
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface-active ionic liquids in micellar catalysis: impact of anion selection on reaction rates in nucleophilic substitutions†
Alice
Cognigni
a,
Peter
Gaertner
a,
Ronald
Zirbs
b,
Herwig
Peterlik
c,
Katharina
Prochazka
c,
Christian
Schröder
d and
Katharina
Bica
*a
aInstitute of Applied Synthetic Chemistry, Vienna University of Technology, Getreidemarkt 9/163, 1060 Vienna, Austria. E-mail: katharina.schroeder@tuwien.ac.at; Fax: +43 1 58801 16360; Tel: +43 1 58801 163601
bGroup for Biologically Inspired Materials, Institute of Nanobiotechnology (DNBT), University of Natural Resources and Life Sciences, Muthgasse 11, 1190 Vienna, Austria
cDynamics of Condensed Systems, University of Vienna, Boltzmanngasse 5, 1090 Vienna, Austria
dInstitute of Computational Biological Chemistry, University of Vienna, Währingerstrasse 17, 1090 Vienna, Austria
First published on 28th April 2016
Abstract
A series of surface-active ionic liquids based on the 1-dodecyl-3-methylimidazolium cation and different anions such as halides and alkylsulfates was synthesized. The aggregation behavior of these ionic liquids in water was characterized by surface tension, conductivity measurements and UV-Vis spectroscopy in order to determine the critical micelle concentration (CMC) and to provide aggregation parameters. The determination of surface activity and aggregation properties of amphiphilic ionic liquids was accompanied by SAXS studies on selected surface-active ionic liquids. The application of these surface-active ionic liquids with different anions was tested in nucleophilic substitution reactions for the degradation of organophosphorus compounds. Kinetic studies via UV-Vis spectrophotometry showed a strong acceleration of the reaction in the micellar system compared to pure water. In addition, an influence of the anion was observed, resulting in a correlation between the anion binding to the micelle and the reaction rate constants, indicating that the careful choice of the surface-active ionic liquid can considerably affect the outcome of reactions.
Introduction
In recent years the scope of ionic liquids as functional fluids has been expanded to include their mixtures with water for multiple applications.1 The microstructure and heterogeneity in the liquid state, fundamental aspects of pure ionic liquids, continue to exist in aqueous solution, and several reviews have been published focusing on the structure and behaviour of ionic liquid clusters in water.2 The self-assembly of ionic liquids in aqueous solution is particularly well investigated for long alkyl chain ionic liquids that have been found to display amphiphilic character analogous to conventional cationic surfactants.3 The co-existence of an ionic hydrophilic head group and a long hydrophobic tail results in the formation of aggregates in water; however, as a general rule the alkyl chain must contain at least eight carbon atoms for aggregation in water.4 A number of computational and experimental techniques, including molecular dynamics simulations, surface tension and conductivity measurements, potentiometry, UV-Vis spectroscopy, fluorescence probes, NMR spectroscopy, mass spectrometry, isothermal titration calorimetry, light scattering and small-angle X-ray and neutron scattering (SAXS and SANS) have been applied to understand the self-aggregation of surface-active ionic liquids in water.4–11While traces of water in ionic liquids are often considered as problematic due to the difficulty in their removal and their large impact on physical properties, the deliberate combination of these two media in well-defined mixtures may result in unique properties that can be fully exploited by varying their composition.12 For example, the limited solubility of many organic compounds in water could be enhanced with ionic liquids, whereas price or viscosity issues of ionic liquids could be balanced when used in aqueous solution. At the same time the specific properties of the highly tunable ionic liquids and the strong hydrogen bond of water can be combined to afford a reaction environment with new properties. Applications of well-defined ionic liquid/water mixtures have already been identified and range from biomolecule stabilization towards their application in catalysis or in novel separation processes. The impact of self-assembly of ionic liquids in aqueous solution has also been exploited by our group, showing that surface-active ionic liquids can be efficiently applied in micellar catalysis of Diels Alder reactions, for asymmetric transfer hydrogenations in water, and also for extraction of natural products.13,14
Studies on the self-assembly and structural organization of ionic liquids in water have so far dealt mainly with monocationic alkyl imidazolium based ionic liquids, however also other structures have already been investigated. Full critical micellar concentration (CMC) characterization and extensive thermodynamic studies allow identifying specific features that influence the properties of these ionic liquids. Several studies showed that the CMC is strongly influenced by the structure of the ionic liquid, in particular in terms of the alkyl chain length of the hydrophobic tail unit.3–6,15–19 As for traditional cationic surfactants, the Klevens equation was found to be valid.20 A linear relationship between the number of carbon atoms on the alkyl chain and the logarithm of the CMC has been verified19 which can be applied for predicting the CMC for a homologous series of linear-single chain amphiphiles at a fixed temperature. Cations with different structures have also been reported.5,6,21 Blesic et al.21 investigated the effect of different cationic rings by comparing alkylmethylimidazolium with alkylmethylpyridinium, alkylmethylpiperidinium and alkylmethylpyrrolidinium core groups. The results show that pyrrolidinium has the lowest CMC while the others present an almost identical surface tension profile, however due to the very complex interactions a univocal explanation based on geometry and/or electronic distribution could not be provided. This is in accordance with a study of Cornellas et al.5 where no substantial difference was identified in the behaviour for surface-active ionic liquids with imidazolium and pyridinium head groups. The introduction of an ester or amide functionality in the side chain of these head groups resulted in improved surface activity according to the studies of Comelles et al.22,23; moreover biodegradability was improved by the ester functionality, while the amide group provided a higher thermal stability. On the other hand El Seoud et al.24 showed that the introduction of a vinyl moiety on the imidazolium cation resulted in higher CMC values compared to the corresponding ethyl analogues due to the less hydrophobic character of the vinyl group. Baltazar et al.25 also investigated the aggregation behaviour of dicationic alkylimidazolium ionic liquids, and CMC values were found to depend not only on the chain length of the imidazolium cation, but also on the length of the linkage chain between the two head groups. Similar results were also obtained by Anderson et al.26 who investigated tricationic surface active ionic liquids. Besides showing a better surface activity compared to dicationic ones, this could be further improved by increasing the size and aromaticity of the internal linker. In addition, surface-active ionic liquids with different anions have been investigated, but studies on the role of the anion are less systematic. Blesic et al.4 compared the aggregation behaviour of 1-dodecycl-3-methylimidazolium ([C12mim]+) based ionic liquids with Cl−, [PF6]−, and [NTf2]− as counterions. Aggregates in aqueous solution were found only in the case of chloride anions, whereas the low solubility of the ionic liquids containing the [PF6]− and [NTf2]− anions resulted in phase separation prior to the formation of aggregates. In a different study on different halide counterions Kim et al. reported higher surface activity for [C12mim]I than for the same ionic liquids containing Br− or Cl− as a counter anion; this was explained by the higher polarizability and binding to the micelles of iodide ions.27
In a study conducted by Frizzo et al.28 concerning dicationic imidazolium based surface-active ionic liquids dissolved in 4.75% ethanol–water solution, a correlation between the aggregation and the anion hydrophobicity was identified. Recently Nowicki et al.29 reported a series of alkylimidazolium surface-active ionic liquids with HSO4− as a counterion. These ionic liquids had a key role as co-catalysts in an oxirane ring opening reaction of epoxidized methyl oleate, and their performance was dependent on the length of the alkyl chain attached to the imidazolium cation. Using a bulky amphiphilic anion such as ibuprofenate allows detection of a critical aggregation concentration even when coupled with [C4mim]+ or [C6mim]+ cations; moreover it was suggested that ionic liquids containing an active pharmaceutical ingredient can be incorporated into ionogels and used for drug delivery applications.30
In contrast, fewer studies exist on ionic liquids with surface-active anions in the literature. Early examples report surface-activity of octylsulfate combined with [C4mim]+, which also contributes to a decrease in the toxicity of the ionic liquid.10,31 Comprehensive work was conducted by Blesic et al.32,33 on alkylimidazolium alkylsulfonate ionic liquids. A matrix of ionic liquids with variable chain length in both the cation and the anion was synthesized and the physico-chemical properties were determined. Again, a strong dependence of the CMC on the chain length of the cation was identified, but the surface activity could be considerably improved by simultaneously introducing a long alkyl chain in the anion. A series of protic ionic liquids based on long-alkyl chain carboxylates was investigated with different cations such as imidazolium and pyrrolidinium. These ionic liquids not only showed a higher surface activity than cationic surfactants, but also performed better than the corresponding surfactants with inorganic cations.34 When diisopropylethylammonium was used as a cation, the formation of bilayer vesicle-like aggregates was also demonstrated.35
Eventually, fluorinated surface-active ionic liquids composed of perfluorinated anions have only recently been investigated.36,37 An extensive study by Pereiro et al. showed that perfluorinated alkylsulfonates strongly decrease the CMC in comparison to the corresponding halide-based ionic liquids, and even for [C2mim][C4F9SO3] and [C2mim][C8F17SO3] a critical aggregation concentration could be detected.
