized as described in a previous paper (see ESI†).31 Counter-ion exchange was carried out in order to eliminate the heavy metal effect of I−. The purity of TC was verified by 1H–NMR and FT-IR analyses.
First, we attempted to synthesize gold nanoclusters by the simple reduction of HAuCl4(Au) with excess NaBH4 in the presence of TC. The Au-to-TC mol ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, 1:5, or 1:7. For our typical synthesis (Au:TC = 1:7 (mol/mol)), we dissolved 0.0218 g of thiocholine chloride in 1 mL of methanol followed by addition of HAuCl4 stock solution (20 mmol dm−3, 1 cm3). It was then diluted with 8 cm3 of water–methanol solvent (1:1 vol/vol) and sonicated for 30 min prior to the addition of aqueous NaBH4 (2 mol dm−3, 50 mm3), the resulting solution was then sonicated for 90 min to ensure complete reduction of Au3+. The black line in Fig. 1 shows the extinction spectrum, including both absorption and scattering from nanoparticles, immediately after the synthesis at the Au:TC = 1:5 (mol/mol). From this result, it could be seen that the samples exhibited plasmon absorption at around 540 nm. Transmission electron microscopy (TEM) images of the representative samples are shown in Fig. S1 (ESI†), where the particle sizes are seen to exceed 4 nm, indicating that gold nanoclusters were not produced. As mentioned above, the strong repulsion between the TC ligands in the “nucleation stage” clusters hinders the formation of nanoclusters; the clusters coalesce spontaneously to form rather large nanoparticles. It is widely known that the surface charges on the gold clusters (mainly Au25 clusters32) could decompose the cluster structure. This would be the critical reason for the formation of rather nanoparticles with TC in the current system. The use of a higher ligand ratio did not solve this problem.
:3, 1:5, or 1:7. For our typical synthesis (Au:TC = 1:7 (mol/mol)), we dissolved 0.0218 g of thiocholine chloride in 1 mL of methanol followed by addition of HAuCl4 stock solution (20 mmol dm−3, 1 cm3). It was then diluted with 8 cm3 of water–methanol solvent (1:1 vol/vol) and sonicated for 30 min prior to the addition of aqueous NaBH4 (2 mol dm−3, 50 mm3), the resulting solution was then sonicated for 90 min to ensure complete reduction of Au3+. The black line in Fig. 1 shows the extinction spectrum, including both absorption and scattering from nanoparticles, immediately after the synthesis at the Au:TC = 1:5 (mol/mol). From this result, it could be seen that the samples exhibited plasmon absorption at around 540 nm. Transmission electron microscopy (TEM) images of the representative samples are shown in Fig. S1 (ESI†), where the particle sizes are seen to exceed 4 nm, indicating that gold nanoclusters were not produced. As mentioned above, the strong repulsion between the TC ligands in the “nucleation stage” clusters hinders the formation of nanoclusters; the clusters coalesce spontaneously to form rather large nanoparticles. It is widely known that the surface charges on the gold clusters (mainly Au25 clusters32) could decompose the cluster structure. This would be the critical reason for the formation of rather nanoparticles with TC in the current system. The use of a higher ligand ratio did not solve this problem.
