
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced anion binding by heteroatom replacement in bambusurils†
Ephrath
Solel
a,
Mandeep
Singh
b,
Ofer
Reany
*b and
Ehud
Keinan
*ac
aSchulich Faculty of Chemistry, Technion - Israel Institute of Technology, Technion City, Haifa 32000, Israel. E-mail: Keinan@technion.ac.il
bDepartment of Natural Sciences, The Open University of Israel, 1 University Road, P.O. Box 808, Ra'anana 43107, Israel. E-mail: oferre@openu.ac.il
cGuangdong Technion Israel Institute of Technology and Department of Chemistry, Shantou University, Guangdong 515063, PR China
First published on 9th March 2016
Abstract
This study was driven by the hypothesis that heteroatom replacement in bambusurils could significantly modify their anion binding properties. Indeed, calculations with various glycoluril and bambusuril analogs predict that such replacements significantly alter their molecular electrostatic potential and binding properties. Both polarization and electrostatic interactions contribute to anion binding, leading to a general trend of affinity among the neutral molecules: X = S > O > NH. In bambusurils the heteroatom replacement at the portal carbonyls affect the induced dipole more significantly than replacements at the equatorial carbonyls. The stronger polarization and stronger anion binding manifest the increased aptitude of the portal heteroatoms as electron sinks. Notably, this study predicts that protonated aza-bambusurils would not only bind multiple anions along their main axis, but could also function as synthetic anion channels.
Introduction
The glycoluril molecule, 1, has a unique multifunctional structure that renders it an increasingly recognized building block for supramolecular architecture, as demonstrated by the two rapidly growing families of cucurbiturils1–4 and bambusurils.5–7 Although both macrocycles are cycloolygomers of glycolurils connected by methylene bridges, they exhibit diametrically different host–guest chemistry. While the cucurbiturils efficiently bind cations at their portals and hydrophobic guests at their interior,8,9 the bambusurils strongly bind anions at their interior.10–15 Clearly, the relative orientation of the glycoluril units with respect to the guest molecules is key to their binding preferences. While cucurbiturils present to their guest molecules the concave, hydrophobic face of their glycoluril units, the bambusurils present their convex, electron-poor faces, rendering them anion binders.16Realizing that these dramatic differences reflect a highly diverse electrostatic landscape on the glycoluril molecular surface, we envisioned that the replacement of either one oxygen atom or both by another heteroatom, such as sulfur or nitrogen (e.g.2, Fig. 1) would significantly alter its molecular electrostatic potential and binding properties. Hence, the appropriately modified cucurbiturils and bambusurils could offer novel binding opportunities. Unfortunately, although predicted to be thermodynamically stable,17,18 thio-cucurbiturils could not be synthesized to date.19,20
 | ||
| Fig. 1 Glycoluril 1, glycoluril derivatives 2 and bambus[6]urils 3. | ||
With bambusurils, however, our efforts to prepare heteroatomic analogs turned out to be more successful.21 We took advantage of the relative stability of substituted monothio-glycolurils to synthesize semithio-bambusuril, 3b, which exhibited strong binding to a broad variety of anions in its interior. Interestingly, although sulfur is less electronegative than oxygen, 3b was found to be a generally stronger anion binder than 3a.22 As revealed by their crystal structures with halide guests, the anion-binding site in both 3a and 3b is located at the center of the macrocycle, far away from the polar groups. In all cases the halide anion is held in an electrostatic focal point with close contacts to 12 methine hydrogen atoms (Fig. 2).5,21
 | ||
| Fig. 2 Left: X-ray structure of 3b with a bromide anion guest. Right: A truncated structure of the complex showing some short contacts (color code: oxygen, red; nitrogen, blue; carbon, grey; bromine, brown; hydrogen, white).21 | ||
We hypothesized that heteroatom replacement in bambusuril could significantly modify its anion binding properties. Here we report that DFT calculations indeed predict that the anion binding ability of either glycolurils, or bambusurils, are strongly affected by the nature of their heteroatoms, following a general trend among the neutral molecules: X = S > O > NH. The results also suggest that the most positive electrostatic potentials occur at the vicinity of the methine hydrogens and the most negative potentials reside at the portal heteroatoms, and not at the equatorial ones. Most importantly, this study predicts that protonated semiaza-bambusurils would function as multiple anion binders and could even serve as anion channels.