Herein, we intend to present further insights into the nature of self-organization of ionic liquids in water and its impact on micellar catalysis of nucleophilic substitution reactions in water. In particular, we focused on the role of the counteranion and aimed for a correlation between anion binding to the micelle and reaction rate constants, suggesting that the careful choice of the surface-active ionic liquid can considerably affect the outcome of reactions.
Results and discussion
Synthesis of surface-active ionic liquids
We focused on 1-dodecycl-3-methylimidazolium-based ionic liquid [C12mim]X, as this is a known and already characterized surface-active cation and we functionalized it with different anions. Our synthetic route is based on two different approaches (Scheme 1). In the case of the surface active [C12mim]Cl 1, [C12mim]Br 2 and [C12mim]I 3 the classical alkylation of N-methylimidazole with the corresponding dodecyl halides was performed. In contrast, surface-active ionic liquids [C12mim]OMs 4 and [C12mim]OTs 5 and [C12mim]OTf 6 were prepared in a two-step synthesis route, including the initial synthesis of N-dodecylimidazole by alkylation of imidazole. Afterwards the final ionic liquids were obtained via direct alkylation of N-dodecylimidazole with the methyl esters of the corresponding anions. This second route offers a halide free pathway for the synthesis of ionic liquids. In any case, good yields between 83 and 98% were obtained. The final products were purified by repeated crystallization from organic solvents to isolate all ionic liquids as colourless crystals in high purity. In contrast, crystallization was not possible for [C12mim]OTf 6; however, the highly volatile anion precursors could be easily removed under reduced pressure, allowing isolation of the ionic liquid as a low-melting colourless solid.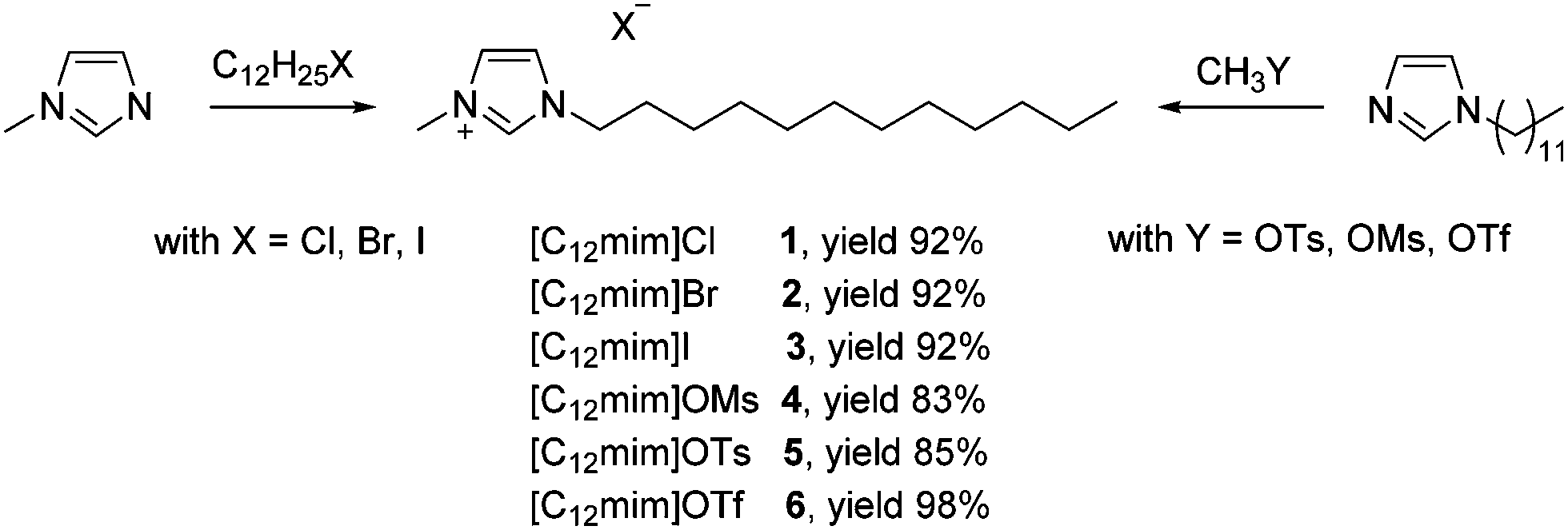 | ||
| Scheme 1 Synthesis with surface-active ionic liquids based on the 1-dodecycl-3-methylimidazolium cation with variable anions. | ||
Characterization of surface-activity
Prior to their application in micellar catalysis all ionic liquids were characterized by different experimental techniques in order to investigate the formation of aggregates in water. Several techniques can and have been used for the determination of the CMC of alkyl methyl imidazolium-based ionic liquids, and the reported literature values for a specific ionic liquid vary considerably according to the different techniques as can be seen from the ionic liquid [C14mim]Cl, where CMC values ranging from 2.98–4.6 mM have been reported. Herein, we selected three different techniques, including surface tension, conductivity and UV-Vis spectroscopy. The results are summarized in Table 1 and will be discussed in the following paragraphs in detail.| Ionic liquida | Surface tensionb | Conductivityc | UV-Visd | |||||
|---|---|---|---|---|---|---|---|---|
| CMC (mM) | Π CMC (mN m−1) | A min (nm2) | P | CMC (mM) | β | ΔG0mic (kJ mol−1) | CMC (mM) | |
| a Solutions were prepared with doubly-distilled Millipore Milli-Q water and left under shaking with 360 min−1 for 24 h to equilibrate. Samples were equilibrated at 25.0 ± 0.1 °C using a HAAKE K15 thermostat before measurements. b Surface tension was determined using the Du Noüy ring method on a Krüss tensiometer. c Conductivity measurement were performed at 25 °C using a Mettler Toledo SevenExcellence InLAB® 741-ISM electrode. d UV-Vis measurements were performed at 25 °C, following the absorbance at 312 nm using benzoylacetone. ΠCMC is the effectiveness of the surface tension reduction, Amin is the area per molecule residing at the surface, P is the packing parameter, β is the degree of counterion binding, ΔG0mic is Gibbs energy of micellization (for details see the ESI). | ||||||||
| [C12mim]Cl 1 | 13.25 (Lit. 13.17)6 | 28.6 | 0.72 | 0.29 | 14.53 | 0.43 | −29.1 | — |
| [C12mim]Br 2 | 9.29 | 29.3 | 0.71 | 0.30 | 10.29 | 0.70 | −36.2 | 10.24 |
| [C12mim]I 3 | 4.76 | 37.7 | 0.62 | 0.34 | 5.19 | 0.84 | −42.3 | 4.12 |
| [C12mim]OMs 4 | 12.50 | 29.4 | 1.13 | 0.18 | 14.71 | 0.45 | −29.5 | 15.26 |
| [C12mim]OTf 5 | 2.37 | 40.2 | 0.55 | 0.38 | 3.31 | 0.73 | −41.7 | 2.69 |
| [C12mim]OTs 6 | 2.25 | 38.3 | 0.65 | 0.32 | 3.44 | 0.89 | −45.4 | — |
Conductivity measurements