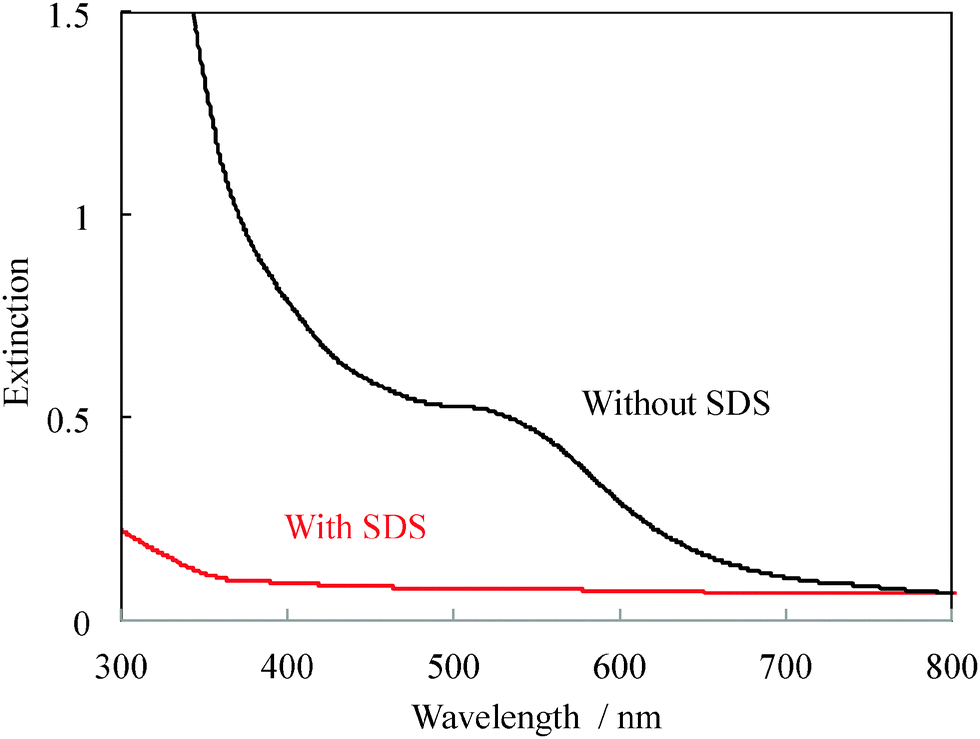 | ||
| Fig. 1 UV-Vis extinction spectra of Au nanoparticles and nanoclusters synthesized in the absence or presence of SDS (Au:TC:SDS = 1:5:0 and 1:5:5 (mol/mol/mol) for black and red spectra). | ||
In order to suppress the electrostatic repulsion between TC ligands on the surface of gold nanoclusters during the nucleation, we added a negatively charged surfactant, sodium dodecylsulfate (SDS), to form a Au(I)–TC–SDS complex before the reduction, which may neutralize the cation on TC and minimize the repulsion between the TC molecules. Synthesis of Au nanoparticles with SDS alone without thiocholine gave only large plasmonic ones (∼20 nm of diameter, Fig. S4, ESI†). To verify this concept, we synthesized gold nanoclusters at Au:TC:SDS molar ratios of 1:3:3, 1:5:5, and 1:7:7 (mol/mol/mol). In our typical experiment (HAuCl4:TC:SDS = 1:7:7 (mol/mol/mol)), we dissolved 0.0218 g of TC and 0.0404 of SDS in 5 cm3 of methanol followed by the addition of HAuCl4 stock aqueous solution (20 mmol dm−3, 1 cm3). The resulting solution was diluted with 3 cm3 of water and sonicated for 60 min until the solution became white indicating the formation of Au–thiolate complexes. The colour change from yellow to pale yellow/white indicated the reduction of Au(III) to Au(I) by the excess thiol compounds (Fig. S5, ESI†). Aqueous NaBH4 (20 mmol dm−3, 1 cm3) was then injected and the resulting solution was sonicated for another 60 min. The red line in Fig. 1 shows the UV-Vis extinction spectra of the gold nanoclusters synthesized at the Au:TC:SDS molar ratio of 1:5:5. This result suggested that in the presence of SDS, non-plasmonic particles were formed. We then observed the TEM images to determine the particle sizes (Fig. 2). The sample showing plasmon absorption (Au:TC:SDS = 1:3:3 (mol/mol/mol), see Fig. S2 (ESI†) for the spectrum) had particles with diameters larger than 3 nm. The samples that did not exhibit plasmon absorption (Au:TC:SDS = 1:5:5 and 1:7:7 (mol/mol/mol)), on the other hand, had particles with diameters well below 2 nm, indicating successful gold nanocluster formation. It is difficult to determine the exact diameter of such small clusters (∼1 nm of diameter) by conventional TEM; however, we observed a clear decreasing trend in the particle size with an increase in the mol ratios of TC and SDS.
 | ||
| Fig. 2 TEM images and particle size distributions of Au nanoparticles or nanoclusters synthesized at Au:TC:SDS mol ratios of (a) 1:3:3, (b) 1:5:5, and (c) 1:7:7 (mol/mol/mol). For the histograms, 200 particles were counted from several enlarged TEM images. | ||
Judging from the TEM and extinction spectra as well as previous data,9,17,33–35 we expected our gold nanoclusters to show fluorescence in the visible region. In order to increase the solubility of obtained gold nanoclusters, we introduced NaOH so that SDS attached to the surface of the TC-stabilized clusters can be removed (i.e. de-neutralization, see the detailed procedure outlined in ESI†). As shown in Fig. S3 (ESI†), after NaOH addition, an absorption shoulder appears at around 350 nm, which could be attributed to gold nanoclusters; a similar peak at this position was observed by other researchers, which was attributed to small gold nanoclusters consisting of 11 gold atoms or fewer9 (will be discussed later with fluorescence spectrum).