Results and discussion
In order to estimate the binding properties of the various bambusuril analogs we had to understand the electrostatic landscape of the monomeric unit, free of the steric constraints exerted by the entire macrocycle. Nevertheless, while embedded within the bambusuril framework the glycoluril unit becomes quite rigid. Therefore, in order to best represent this situation we carried out all computations on the more rigid derivative, 2, where all nitrogen atoms are locked within a tetracyclic system with restricted conformational freedom. Furthermore, in all calculations we replaced only one of the glycoluril oxygen atoms by other heteroatoms in order to use the unmodified urea as an internal reference for comparison with the modified one.To calculate the structures of the complexes formed by 2a–e with chloride anions as well as their energies we used three different methods: B3LYP/6-311+G** with and without the D3 version of Grimme's dispersion,23 and M062X/6-311+G**.24,25 We have taken this approach in order to evaluate the dispersion forces, which could have a significant effect on the geometry and energy of the complexes. Since the M06-2X functional has proven useful for predicting non-covalent interactions we compared the electrostatic potential maps, structure of the HOMO and LUMO and binding geometries of the different complexes (Fig. 3) as calculated with either M06-2X or with the two versions of B3LYP.
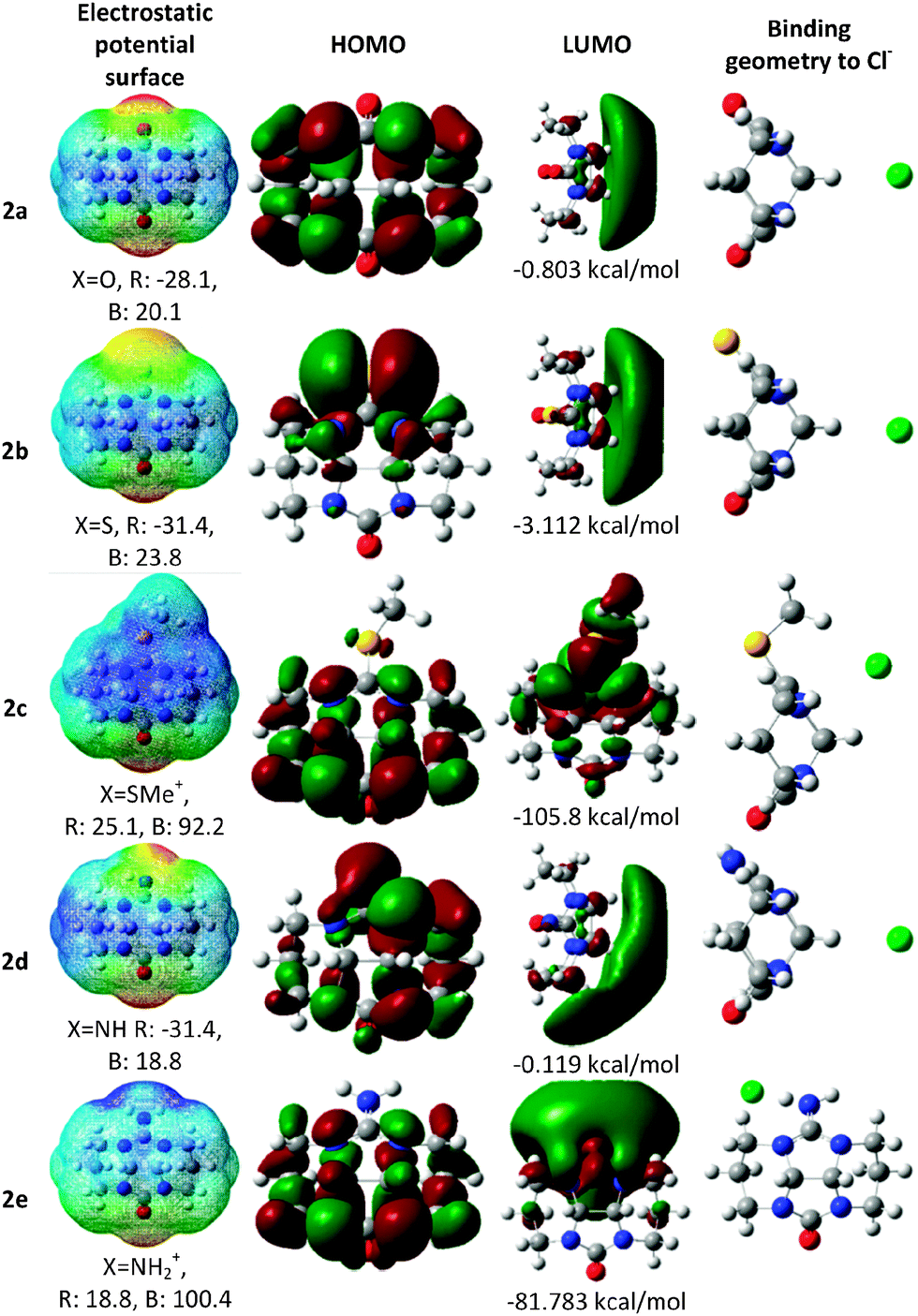 | ||
| Fig. 3 Calculated electronic properties of glycoluril analogs, 2a–e at the M06-2X/6-311+G** level of theory. The electrostatic potential surfaces are presented with their energy scale (kcal mol−1), ranging from red (R) to blue (B). | ||
Since in bambusurils the guest anion is held at the center of the macrocycle with close contacts to the methine hydrogens we checked whether this would also be the case with the monomeric model, 2. Indeed, our calculations show that the same interactions are dominant in the cases of all neutral molecules, 2a, 2b and 2d (X = O, S, NH, Fig. 3). In the case of the charged analogs the binding geometries reflect different interactions.