Conductivity measurements can be utilized for the characterization of ionic surfactants due to the different mobility of the individual ions and their aggregates. Curves of the specific conductivity (κ) versus the ionic liquid concentration are reported in Fig. 1 (left). Two different linear regimes could be typically identified that allowed calculating the CMC values via their breaking point. In principle, two phenomena are responsible for this curve shape: formed micelles have a lower mobility than single ions. Moreover, parts of the anions are adsorbed on the surface of the aggregates, decreasing the total species contributing to the solution conductivity upon formation of micelles. The degree of counterion binding (β) can be estimated from the ratio of the slopes27 and indicates the amount of anions on the surface of the micelles. The observed CMC values for ionic liquids already reported are in agreement with the results obtained by other research groups.27,32 A clear trend can be seen in the halide series, and CMC values decrease in the order Cl > Br > I. Similarly, a trend is visible for sulfonates where tosylate (OTs−) and triflate (OTf−) based ionic liquids have a lower CMC than [C12mim]OMs 4. A larger and more hydrophobic anion such as OTs− tends to adsorb on the micellar surface instead of being hydrated, and consequently favours the formation of aggregates by shielding the electrostatic repulsion forces of the charged headgroups.38 This behaviour is also supported by the degree of counterion binding (β): a higher value corresponds to a lower CMC due to the higher amount of anions bound to the micellar surface. Additionally, the Gibbs energy of micellization (ΔG0mic) could be calculated from the conductivity data, proving that the formation of micelles is a spontaneous process that is accompanied by a negative free energy change (see the ESI† for details).39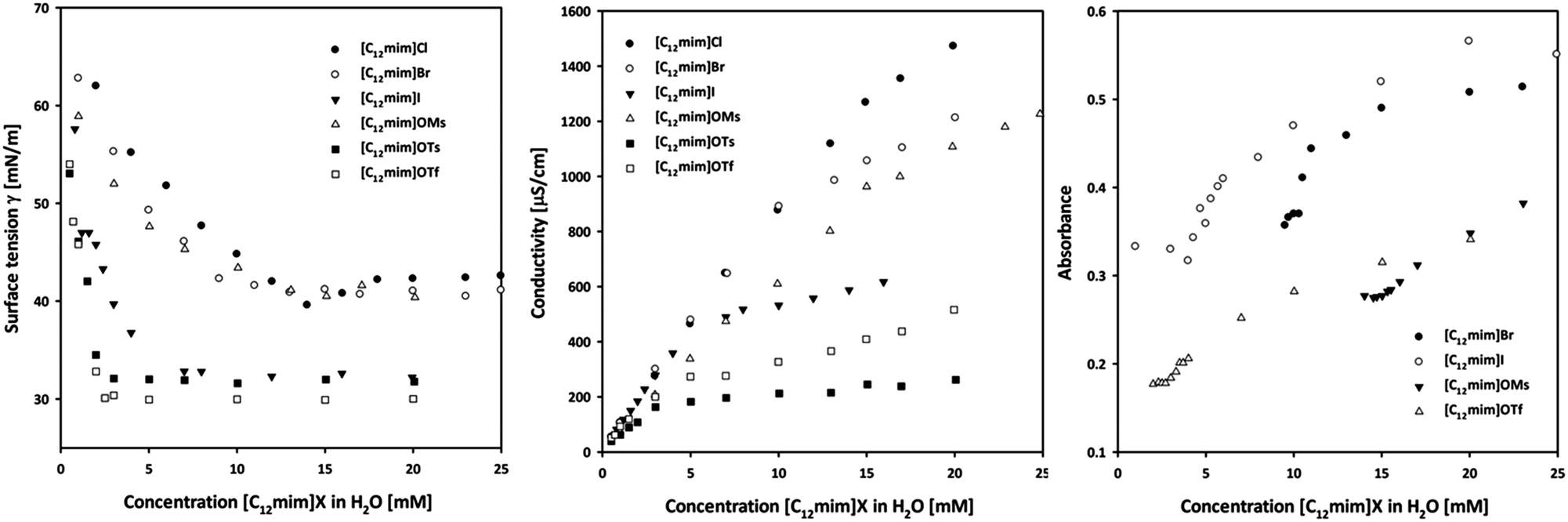 | ||
| Fig. 1 Surface tension (left), conductivity (center) and UV-Vis profiles (right) of the investigated ionic liquids at 25 °C. | ||
Surface-tension measurements
Fig. 1 (center) displays the surface tension (γ) curves as a function of the concentration of ionic liquids. As a consequence of absorption of ionic liquids at the surface, the surface tension decreases with increasing ionic liquid concentration, until a stable value is obtained after surface saturation. The breaking point of the curve can be used to determine the CMC value of the respective surfactant (see the ESI† for details). This characteristic behaviour allowed calculating CMC values for all ionic liquids. In general, the obtained CMC values were systematically slightly lower than those measured via the conductivity, however the same trend can be observed for the anions. According to the effectiveness of the surface tension reduction (ΠCMC) all investigated ionic liquids reduce the water surface tension of 30–40 mN m−1. The effectiveness can also be related to the degree of packing of adsorbed surfactant molecules at the interface. The Gibbs adsorption isotherm equation can be used to estimate the average area per molecule residing at the surface (Amin), assuming that the structure pattern corresponds to a monolayer.40 A lower Amin value corresponds to a closer packing of monomers at the interface. This value could be used to estimate the shape of the formed aggregates according to the packing parameter41 (P) which includes the volume (v) of the hydrocarbon chain embedded in the hydrocarbon core of the aggregate and the maximum effective length (lc) that the chain can assume, calculated according to the Tanford equation (see the ESI† for details).42 In general, a packing parameter P < 0.33 corresponds to spherical micelles, an intermediate value of 0.33 < P < 0.5 corresponds to non-spherical, ellipsoidal micelles, while higher values correspond to rod-like micelles and various interconnected structures.41 The results in Table 1 show that both spherical and ellipsoidal aggregates are presumably formed and depend on the anion size. According to this methodology, smaller anions such as bromide or mesylate form spherical micelles, while bigger anions, e.g. iodide or tosylate, favor the formation of ellipsoidal aggregates. This is to some extent in agreement with a study on the shape of alkyl imidazolium bromides by Goodchild et al.,9 who performed SANS experiments and found that [C12mim]Br forms spherical particles at low concentration near the CMC, while the shape tends to turn into elongated micelles at higher concentrations.UV-Vis measurements
The determination of the CMC of surfactants using UV-Vis spectroscopy is based on the ketoenolic equilibrium of benzoylacetone. The enolic form can be stabilized by intramolecular hydrogen bonding forming a 6-membered ring. This structure is favoured inside the micelles where no competition for hydrogen bonding with the solvent is present. Consequently the formation of micelles with increasing ionic liquid concentration will shift the ketoenolic equilibrium towards the enol form. This results in an increase of the absorbance at 312 nm which can be used to calculate the CMC value from the breaking point of the curves.43,44 The values depicted in Fig. 1 (right) are in good agreement with those obtained using the previous techniques, with the exception of [C12mim]Cl 1, where we were not able to find an unambiguous break point. However, we could not perform measurements for [C12mim]OTs due to the overlap of the absorption spectrum of the ionic liquids with that of benzoylacetone.Small-angle X-ray scattering