Under irradiation at 300 nm, we observed blue fluorescence, as shown in Fig. 3 (see images of samples under UV irradiation in the inset). The maximum excitation wavelength was around 357 nm, which was in good agreement with that for gold nanoclusters comprising 11 gold atoms and fewer, particularly those reported by Tran17 and Jin34 for Au4 (∼313 nm) and Au3 (∼303 nm), respectively, and Zheng35 for Au5 (330 nm) and Au8 (∼384 nm). Moreover, the maximum fluorescence wavelength (∼448 nm) agreed well with that of the Au8 cluster (455 nm).35 Hence, we hypothesized that the observed emission wavelength mainly originates from the Au8 nanocluster or a gold nanocluster with a similar size. Other smaller or larger nanoclusters (e.g. Au12, Au10, Au5, or Au4 which fluoresce at ∼630 nm,36 ∼523 nm,37 ∼385 nm,35 or ∼371 nm,17 respectively) could be minor contributors to the observed emission, and consequently, our gold nanoclusters showed relatively broad fluorescence.
 | ||
| Fig. 3 Fluorescence emission (blue) and excitation (black) spectra of synthesized gold nanoclusters (excitation wavelength = 300 nm, Au:TC:SDS = 1:7:7 (mol/mol/mol)). | ||
High-angle annular dark-field (HAADF) images of scanning transmission electron microscopy (STEM) were recorded in order to confirm the existence of very small nanocluster components (Fig. 4). Small gold nanoclusters are unstable and easily coalesce under strong electron beam irradiation; however, very small clusters composed only of Au atoms were observed in our experiments, consistent with our speculation based on fluorescence measurements.
 | ||
| Fig. 4 STEM-HAADF image of obtained Au nanoclusters. | ||
In summary, fluorescent gold nanoclusters were successfully prepared for the first time using the shortest cationic thiol, TC. By introducing SDS to minimize electrostatic repulsion between the TC ligands on the surface of the gold nanoclusters during nucleation, we could obtain non-plasmonic particles, which showed blue emission at around 448 nm. Without SDS, on the other hand, only large non-fluorescent gold nanoparticles were obtained. The method developed in this study is expected to be beneficial to the field of bioimaging and biosensing and be of interest to scientists pursuing research on nanoclusters.
This work was partially supported by KAKENHI (24241041 to TY) from JSPS, Japan and Murata Foundation (to TY). YI acknowledges the financial support from Building of Consortia for the Development of Human Resources in Science and Technology of Hokkaido University, The Suhara Memorial Foundation, Iketani Science and Technology Foundation, Toyota Physical & Chemical Research Institute Scholars, The Foundation for The Promotion of Ion Engineering, and Foundation Advanced Technology Institute. RDC thanks JICA, Japan, for the financial support during his stay in Sapporo. We also thank Dr Y. Wang (Hokkaido Univ.) for his valuable experimental assistance.
Notes and references
- R. Jin, Nanoscale, 2015, 7, 1549–1565 RSC.
- Y. Yamanoi, N. Shirahata, T. Yonezawa, N. Terasaki, N. Yamamoto, Y. Matsui, K. Nishio, H. Masuda, Y. Ikuhara and H. Nishihara, Chem. – Eur. J., 2006, 12, 314–323 CrossRef CAS PubMed.
- V. Malgras, Q. Ji, Y. Kamachi, T. Mori, F. Shieh, K. C. W. Wu, K. Ariga and Y. Yamauchi, Bull. Chem. Soc. Jpn., 2015, 88, 1171–1200 CrossRef CAS.
- K. Ariga, A. Vinu, Y. Yamauchi, Q. Ji and J. Hill, Bull. Chem. Soc. Jpn., 2012, 85, 1–32 CrossRef CAS.
- K. Ariga, Y. Yamauchi, G. Rydzek, Q. Ji, Y. Yonamine, K. C. W. Wu and J. Hill, Chem. Lett., 2014, 43, 36–68 CrossRef CAS.
- L. Wang, C. Liu, Y. Nemoto, N. Fukata, K. C. W. Wu and Y. Yamauchi, RSC Adv., 2012, 2, 4608–4611 RSC.