In the case of 2c (X = SMe+) we examined two conformers with the methyl group residing on either the concave or convex face of the molecule. The latter conformer, shown in Fig. 3, is more stable and also binds the chloride anion more strongly. The anion interacts with the π system of the thiourea unit, thus maintaining contacts with both the methine hydrogens and the methyl group (Fig. 3). Expectedly, with 2e (X = NH2+), the preferred anion binding site lies near the cationic group with close charge–charge interaction as well as hydrogen bonding with both the N–H group and the neighbouring methylene.
Remarkably, the HOMO of 2b is located mostly on the sulfur atom rather than on the oxygen, even though the latter is more electronegative. This observation leads to the prediction that semithio-glycolurils would preferentially bind metal cations at the sulfur atom rather than at oxygen. Indeed, we have already shown that semithio-bambus[4]uril strongly binds mercuric chloride and palladium(II) acetate at its sulfur atoms.21
Table 1 presents the electronic energy, binding enthalpy and Gibbs free energy of complexation (calculated with all three functionals) between the glycoluril analogs, 2, and a chloride anion. The inclusion of dispersive interactions (with B3LYP D3 and M06-2X) produced more stable structures, about 2 kcal mol−1 more negative than with B3LYP (except for the case of 2c). The binding energies with all three methods were found to follow the same trend with the positively charged derivatives, 2c and 2e, being the strongest binders, whereas for the neutral molecules the binding increases with S > O > NH.
| X | Methoda | ΔE | ΔE + zpe | ΔH | ΔG | |
|---|---|---|---|---|---|---|
| a Calculations using three different DFT methods with the 6-311+G** basis set. | ||||||
| 2a | O | B3LYP | −15.54 | −15.46 | −15.39 | −9.75 |
| B3LYP D3 | −17.75 | −17.64 | −17.59 | −11.66 | ||
| M06-2X | −17.62 | −17.44 | −17.38 | −11.41 | ||
| 2b | S | B3LYP | −18.22 | −18.01 | −17.95 | −12.19 |
| B3LYP D3 | −20.48 | −20.35 | −20.27 | −14.42 | ||
| M06-2X | −20.44 | −20.30 | −20.23 | −14.18 | ||
| 2c | SMe+ | B3LYP | −89.18 | −88.87 | −89.01 | −81.80 |
| B3LYP D3 | −94.10 | −93.71 | −93.92 | −86.48 | ||
| M06-2X | −96.71 | −96.23 | −96.39 | −88.90 | ||
| 2d | NH | B3LYP | −14.18 | −14.14 | −14.05 | −8.36 |
| B3LYP D3 | −16.36 | −16.30 | −16.22 | −10.49 | ||
| M06-2X | −16.12 | −16.02 | −15.95 | −10.01 | ||
| 2e | NH2+ | B3LYP | −98.35 | −99.32 | −99.72 | −92.73 |
| B3LYP D3 | −101.26 | −102.17 | −102.58 | −96.80 | ||
| M06-2X | −101.93 | −103.33 | −103.79 | −96.46 | ||
Natural bond orbital (NBO) analysis allowed for assessing the natural atomic charges before and after complexation. Results of these calculations reflect the charge transferred from the chloride ion to 2a, 2b, 2d and 2e and the induced change in electronic charge distribution in the glycoluril derivatives as a result of interaction with the electrostatic field of the chloride anion (Table 2). The calculations show that the binding energies are strongly affected by the nature of the heteroatom X, although it is positioned far away from the anion. Most of the positive electron density resides on the methine hydrogens and its amount is larger with 2b than with 2a and 2d (Table 2 and Fig. 4).