The determination of the surface activity and aggregation properties of amphiphilic ionic liquids was accompanied by SAXS studies. As the SAXS measurements and the evaluation were more time consuming, we decided to select three representative ionic liquids 1, 2 and 6, which cover the observed range of different conductivities (high, medium, and low conductivity in Fig. 1, center).The SAXS intensities (Fig. 2) were fitted using a model describing spherical micelles with a radius of the core Rc, a Schulz distribution for the radii and a shell thickness δ. The core radius is about 1 nm and exhibits a more uniform and narrow size distribution with increasing concentration (see Fig. S1a to c in the ESI†). The shell has a similar size, about 1 nm for [C12mim]Cl 1 and slightly higher for [C12mim]Br 2 and [C12mim]OTs 6. The most important information is the parameter γ, which describes the electron differences between the core, shell and surrounding medium45 and is defined in the ESI:† it decreases with increasing concentration as well as it is significantly lower for [C12mim]Cl (even close to zero for [C12mim]Cl at 100 mM concentration) than for both other ionic liquids. As a value of γ = 0 means that the scattering only arises from the core45 and the density of the shell is the same as the density of the surrounding solution, one may conclude that Cl is mobile and distributed in the solution, whereas OTs is only very weakly affected by different concentrations (Table 2). The inset in Fig. 2 shows the scattering intensities towards very low q-values. A weak maximum is also visible, which shifts towards smaller q-values for higher concentrations. This can be interpreted as a weak tendency for agglomeration of micelles with a cluster size of approximately 15 nm for the 25 mM concentration and considerably larger for higher concentration.
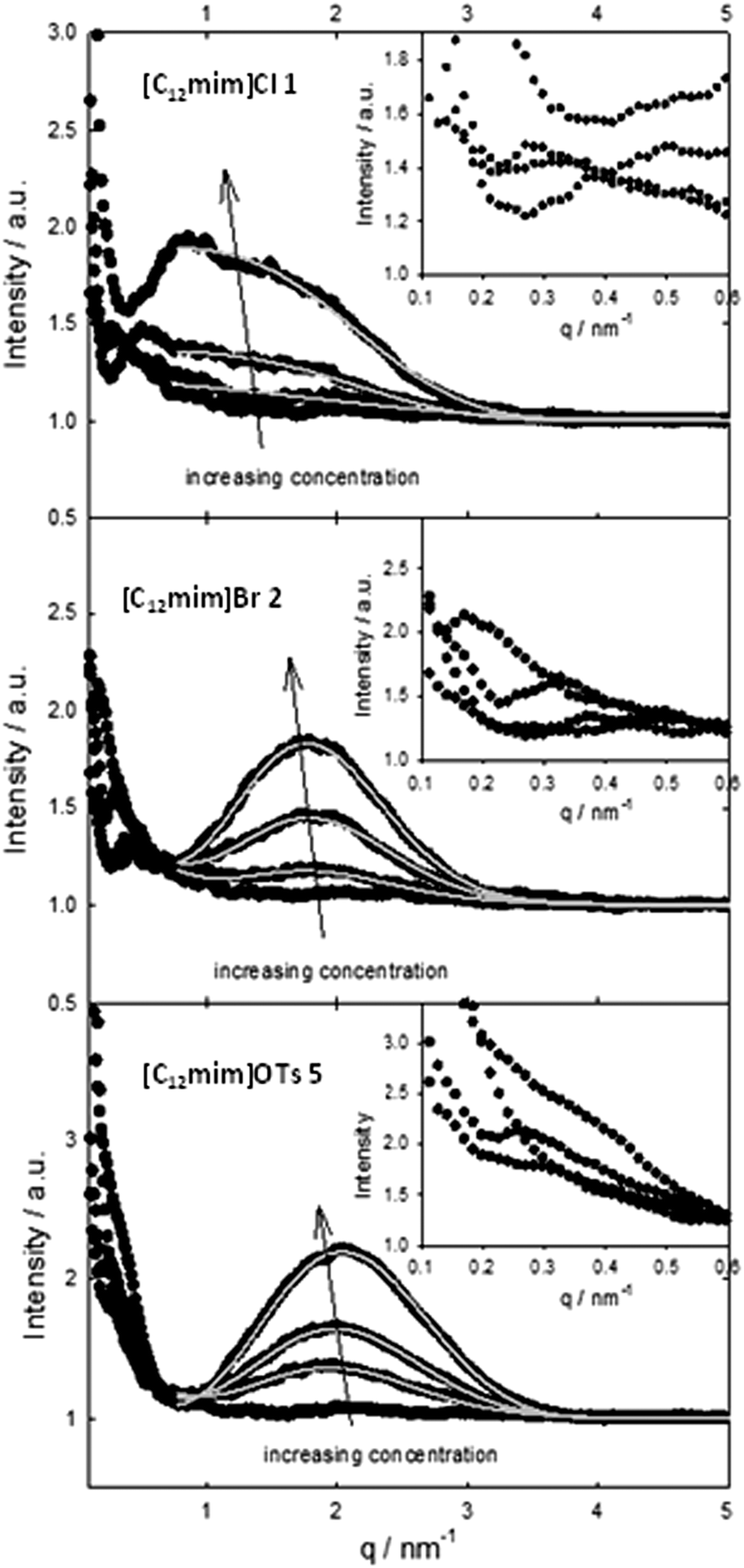 | ||
| Fig. 2 Small-angle X-ray intensities for the different ionic liquids. The experimental intensities are shown as filled circles, the fit from the core–shell model with grey fit lines and the insets show the scattering intensities towards very low q-values. | ||
| Ionic liquid | Concentration/mM | Core radius Rc/nm | Z | Shell thickness δ/nm | γ |
|---|---|---|---|---|---|
| [C12mim]OTs | 25 | 0.95 | 15 | 1.23 | 0.16 |
| [C12mim]OTs | 50 | 0.97 | 28 | 1.26 | 0.13 |
| [C12mim]OTs | 100 | 1.02 | 84 | 1.22 | 0.12 |
| [C12mim]Br | 25 | 0.97 | 8.3 | 1.27 | 0.20 |
| [C12mim]Br | 50 | 0.97 | 13 | 1.39 | 0.13 |
| [C12mim]Br | 100 | 0.98 | 25 | 1.59 | 0.09 |
| [C12mim]Cl | 25 | 1.14 | 52 | 0.95 | 0.07 |
| [C12mim]Cl | 50 | 1.18 | 250 | 0.94 | 0.06 |
| [C12mim]Cl | 100 | 1.14 | 51 | 0.94 | 0.007 |
Application in nucleophilic substitutions
Micellar catalysis has already been proven to be a very powerful tool to overcome issues of conventional synthesis in both pure organic solvents and water, and recent developments in the fields have been thoroughly reviewed.46–48 The problems created by the massive use of volatile, toxic and flammable organic solvents in industrial processes have increased the demand for more benign reaction media, and water is a prime candidate here. However, issues related to the poor solubility of organic compounds in water are well known, a problem that can be overcome via the addition of surfactants. It has already been reported that surface active ionic liquid aggregates can increase the solubility of apolar organic compounds that exhibit very poor solubility in water.49 Furthermore micelles have the capability to drastically alter reaction rates of organic reactions, as the local concentration experienced by the reactants at/in the micellar zone is higher than in a bulk solution. Surfactants based on ionic liquids can also be finely tuned by adjustments on the head group, alkyl chain or counterion structure in order to offer an optimal reaction environment increasing parameters such as yield, rate and selectivity. Successful results were already obtained in our previous research on Diels–Alder reactions.14 The highest reaction rate occurred above the CMC, clearly showing the positive effect of ionic liquid-derived micelles.Nucleophilic substitutions of organophosphorus compounds can be accelerated by cationic surfactants and are strongly influenced by different anions present in the reaction mixture.50 Optimal conditions for the substitution of organophosphorus compounds are obtained with α-nucleophiles, i.e. nucleophiles possessing a heteroatom with an unshared electron pair adjacent to the nucleophilic centre that exhibit a higher nucleophilic reactivity compared with common nucleophiles of similar basicity, a concept that is also known as the “α-effect”. More specifically, oximates have been shown to efficiently promote the hydrolysis of phosphate triesters as they prevent the competition with other nucleophiles such as OH− or halides.51–53 Studies on the effect of several oximes54 and substrate features51,53 have been reported.