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- T. Yonezawa, T. Tominaga and D. Richard, J. Chem. Soc., Dalton Trans., 1996, 783–789 RSC.
- J. Zheng, C. W. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 0077402 CrossRef PubMed.
- S. K. Ghosha and T. Pal, Phys. Chem. Chem. Phys., 2009, 11, 3831–3844 RSC.
- R. P. Gotor and E. Grueso, Phys. Chem. Chem. Phys., 2011, 13, 1479–1489 RSC.
- A. Das, T. Li, K. Nobusada, Q. Zeng, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2012, 134, 20286–20289 CrossRef CAS PubMed.
- Y. Tao, M. Li, J. Rena and X. Qu, Chem. Soc. Rev., 2015, 44, 8636–8663 RSC.
- M. Bottrill and M. Green, Chem. Commun., 2011, 47, 7039–7050 RSC.
- N. Khlebtsov and L. Dykmana, Chem. Soc. Rev., 2011, 40, 1647–1671 RSC.
- S. Kanaoka, N. Yagi, Y. Fukuyama, S. Aoshima, H. Tsunoyama, T. Tsukuda and H. Sakurai, J. Am. Chem. Soc., 2007, 129, 12060–12061 CrossRef CAS PubMed.
- M. L. Tran, A. V. Zvyagin and T. Plakhotnika, Chem. Commun., 2006, 2400–2401 RSC.
- G. Liu, Y. Shao, K. Ma, Q. Cui, F. Wu and S. Xu, Gold Bull., 2012, 45, 69–74 CrossRef CAS.
- J. M. Pettibonea and J. W. Hudgens, Phys. Chem. Chem. Phys., 2012, 14, 4142–4154 RSC.
- R. S. Ingram, M. J. Hostetler, R. W. Murray, T. G. Schaff, J. Khoury, R. L. Whetten, T. P Bigioni, D. K. Guthrie and P. N. First, J. Am. Chem. Soc., 1997, 119, 9279–9280 CrossRef CAS.
- A. Kyrychenko, G. V. Karpushina, D. Svechkarev, D. Kolodezny, S. I. Bogatyrenko, A. P. Kryshtal and A. O. Doroshenko, J. Phys. Chem. C, 2012, 116, 21059–21068 CAS.
- A. P. Gies, D. M. Hercules, A. E. Gerdon and D. E. Cliffel, J. Am. Chem. Soc., 2007, 129, 1095–1104 CrossRef CAS PubMed.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- T. Das, P. Ghosh, M. S. Shanavas, A. Maity, S. Mondala and P. Purkayastha, RSC Adv., 2012, 2, 12210–12215 RSC.
- K. Kobayashi, J. Wei, R. Iida, K. Ijiro and K. Niikura, Polym. J., 2014, 46, 460–468 CrossRef CAS.
- T. Yonezawa, S. Onoue and N. Kimizuka, Chem. Lett., 2002, 1172–1173 CrossRef CAS.
- T. Yonezawa, S. Onoue and T. Kunitake, Kobunshi Ronbunshu, 1999, 56, 855–859 CrossRef CAS.
- T. Yonezawa, H. Genda and K. Koumoto, Chem. Lett., 2003, 32, 194–195 CrossRef CAS.
- M. Hosogi, G. Hashiguchi, M. Haga, T. Yonezawa, K. Kakushima and H. Fujita, Jpn. J. Appl. Phys., 2005, 44, L955 CrossRef CAS.
- Y. Ishida, T. Jirasupangkul and T. Yonezawa, New J. Chem., 2015, 39, 4214–4217 RSC.
- Y. Ishida, C. Lee and T. Yonezawa, Sci. Rep., 2015, 5, 15372 CrossRef CAS PubMed.
- X. Yuan, N. Goswami, I. Mathews, Y. Yu and J. Xie, Nano Res., 2015, 8, 3488–3495 CrossRef CAS.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- R. Jin, S. Egusa and N. F. Scherer, J. Am. Chem. Soc., 2004, 126, 9900–9901 CrossRef CAS PubMed.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402 CrossRef PubMed.
- Y. Negishi and T. Tsukuda, Chem. Phys. Lett., 2004, 383, 161–165 CrossRef CAS.
- P. Yu, X. Wen, Y. R. Toh and J. Tang, J. Phys. Chem. C, 2012, 116, 6567–6571 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S3, experimental details. DOI: 10.1039/c6cp00538a |
| This journal is © the Owner Societies 2016 |