 | ||
| Fig. 4 Glycoluril derivatives 2–5 with selected close contacts (Å) of the chloride ion with hydrogen atoms. | ||
| 2a | 2a&Cl− | Diff. | 2b | 2b&Cl− | Diff. | 2d | 2d&Cl− | Diff. | 2e | 2e&Cl− | Diff. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a For clarity, atoms with asterisk do not involve a plane of symmetry as shown in Fig. 4. | ||||||||||||
| X | −0.649 | −0.691 | −0.042 | −0.213 | −0.312 | −0.099 | −0.769 | −0.814 | −0.045 | −0.767 | −0.798 | −0.031 |
| H(NH) | 0.341 | 0.330 | −0.011 | 0.429 | 0.417 | −0.012 | ||||||
| H*(NH) | 0 | 0.429 | 0.399 | −0.030 | ||||||||
C(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X) X) |
0.868 | 0.868 | 0 | 0.329 | 0.360 | 0.031 | 0.660 | 0.669 | 0.009 | 0.747 | 0.741 | −0.006 |
| N | −0.572 | −0.563 | 0.009 | −0.547 | −0.535 | 0.012 | −0.571 | −0.562 | 0.009 | −0.498 | −0.510 | −0.012 |
| N* | −0.562 | −0.554 | 0.008 | −0.498 | −0.533 | −0.035 | ||||||
| C(CH) | 0.199 | 0.177 | −0.022 | 0.198 | 0.176 | −0.022 | 0.200 | 0.176 | −0.024 | 0.200 | 0.201 | 0.001 |
| C* | 0.200 | 0.182 | −0.018 | 0.200 | 0.200 | 0 | ||||||
| H(CH) | 0.210 | 0.278 | 0.068 | 0.215 | 0.284 | 0.069 | 0.210 | 0.282 | 0.072 | 0.236 | 0.220 | −0.016 |
| H*(CH) | 0.210 | 0.272 | 0.062 | 0.236 | 0.218 | −0.018 | ||||||
| N′ | −0.572 | −0.563 | 0.009 | −0.572 | −0.562 | 0.01 | −0.574 | −0.564 | 0.01 | −0.567 | −0.569 | −0.002 |
| N′* | −0.572 | −0.563 | 0.009 | −0.567 | −0.575 | −0.008 | ||||||
| C(CO) |
0.868 | 0.868 | −0.001 | 0.870 | 0.870 | 0 | 0.869 | 0.868 | −0.001 | 0.867 | 0.869 | 0.002 |
| O | −0.649 | −0.691 | −0.042 | −0.644 | −0.684 | −0.04 | −0.653 | −0.693 | −0.04 | −0.602 | −0.631 | −0.029 |
| Cl | −0.974 | −0.970 | −0.974 | −0.748 | ||||||||
The enhanced positive electron density at the methine area of 2b relative to either 2a or 2d can be understood in terms of the greater charge transfer from the lone pairs of the ureido nitrogen atoms to the π*(CX) orbital in 2b (XS) than in 2a (XO) and 2d (XNH). As observed with thioamide and amide groups,26 this effect reflects the greater polarizability of the sulfur atom, awarding the nitrogen atoms in 2b with larger positive charge.
Expectedly, upon binding to the chloride anion the X atom and the methine carbon become more negative whereas the hydrogen atoms of the methine groups become more positive, all testify for the formation of an induced dipole moment upon binding. In the case of 2d the charge change on either O or NH is comparable whereas in 2b the charge modification on S is greater than on O, reflecting greater polarizability of sulfur in comparison to oxygen.27,28 Thus, although oxygen is more electronegative than sulfur, the greater polarizability of the latter contributes to the stronger binding to anions.
Thus, the greater polarizability allows, not only for stronger dispersion forces, but also for stronger electrostatic interactions, both from the initial electrostatic charges on the glycolurils and from the greater induced dipole moment. The relative contribution of the dispersion interaction in the complexes can be deduced from the energy differences calculated with either B3LYP or B3LYP-D3.