In this study, we investigated the reaction between 4-nitrophenyl diphenyl phosphate (PNPDPP) and acetaldoxime (pKa = 11.82) in different surface-active ionic liquid/water mixtures (Scheme 2). PNPDPP is a highly lipophilic substrate which tends to interact strongly with micellar aggregates in aqueous solution. Moreover, it has an analogous structure to pesticides such as paraoxon or fenitron, both being toxic organophosphorous compounds that are widely used as pesticides in agriculture for field crop and fruit protection against a variety of insects and show considerable toxicity towards human and other mammals.54 In the literature second order kinetic constants of the reaction occurring in the micellar phase and in the bulk water are reported to be very similar, consequently, we were only interested in the observed pseudo first order kinetic constant that shows the overall reaction rate acceleration.53,54 The substitution reaction was conducted under pseudo-first order conditions where the oxime was used in 100-fold excess – an oxime concentration high enough to act as a buffer itself. Different possible reaction pathways have been reported for nucleophilic substitutions of organophosphorus compounds. In our case the only possible pathway is the attack of the nucleophile at the phosphorous centre, which follows a SN2(P) mechanism,54 while the attack on the aromatic carbons is very unlikely.53,55,56 In principle, two leaving groups are possible, either the favored p-nitrophenolate, whose formation can be observed at 402.5 nm or phenolate, which cannot be seen due to the spectral overlap with other components in the reaction mixture. We observed that the final absorbance reached during the kinetic runs did not correspond to the maximum theoretical one, suggesting the formation of a by-product. This was verified through 31P experiments, where the formation of both p-nitrophenyl phenyl phosphate and diphenyl phosphate as by-products was observed, although the formation of the latter one was considerably slower. Eventually, rate constants for both the desired reaction and the by-product formation were calculated through the least-squares exponential fit of the experimental data: results reported here as observed reaction rates kobs refer to the desired nucleophilic reaction only (see the ESI† for details).
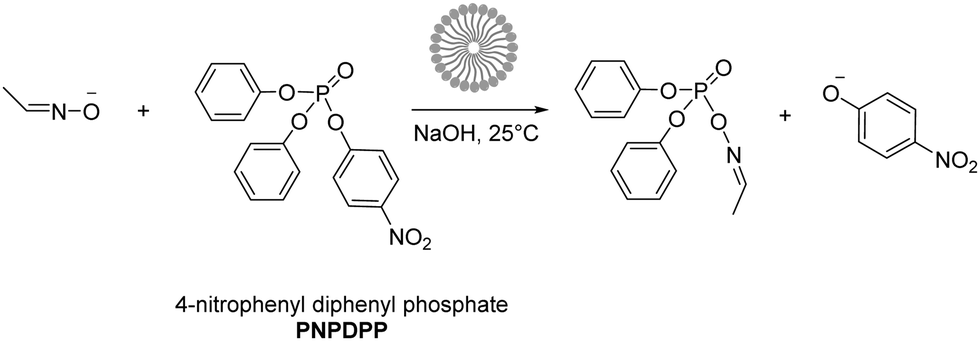 | ||
| Scheme 2 Nucleophilic substitution of 4-nitrophenyl diphenyl phosphate (PNPDPP) with acetaldoxime. | ||
In order to investigate the effect of our IL anions we initially investigated the reaction in all halide based ionic liquids [C12mim]X with X = Cl, Br and I in the concentration range from 0 to 100 mM. The observed reaction rates are shown in Fig. 3, and three interesting factors can be identified: in all cases a strong increase in the reaction rate was observed with increasing concentration of ionic liquids, resulting in strongly enhanced reaction rates compared to pure water. This rate enhancement in the micellar medium arises from two effects: on the one side the lipophilic substrate accumulates in the micelles, and at the same time the electrostatic attraction of the cationic head groups to the negatively charged nucleophile leads to the increment of its concentration at the micellar interface where the reaction takes place.
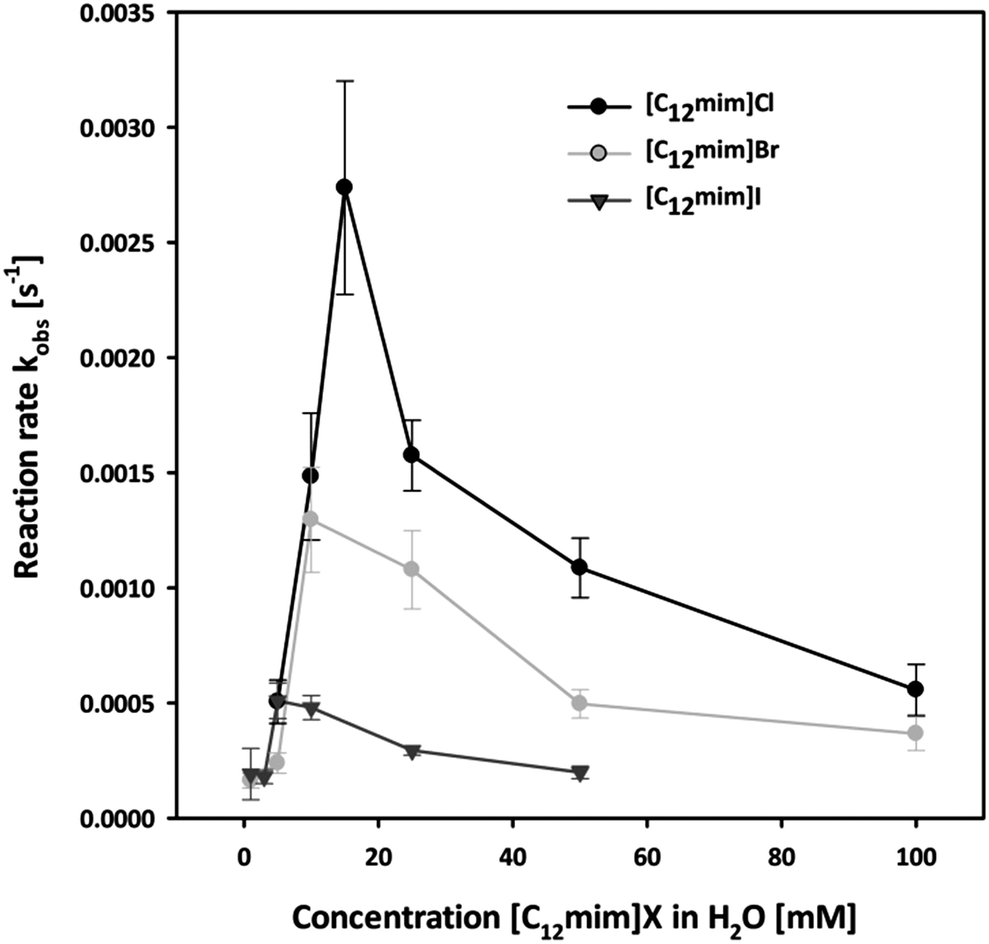 | ||
| Fig. 3 Pseudo first order reaction rate constant of the nucleophilic substitution of nitrophenyl diphenyl phosphate (PNPDPP) with acetaldoxime as a function of [C12mim]X with X = Cl, Br and I in water. | ||
Moreover, it can be observed that in all three cases the maximum acceleration for each ionic liquid corresponds to their CMC. Below the CMC the formation of aggregates induced by the presence of the solute causes the rate enhancement compared to water, while above the CMC most probably both a dilution effect over more micelles and a higher competition with the higher amount of inert anions take place. This effect is in accordance with the concept of micellar catalysis and has also been observed in our previous work on Diels–Alder reaction in surface-active ionic liquid/water mixtures.14
Most importantly, the highest enhancement of the reaction rate was observed with chloride-based ionic liquids, suggesting that the choice of anion plays a fundamental role. It has been reported that the presence of the inert anions influences the reaction rate due to a competition at the micellar interface with the reactive oximate.53,57 As a consequence different rate enhancement will take place depending on the extent to which the inert anions are bound to the micellar surface.50,58 The observed reaction rates are therefore in accordance with the physico-chemical data of the respective ionic liquid, as the chloride anion is less bound to the micellar surface and hence more readily exchangeable compared to bromide or iodide as it can also be seen from the β value calculated from the conductivity data.
This trend also exists when kinetic studies were extended to all surface-active ionic liquids involved in this study. Table 3 reports the maximum rate constants for all ionic liquids [C12mim]X 1–6 at a concentration close to their respective CMC, as this should correspond to the highest rate constants.
The results reported in Table 2 show again the dependence between the CMC and the rate enhancement, as ionic liquids with lower CMC show poorer performance than those with higher CMC, due to the ability of the anion to exchange with the nucleophile. Moreover, the correlation between the degree of anion binding to the micelle and the reaction rate constants exists also in the sulfonate-based series with [C12mim]X 4–6. The highest rate among these surface-active ionic liquids was observed for [C12mim]OMs, indicating again that the careful choice of the surface-active ionic liquid can considerably affect the outcome of reactions. In fact, this suggests that it would be possible to govern the rate of a desired reaction by applying a definite micellar system with targeted surface-active ionic liquids, and various projects in this regard are currently investigated in our lab.
| Ionic liquid | Concentration (mM) | Rate constant 103kobs (s−1) |
|---|---|---|
| Kinetics experiments were done using UV-Visible spectroscopy on a Shimadzu UV1800 spectrometer at 25 °C. All kinetic runs were performed under pseudo first order conditions, the initial concentrations of PNPDPP and oxime in the cuvette were 2.3 × 10−5 M and 2.3 × 10−3 M respectively. The appearance of the product p-nitrophenolate was monitored at 402.5 nm. | ||
| [C12mim]Cl 1 | 15 | 2.74 ± 0.46 |
| [C12mim]Br 2 | 10 | 1.30 ± 0.23 |
| [C12mim]I 3 | 5 | 0.51 ± 0.08 |
| [C12mim]OMs 4 | 15 | 1.68 ± 0.29 |
| [C12mim]OTs 5 | 3 | 0.42 ± 0.07 |
| [C12mim]OTf 6 | 2.5 | 0.18 ± 0.03 |
| Water | — | 0.08 ± 0.01 |
Experimental
Materials and methods
Commercially available reagents and solvents were used as received from Sigma Aldrich unless otherwise specified. Doubly-distilled deionized water was obtained from a Millipore Milli-Q water purification system (Millipore, USA). 1H and 13C NMR spectra were recorded on a Bruker AC 200 at 200 MHz, using the solvent peak as reference. N-Dodecylimidazole was synthesized according to a procedure already reported in the literature and distilled before use.59 All ionic liquids were dried for at least 48 h at room temperature and 0.01 mbar before use and were stored under an argon atmosphere.Synthetic procedures
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1163 (C–N). 1H NMR (200 Hz, CDCl3): δ (ppm) = 0.82 (3H, m, –C11H22–CH3), 1.19 (18H, m, –C2H4– C9H18–CH3), 1.88 (2H, m, –CH2– CH2–C10H21), 4.08 (3H, s, N–CH3), 4.27 (2H, t, J = 7.36 Hz, –CH2–C11H23), 7.45 (1H, s, –N–CH–CH–), 7.59 (1H, s, –N–CH–CH–), 9.94 (1H, s, –N–CH–N–). 13C NMR (200 Hz, CDCl3): δ (ppm) = 136.59, 123.84, 122.18, 50.20, 37.07, 31.82, 30.23, 29.52 (2C), 29.44, 29.32, 29.25, 28.93, 26.18, 22.60, 14.06.
C), 1498 (SO), 1194 (Ar–C–C), 1034 (C–N). 1H NMR (200 Hz, CDCl3): δ (ppm) = 0.81 (3H, t, J = 12.8 Hz, –C11H22–CH3), 1.16 (18H, m, –C2H4– C9H18–CH3), 1.71 (2H, t, J = 6.55 Hz, –CH2– CH2–C10H21), 2.27 (3H, s, –C6H6–CH3), 3.91 (3H, s, N–CH3), 4.09 (2H, t, J = 14.87 Hz, –CH2–C11H23), 7.07 (2H, d, J = 7.98 Hz, aromatic proton), 7.18 (1H, s, –N–CH–CH–), 7.32 (1H, s, –N–CH–CH–), 7.71 (2H, d, J = 8.12 Hz, aromatic proton), 9.78 (1H, s, –N–CH–N–). 13C NMR (50 Hz, CDCl3): δ (ppm) = 143.91, 139.25, 137.6, 137.44, 128.60, 125.73, 123.79, 122.01, 49.74, 36.15, 31.83, 30.87, 30.14, 29.55–29.48 (3C), 29.34–29.27 (2C), 28.93, 26.13, 22.61, 21.21, 14.06.
C), 1163 (C–N). 1H NMR (200 Hz, CDCl3): δ (ppm) = 0.82 (3H, m, –C11H22–CH3), 1.19 (18H, m, –C2H4– C9H18–CH3), 1.88 (2H, m, –CH2– CH2–C10H21), 4.08 (3H, s, N–CH3), 4.27 (2H, t, J = 7.36 Hz, –CH2–C11H23), 7.45 (1H, s, –N–CH–CH–), 7.59 (1H, s, –N–CH–CH–), 9.94 (1H, s, –N–CH–N–). 13C NMR (200 Hz, CDCl3): δ (ppm) = 136.59, 123.84, 122.18, 50.20, 37.07, 31.82, 30.23, 29.52 (2C), 29.44, 29.32, 29.25, 28.93, 26.18, 22.60, 14.06.