Apparently, the dispersion contribution is rather small, in the order of 2–3 kcal mol−1 for all neutral complexes, whereas for 2b this contribution is slightly larger than for 2a and 2d. Remarkably, calculations with M06-2X show that 2e can bind more than one anion. In addition to the central binding site, a second chloride anion can form hydrogen bonds with either the N–H* (Fig. 4) and the nearby CH2 or with the two methine hydrogen atoms. The binding energies of these complexes (relative to the monoanionic complex) were found to be −31.89 and −29.44 kcal mol−1, respectively. Adding a third chloride anion to the more stable dichloride complex would cost 16.30 kcal mol−1.
In order to identify the relative magnitude of the various contributions to the complexation energy other than dispersion interactions, we performed natural energy decomposition analysis (NEDA)29,30 at the B3LYP/6-311+G** level (Table 3). The results suggest that the main driving force for the chloride ion binding are electric interactions (EL), which include electrostatic interactions that originate from the charge distributions in each component before complexation (ES) and the interactions that result from the induced dipoles upon complexation (POL). The charge transfer interaction (CT), which reflects electron delocalization between filled orbitals on one of the units and unfilled orbitals on the other unit, plays a smaller but still significant part of the overall interaction with the value of CT reaching half the value of either ES or POL.
| Complex | 2a&Cl− | 2b&Cl− | 2d&Cl− |
|---|---|---|---|
| a Energy components: charge transfer (CT), electrostatic (ES), polarization (POL), exchange correlation (XC), deformation of the electron density (DEF), self-energy (SE), electrical (EL), core repulsions (core). | |||
| CT | −9.21 | −10.35 | −9.14 |
| ES | −18.23 | −21.25 | −16.62 |
| POL | −16.58 | −17.53 | −16.62 |
| XC | −4.00 | −4.27 | −3.91 |
| 2 DEF | 18.37 | 19.29 | 18.17 |
| 2 SE | 9.72 | 10.31 | 9.69 |
| Cl− DEF | 13.61 | 15.29 | 13.40 |
| Cl− SE | −1.66 | −1.79 | −1.61 |
| EL (ES + POL + SE) | −26.74 | −30.26 | −25.17 |
| Core (XC + DEF − SE) | 19.91 | 21.79 | 19.59 |
| Total (CT + EL + core) | −16.04 | −18.82 | −14.72 |
The NBO analysis (Table S2 in ESI†) also indicates that the major charge transfer component between the chloride ion and the neutral derivatives, 2a, 2b and 2d, arises from the interaction between the chloride filled orbitals and the σ* of the methine C–H bond, and, to a lesser extent, from interactions of the chloride orbitals with the σ* of the methine C–N bonds.
Notably, all three electrostatic components, ES, POL and CT are larger for sulfur (2b) than for oxygen (2a), reflecting the greater polarizability of sulfur. The largest difference is seen in the ES component, originating from the differences in the initial charges, as shown and discussed above (Table 2). Interestingly, the comparison between oxygen (2a) and nitrogen (2d) shows that all components are very similar except for the ES term, which is more negative for oxygen than for nitrogen.
This indicates that the main difference between them stems from the initial charge distribution before complexation, which reflects the higher electronegativity of oxygen relative to nitrogen whereas the polarization is similar.
Our next step was calculating the binding geometry of a chloride ion to tetramethylglycoluril, 4 (Fig. 4), which mimics the immediate environment of the glycoluril units within the bambusuril framework. With this model system the preferred position of the anion was found to be above the center of one of the five-membered rings with close contacts to two methine groups and two methyl groups, (Fig. 4, highlighted in blue, and Fig. S5, ESI†). Another binding mode, 2.3 kcal mol−1 higher in energy, shows the chloride anion binding to two methyl groups and only one methine hydrogen (Fig. S5, ESI†). Experimental verification of the above predictions was provided by the crystal structures of the triflate salts of 5b and 5c. (Table S1, ESI† and Fig. 5). As seen in Fig. 5, all oxygen atoms of the triflate anion maintain close contacts with four molecules of 5b or three molecules of 5c. Two molecules approach the anion their methine and methyl hydrogens whereas the others maintain close contacts with the anion by their guanidinium groups.