C), 1498 (SO), 1194 (Ar–C–C), 1034 (C–N). 1H NMR (200 Hz, CDCl3): δ (ppm) = 0.81 (3H, t, J = 12.8 Hz, –C11H22–CH3), 1.16 (18H, m, –C2H4– C9H18–CH3), 1.71 (2H, t, J = 6.55 Hz, –CH2– CH2–C10H21), 2.27 (3H, s, –C6H6–CH3), 3.91 (3H, s, N–CH3), 4.09 (2H, t, J = 14.87 Hz, –CH2–C11H23), 7.07 (2H, d, J = 7.98 Hz, aromatic proton), 7.18 (1H, s, –N–CH–CH–), 7.32 (1H, s, –N–CH–CH–), 7.71 (2H, d, J = 8.12 Hz, aromatic proton), 9.78 (1H, s, –N–CH–N–). 13C NMR (50 Hz, CDCl3): δ (ppm) = 143.91, 139.25, 137.6, 137.44, 128.60, 125.73, 123.79, 122.01, 49.74, 36.15, 31.83, 30.87, 30.14, 29.55–29.48 (3C), 29.34–29.27 (2C), 28.93, 26.13, 22.61, 21.21, 14.06.
Characterization of surface-activity
Solutions at various concentrations of the ionic liquids were prepared with doubly-distilled Millipore Milli-Q water and left under shaking at 360 min−1 for 24 h at RT to equilibrate.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ in the range from q = 0.1 to 20 nm−1, with 2θ being the scattering angle. In the figures, only the range up to q = 5 nm−1 is shown for clarity.
:oxime ratio of 1:2.
θ in the range from q = 0.1 to 20 nm−1, with 2θ being the scattering angle. In the figures, only the range up to q = 5 nm−1 is shown for clarity.
:oxime ratio of 1:2.
The reported values are an average of at least 3 runs. The values of the rate constant of the parallel reaction k′ were always at least one order of magnitude smaller than kobs.
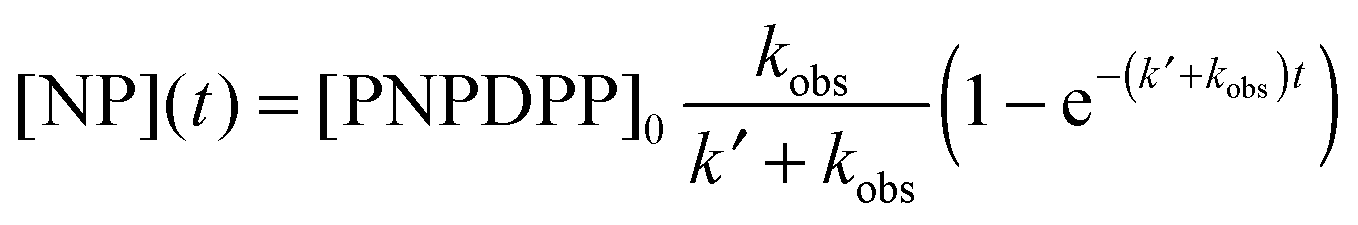 :90 (w/w)] were measured containing either the starting material or the reaction mixture. The peaks were referred to a capillary with H3PO4 as external standard. The obtained chemical shifts are in good agreement with those reported in the literature, and no other products were identified.
:90 (w/w)] were measured containing either the starting material or the reaction mixture. The peaks were referred to a capillary with H3PO4 as external standard. The obtained chemical shifts are in good agreement with those reported in the literature, and no other products were identified.
Conclusions
We have reported a study on the counterion role in surface-active ionic liquids based on 1-dodecyl-3-methylimidazolium. The two investigated series based on halides and sulfonate anions show a correlation between the tendency of the anions to reside closer to the micellar interface and their surface activity. These properties were reflected also when the surface-active ionic liquids were implied in micellar catalysis, in fact, a different rate acceleration was obtained using the different ionic liquids in nucleophilic substitutions in micellar media.Acknowledgements
Financial support from the Austrian Science Fund (FWF project P25504-N28) is gratefully acknowledged.Notes and references
- Y. Kohno and H. Ohno, Chem. Commun., 2012, 48, 7119 RSC.
- S. Chen, S. Zhang, X. Liu, J. Wang, J. Wang, K. Dong, J. Sun and B. Xu, Phys. Chem. Chem. Phys., 2014, 16, 5893 RSC.
- J. Łuczak, J. Hupka, J. Thöming and C. Jungnickel, Colloids Surf., A, 2008, 329, 125 CrossRef.
- M. Blesic, M. H. Marques, N. V. Plechkova, K. R. Seddon, L. P. N. Rebelo and A. Lopes, Green Chem., 2007, 9, 481 RSC.
- A. Cornellas, L. Perez, F. Comelles, I. Ribosa, A. Manresa and M. T. Garcia, J. Colloid Interface Sci., 2011, 355, 164 CrossRef CAS PubMed.
- O. A. El Seoud, P. A. R. Pires, T. Abdel-Moghny and E. L. Bastos, J. Colloid Interface Sci., 2007, 313, 296 CrossRef CAS PubMed.
- J. Sirieix-Plénet, Talanta, 2004, 63, 979–986 CrossRef PubMed.
- J. Bowers, C. P. Butts, P. J. Martin, M. C. Vergara-Gutierrez and R. K. Heenan, Langmuir, 2004, 20, 2191 CrossRef CAS PubMed.
- I. Goodchild, L. Collier, S. L. Millar, I. Prokeš, J. C. D. Lord, C. P. Butts, J. Bowers, J. R. P. Webster and R. K. Heenan, J. Colloid Interface Sci., 2007, 307, 455 CrossRef CAS PubMed.
- Z. Miskolczy, K. Sebők-Nagy, L. Biczók and S. Göktürk, Chem. Phys. Lett., 2004, 400, 296 CrossRef CAS.
- M. Tariq, M. G. Freire, B. Saramago, J. A. P. Coutinho, J. N. C. Lopes and L. P. N. Rebelo, Chem. Soc. Rev., 2012, 41, 829 RSC.
- K. R. Seddon, A. Stark and M.-J. Torres, Pure Appl. Chem., 2000, 72, 2275 CrossRef CAS.
- A. K. Ressmann, R. Zirbs, M. Pressler, P. Gaertner and K. Bica, Z. Naturforsch., B: J. Chem. Sci., 2013, 68b, 1129 Search PubMed.
- K. Bica, P. Gärtner, P. J. Gritsch, A. K. Ressmann, C. Schröder and R. Zirbs, Chem. Commun., 2012, 48, 5013 RSC.
- T. Inoue, H. Ebina, B. Dong and L. Zheng, J. Colloid Interface Sci., 2007, 314, 236 CrossRef CAS PubMed.
- J. Łuczak, C. Jungnickel, M. Joskowska, J. Thöming and J. Hupka, J. Colloid Interface Sci., 2009, 336, 111 CrossRef PubMed.
- C. Jungnickel, J. Łuczak, J. Ranke, J. F. Fernández, A. Müller and J. Thöming, Colloids Surf., A, 2008, 316, 278 CrossRef CAS.
- G. Bai, A. Lopes and M. Bastos, J. Chem. Thermodyn., 2008, 40, 1509 CrossRef CAS.
- R. Vanyúr, L. Biczók and Z. Miskolczy, Colloids Surf., A, 2007, 299, 256 CrossRef.
- P. D. T. Huibers, Langmuir, 1999, 15, 7546 CrossRef CAS.
- M. Blesic, A. Lopes, E. Melo, Z. Petrovski, N. V. Plechkova, J. N. Canongia Lopes, K. R. Seddon and L. P. N. Rebelo, J. Phys. Chem. B, 2008, 112, 8645 CrossRef CAS PubMed.