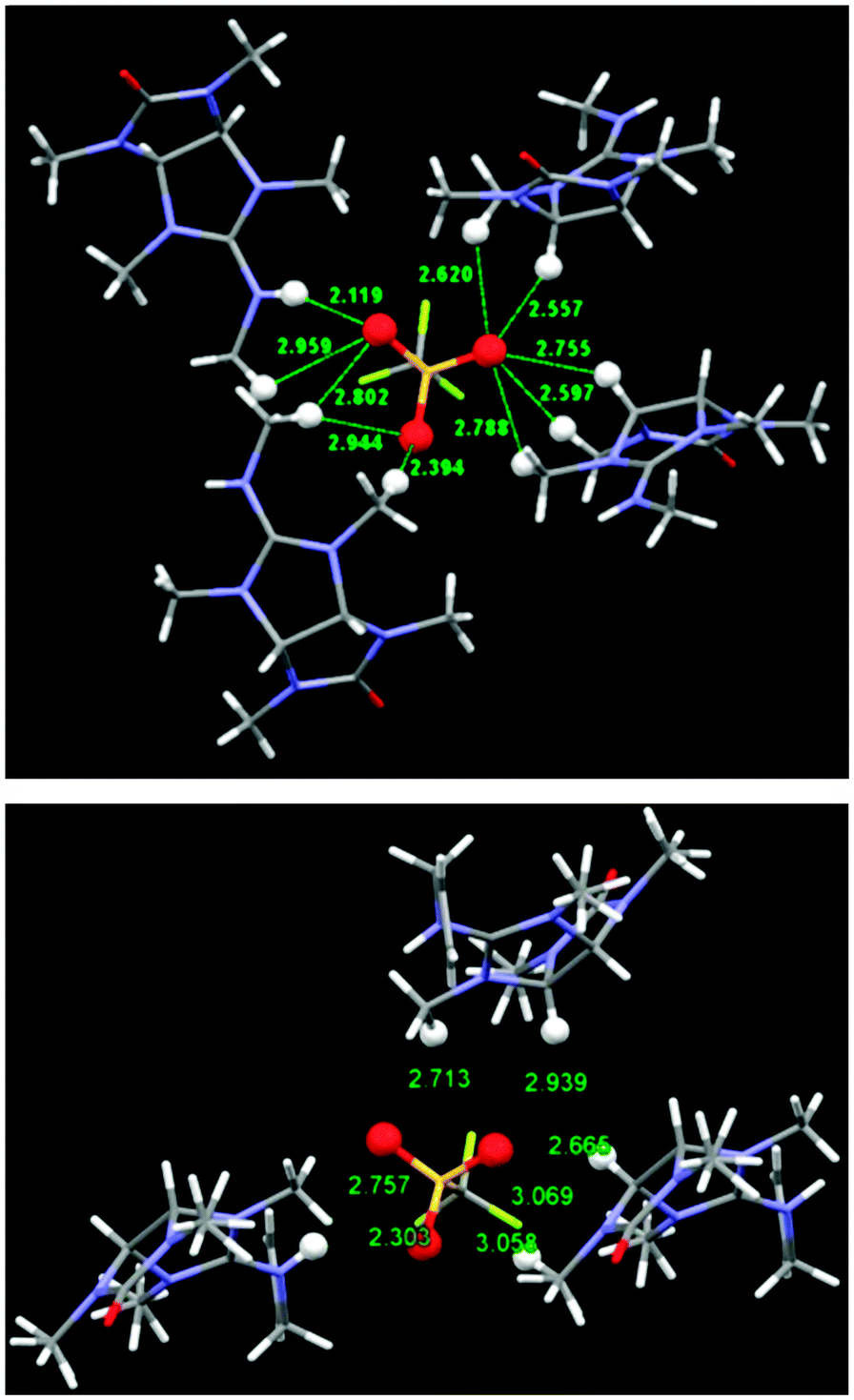 | ||
| Fig. 5 Solid state structures of the triflate salts of 5b (top) and 5c (bottom) showing the close contacts to the triflate oxygen atoms. All relevant atoms are highlighted by Ball & Stick model. | ||
Finally, encouraged by the experimental confirmation of our calculated interactions of the monomeric glycolurils we studied the structure of the hexameric macrocycles, 3, and their anion binding properties. Due to the relatively larger molecular dimensions we carried out these calculations at the M06-2X/6-31G** level. The energies, orbitals and electrostatic potential maps were all calculated at the M06-2X/6-311+G** level (Table 4). The results indicate that all neutral bambusurils, 3a, 3b and 3d, follow the same order of binding strength predicted for the monomeric glycolurils, namely X = S > O > NH. Furthermore, in agreement with the binding properties of 2e, the charged bambusuril, 3e, was also found to be the best anion binder. Likewise, this trend is reflected by the LUMO energies.
| X | Y | E (LUMO) | ΔE | |
|---|---|---|---|---|
| a Optimizations were carried out at the M06-2X/6-31G** level and the energies were calculated for these structures with the M06-2X/6-311+G** method. | ||||
| 3a | O | O | −33.27 | −81.65 |
| 3b | S | O | −41.01 | −86.10 |
| 3c | O | S | −20.62 | −64.27 |
| 3d | NH | O | −26.00 | −71.80 |
| 3e | NH2+ | O | −272.16 | −319.18 |
Since the two dominant contributors to anion binding with the small model molecules, 2, were found to be polarization (POL) and electrostatic interactions (ES), we examined these factors also with the complete macrocycles, 3, as well. The ES could be evaluated from the electrostatic potential values along the z-axis, which is the main symmetry axis of the macrocycle (Fig. 6). We also calculated the electrostatic potential maps of 3 (Fig. S6–S10, ESI†). As can be seen from Fig. 6, anions traversing through the cavity from one portal to the other would be attracted by a shallow double-well potential rather than by a deep, single-well potential. While the neutral molecules follow the above-described trend, X = S > O > NH, the charged derivative, 3e, exhibits a much stronger potential with continuous intensity along the cavity. This, rather unusual shape of electrostatic potential may allow for continuous flow of anions through the macrocycle cavity, suggesting that 3e could not only bind multiple anions along its main axis, but also function as a synthetic anion channel.
 | ||
| Fig. 6 The electrostatic potential energy (EPE) calculated along the z-axis in 3a–e. | ||
The polarization change upon binding to 3a, 3b and 3d can be assessed from the changes in NBO charges (Table S3, ESI†). The results suggest that the main electrostatic changes occur at the vicinity of the methine hydrogens and at the heteroatoms of the portals, but not at the equatorial heteroatoms. Thus, the increased ability of the portal heteroatoms to act as an electron sink is manifested by stronger polarization and stronger anion binding. The prediction that the remote carbonyls at the portals influence the binding properties more strongly than the equatorial carbonyls, which lie closer to the binding site, seems counterintuitive. The remarkably different contribution of either portal or equatorial carbonyls to binding is also supported by previous calculations, which predicted that the change in the C–O stretching vibration frequency upon anion binding to bambus[6]urils would be more pronounced for the portal carbonyls rather than the equatorial carbonyls.15
The dominant effect of the portal carbonyls also explains why 3b is a stronger anion binder than 3a, as predicted by calculations (Table 4) and demonstrated experimentally.21 In 3b both effects operate in the same direction: first, the dipole moment of thiourea is larger than that of urea,31 resulting in greater electrostatic interactions (ES), and second, sulfur is more polarizable than oxygen, resulting in a larger induced dipole (POL) and stronger interactions with this dipole.
Since the orientation of the induced dipole is crucial for binding, the above discussion leads to an interesting prediction that 3b would be a much better anion binder than its isomer, 3c. Although these two isomers differ only in the relative positioning of their sulfur and oxygen atoms, either at the portals or at the equator, their electrostatic potentials and binding energies are diametrically different. Thus, compound 3c has a lower electrostatic potential energy along the z-axis (Fig. 6) and lower binding energy than 3a, 3b and 3d (Table 4), indicating that replacing the portal heteroatoms with a stronger electron sink would enhance anion binding whereas analogous heteroatom replacements at the equator are counterproductive for anion binding.
Conclusions
In summary, this study was driven by the hypothesis that heteroatom replacement in bambusuril could significantly modify its anion binding properties. Indeed, calculations show that the energies of anion binding to either glycolurils, 2, or to bambusurils, 3, are strongly affected by the nature of the remote heteroatom X. Although these computations refer to the gas phase and the binding energies would be different in solution, they reveal trends of a predictive value. The most significant contributor to the general trend of increased anion binding among the neutral molecules: X = S > O > NH, was found to be the electrostatic interactions (ES). The results also suggest that the main electrostatic changes upon chloride binding occur at the vicinity of the methine hydrogens and at the heteroatoms of the portals, but not at the equatorial heteroatoms. Thus, the increased capability of the portal carbonyls/heterocarbonyls to act as an electron sink is manifested by stronger dipole moments, polarizations and stronger anion binding.Acknowledgements
Prof. Ehud Keinan is the incumbent of the Benno Gitter & Ilana Ben-Ami Chair of Biotechnology, Technion. We acknowledge the financial support by the Ministry of Science, Technology and Space (MOST grant No. 3-10855), the Open University of Israel (OUI), the High Level University Project: Chemistry & Materials in Shantou University, Guangdong Province, PR China and Israel Council for Higher Education (PBC Program for international postdoctoral fellowship).Notes and references
- M. J. González-Álvarez, T. Carmona, D. Evren and F. Mendicuti, Supramol. Chem., 2014, 26, 414 CrossRef.