- M. T. Garcia, I. Ribosa, L. Perez, A. Manresa and F. Comelles, Colloids Surf., B, 2014, 123, 318 CrossRef CAS PubMed.
- M. T. Garcia, I. Ribosa, L. Perez, A. Manresa and F. Comelles, Langmuir, 2013, 29, 2536 CrossRef CAS PubMed.
- N. I. Malek, Z. S. Vaid, U. U. More and O. A. El Seoud, Colloid Polym. Sci., 2015, 293, 3213 CAS.
- Q. Q. Baltazar, J. Chandawalla, K. Sawyer and J. L. Anderson, Colloids Surf., A, 2007, 302, 150 CrossRef CAS.
- O. Nacham, A. Martín-Pérez, D. J. Steyer, M. J. Trujillo-Rodríguez, J. L. Anderson, V. Pino and A. M. Afonso, Colloids Surf., A, 2015, 469, 224 CrossRef CAS.
- M. Ao and D. Kim, J. Chem. Eng. Data, 2013, 58, 1529 CrossRef CAS.
- C. P. Frizzo, C. R. Bender, I. M. Gindri, P. R. S. Salbego, M. A. Villetti and M. A. P. Martins, Colloid Polym. Sci., 2015, 293, 2901 CAS.
- J. Nowicki, J. Łuczak and D. Stańczyk, RSC Adv., 2016, 6, 11591 RSC.
- C. Tourné-Péteilh, J.-M. Devoisselle, A. Vioux, P. Judeinstein, M. In and L. Viau, Phys. Chem. Chem. Phys., 2011, 13, 15523 RSC.
- A. Heintz, J. K. Lehmann, S. A. Kozlova, E. V. Balantseva, A. B. Bazyleva and D. Ondo, Fluid Phase Equilib., 2010, 294, 187 CrossRef CAS.
- M. Blesic, M. Swadźba-Kwaśny, J. D. Holbrey, J. N. Canongia Lopes, K. R. Seddon and L. P. N. Rebelo, Phys. Chem. Chem. Phys., 2009, 11, 4260 RSC.
- M. Blesic, M. Swadźba-Kwaśny, T. Belhocine, H. Q. N. Gunaratne, J. N. C. Lopes, M. F. C. Gomes, A. A. H. Pádua, K. R. Seddon and L. P. N. Rebelo, Phys. Chem. Chem. Phys., 2009, 11, 8939 RSC.
- M. Anouti, J. Jones, A. Boisset, J. Jacquemin, M. Caillon-Caravanier and D. Lemordant, J. Colloid Interface Sci., 2009, 340, 104 CrossRef CAS PubMed.
- M. Anouti, P.-Y. Sizaret, C. Ghimbeu, H. Galiano and D. Lemordant, Colloids Surf., A, 2012, 395, 190 CrossRef CAS.
- A. B. Pereiro, J. M. M. Araújo, F. S. Teixeira, I. M. Marrucho, M. M. Piñeiro and L. P. N. Rebelo, Langmuir, 2015, 31, 1283 CrossRef CAS PubMed.
- F. S. Teixeira, N. S. M. Vieira, O. A. Cortes, J. M. M. Araújo, I. M. Marrucho, L. P. N. Rebelo and A. B. Pereiro, J. Chem. Thermodyn., 2015, 82, 99 CrossRef CAS.
- J. Łuczak, M. Markiewicz, J. Thöming, J. Hupka and C. Jungnickel, J. Colloid Interface Sci., 2011, 362, 415 CrossRef PubMed.
- R. Zana, Langmuir, 1996, 12, 1208 CrossRef CAS.
- M. J. Rosen and J. T. Kunjappu, Surfactants and interfacial phenomena, John Wiley & Sons, 4th edn, 2012 Search PubMed.
- J. N. Israelachvili, Intermolecular and Surface Forces, Elsevier, 2011, p. 535 Search PubMed.
- C. Tanford, J. Phys. Chem., 1972, 76, 3020 CrossRef CAS.
- E. Iglesias, J. Phys. Chem., 1996, 100, 12592 CrossRef CAS.
- A. Dominguez, A. Fernandez, N. Gonzalez, E. Iglesias and L. Montenegro, J. Chem. Educ., 1997, 74, 1227 CrossRef CAS.
- P. Bartlett and R. H. Ottewill, J. Chem. Phys., 1992, 96, 3306 CrossRef CAS.
- G. La Sorella, G. Strukul and A. Scarso, Green Chem., 2015, 17, 644 RSC.
- B. H. Lipshutz and S. Ghorai, Green Chem., 2014, 16, 3660 RSC.
- T. Dwars, E. Paetzold and G. Oehme, Angew. Chem., Int. Ed., 2005, 44, 7174 CrossRef CAS PubMed.
- J. Łuczak, C. Jungnickel, M. Markiewicz and J. Hupka, J. Phys. Chem. B, 2013, 117, 5653 CrossRef PubMed.
- C. A. Bunton and L. Robinson, J. Org. Chem., 1969, 34, 773 CrossRef CAS.
- S. Couderc and J. Toullec, Langmuir, 2001, 17, 3819 CrossRef CAS.
- F. Terrier, P. Rodriguez-Dafonte, E. Le Guével and G. Moutiers, Org. Biomol. Chem., 2006, 4, 4352 CAS.
- V. K. Balakrishnan, X. Han, G. W. VanLoon, J. M. Dust, J. Toullec and E. Buncel, Langmuir, 2004, 20, 6586 CrossRef CAS PubMed.
- X. Han, V. K. Balakrishnan, G. W. vanLoon and E. Buncel, Langmuir, 2006, 22, 9009 CrossRef CAS PubMed.
- J. E. Omakor, I. Onyido, G. W. vanLoon and E. Buncel, J. Chem. Soc., Perkin Trans. 2, 2001, 324 RSC.
- B. A. Burnside, L. L. Szafraniec, B. L. Knier, H. D. Durst, R. A. Mackay and F. R. Longo, J. Org. Chem., 1988, 53, 2009 CrossRef CAS.
- C. A. Bunton, F. Nome, F. H. Quina and L. S. Romsted, Acc. Chem. Res., 1991, 24, 357 CrossRef CAS.
- N. Ouarti, A. Marques, I. Blagoeva and M.-F. Ruasse, Langmuir, 2000, 16, 2157 CrossRef CAS.
- B. S. Souza, E. C. Leopoldino, D. W. Tondo, J. Dupont and F. Nome, Langmuir, 2012, 28, 833 CrossRef CAS PubMed.
- B. F. Gilmore, G. P. Andrews, G. Borberly, M. J. Earle, M. A. Gilea, S. P. Gorman, A. F. Lowry, M. McLaughlin and K. R. Seddon, New J. Chem., 2013, 37, 873 RSC.
- A. E. Bradley, C. Hardacre, J. D. Holbrey, S. Johnston, S. E. J. McMath and M. Nieuwenhuyzen, Chem. Mater., 2002, 14, 629 CrossRef CAS.
- A. E. Bradley, C. Hardacre, J. D. Holbrey, S. Johnston, S. E. J. McMath and M. Nieuwenhuyzen, Chem. Mater., 2002, 14, 629 CrossRef CAS.
- S. Jones and D. Selitsianos, Org. Lett., 2002, 4, 3671 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Formulae for calculating aggregation parameters and fitting of kinetic constants and copies of NMR spectra. See DOI: 10.1039/c6cp00493h |
| This journal is © the Owner Societies 2016 |