- S. J. C. Lee, J. W. Lee, H. H. Lee, J. Seo, D. H. Noh, Y. H. Ko, K. Kim and H. I. Kim, J. Phys. Chem. B, 2013, 117, 8855 CrossRef CAS PubMed.
- X.-L. Ni, X. Xiao, H. Cong, L.-L. Liang, K. Cheng, X.-J. Cheng, N.-N. Ji, Q.-J. Zhu, S.-F. Xue and Z. Tao, Chem. Soc. Rev., 2013, 42, 9480 RSC.
- G. Parvari, O. Reany and E. Keinan, Isr. J. Chem., 2011, 51, 646 CrossRef CAS.
- J. Svec, M. Necas and V. Sindelar, Angew. Chem., Int. Ed., 2010, 49, 2378 CrossRef CAS PubMed.
- V. Havel, J. Svec, M. Wimmerova, M. Dusek, M. Pojarova and V. Sindelar, Org. Lett., 2011, 13, 4000 CrossRef CAS PubMed.
- J. Rivollier, P. Thuery and M.-P. Heck, Org. Lett., 2013, 15, 480 CrossRef CAS PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabartiand and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844 CrossRef CAS PubMed.
- W. M. Nau, M. Florea and K. I. Assaf, Isr. J. Chem., 2011, 51, 559 CrossRef CAS.
- M. A. Yawer, V. Havel and V. Sindelar, Angew. Chem., Int. Ed., 2015, 54, 276 CrossRef CAS PubMed.
- V. Havel, M. A. Yawer and V. Sindelar, Chem. Commun., 2015, 51, 4666 RSC.
- P. A. Denis and J. S. Gancheff, Comput. Theor. Chem., 2013, 1023, 5 CrossRef CAS.
- P. Toman, E. Makrlik and P. Vanura, Chem. Phys. Lett., 2012, 547, 63 CrossRef CAS.
- P. Toman, E. Makrlik and P. Vanura, Monatsh. Chem., 2011, 142, 881 CrossRef CAS.
- V. V. Gobre, P. H. Dixit, J. K. Khedkar and S. P. Gejji, Comput. Theor. Chem., 2011, 976, 76 CrossRef CAS.
- V. Havel, V. Sindelar, M. Necas and A. E. Kaifer, Chem. Commun., 2014, 50, 1372 RSC.
- F. Pichierri, Chem. Phys. Lett., 2005, 403, 252 CrossRef CAS.
- F. Pichierri, Chem. Phys. Lett., 2004, 390, 214 CrossRef CAS.
- W. L. Mock, T. Manimaran, W. A. Freeman, R. M. Kuksuk, J. E. Maggio and D. H. Williams, J. Org. Chem., 1985, 50, 60 CrossRef CAS.
- M. Singh, G. Parvari, M. Botoshansky, E. Keinan and O. Reany, Eur. J. Org. Chem., 2014, 933 CrossRef CAS.
- M. Singh, E. Solel, E. Keinan and O. Reany, Chem. – Eur. J., 2015, 21, 536 CrossRef CAS PubMed.
- J. Svec, M. Dusek, K. Fejfarova, P. Stacko, P. Klan, A. E. Kaifer, W. Li, E. Hudeckova and V. Sindelar, Chem. – Eur. J., 2011, 17, 5605 CrossRef CAS PubMed.
- S. J. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Am. Chem. Soc., 2007, 129, 8440 CrossRef CAS PubMed.
- M.-C. Ou, M.-S. Tsai and S.-Y. Chu, J. Mol. Struct., 1994, 310, 247 CrossRef.
- K. B. Wiberg and P. R. Rablen, J. Am. Chem. Soc., 1995, 117, 2201 CrossRef CAS.
- M. K. Kesharwani and B. Gaguly, J. Comput. Chem., 2011, 32, 2170 CrossRef CAS PubMed.
- E. D. Glendening, J. Am. Chem. Soc., 1996, 118, 2473 CrossRef CAS.
- E. D. Glendening, J. Phys. Chem. A, 2005, 109, 11936 CrossRef CAS PubMed.
- W. D. Kumler and G. M. Fohlen, J. Am. Chem. Soc., 1942, 64, 1944 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, computed charges, electrostatic potential surfaces and complex structures. CCDC 1064130 and 1058391. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cp00442c |
| This journal is © the Owner Societies 2016 |