
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigations on HONO formation from photolysis of adsorbed HNO3 on quartz glass surfaces
Sebastian
Laufs
and
Jörg
Kleffmann
*
Physikalische und Theoretische Chemie/Fakultät für Mathematik und Naturwissenschaften, Bergische Universität Wuppertal, 42097 Wuppertal, Germany. E-mail: kleffman@uni-wuppertal.de; Fax: +49 202 439 2505; Tel: +49 202 439 3534
First published on 9th March 2016
Abstract
During the last few decades, nitrous acid (HONO) has attracted significant attention as a major source of the OH radical, the detergent of the atmosphere. However, the different daytime sources identified in the laboratory are still the subject of controversial discussion. In the present study, one of these postulated HONO sources, the heterogeneous photolysis of nitric acid (HNO3), was studied on quartz glass surfaces in a photo flow-reactor under atmospherically relevant conditions. In contrast to other investigations, a very low HNO3 photolysis frequency for HONO formation of J(HNO3 → HONO) = 2.4 × 10−7 s−1 (0° SZA, 50% r.h.) was determined. If these results can be translated to atmospheric surfaces, HNO3 photolysis cannot explain the significant HONO levels in the daytime atmosphere. In addition, it is demonstrated that even the small measured yields of HONO did not result from the direct photolysis of HNO3 but rather from the consecutive heterogeneous conversion of the primary photolysis product NO2 on the humid surfaces. The secondary NO2 conversion was not photo-enhanced on pure quartz glass surfaces in good agreement with former studies. A photolysis frequency for the primary reaction product NO2 of J(HNO3 → NO2) = 1.1 × 10−6 s−1 has been calculated (0° SZA, 50% r.h.), which indicates that renoxification by photolysis of adsorbed HNO3 on non-reactive surfaces is also a minor process in the atmosphere.
1. Introduction
During the last two decades, unexpectedly high nitrous acid (HONO) mixing ratios have been observed during daytime in remote,1–5 semi urban6–9 and urban regions.10–17 These results stimulated laboratory investigations on potential daytime HONO sources, by (i) the reduction of nitrogen dioxide (NO2) in the presence of organic photosensitisers,18–21 (ii) the photolysis of adsorbed nitric acid22–26 and (iii) bacterial production of nitrite in soil27–30 and/or desorption of adsorbed HONO from soil surfaces during daytime.31–33 Another intensely discussed source, the gas-phase reaction of excited NO2 with water,34 was found to be of minor importance as demonstrated by laboratory studies.35,36 Also the photolysis of nitro-phenols or similar compounds is too slow to explain significant HONO levels in the daytime atmosphere.37 Finally, a gas-phase HONO source recently postulated by Li et al.,38i.e. the reaction of HO2·H2O complexes with NO2, could not be confirmed by the same group in simulation chamber experiments39 and is also in conflict with recent aircraft measurements.40In summary, mainly ground surface sources have been identified to date in laboratory studies to explain atmospheric HONO formation during daytime,41 which has also been confirmed by recent flux measurements in the atmosphere.25,42,43
Among the different proposed sources, the photolysis of adsorbed nitric acid (HNO3) has been suggested as an important HONO formation pathway, especially in rural areas.
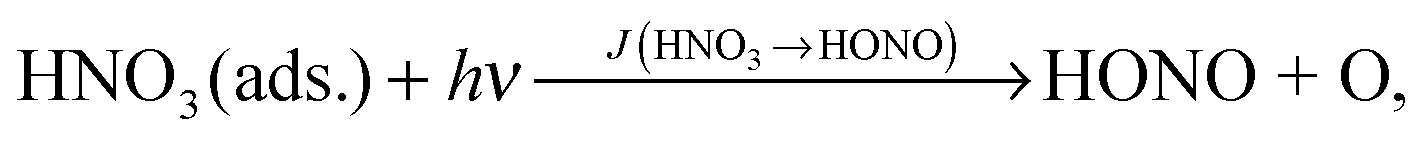 | (R1) |
To elucidate the importance of reaction (R1) as a HONO source in the atmosphere, photolysis of adsorbed HNO3 was studied in a photo flow-reactor under atmospherically relevant concentrations, humidity and actinic flux conditions.
2. Experimental
2.1. Experimental setup
Experiments were performed in a cylindrical flow-reactor (5.0 cm i.d.) made of smooth quartz glass with an illuminated length of 56.5 cm (see Fig. 1). For a few experiments a similar flow reactor made of Pyrex glass was used. The reactor was irradiated with 6 UV-Vis lamps (Philips TL/05 40 Watt, 300–500 nm, λmax ≈ 370 nm; or Wolff System Helarium 40 W, 290–450 nm, λmax ≈ 350 nm), which were placed in an air-cooled (fan), UV-reflecting aluminium box. The actinic flux inside the photoreactor (see Fig. 2) was measured using a spectro-radiometer (Δλ = 1 nm; Metcon Inc., USA). Calculated photolysis frequencies J(NO2) and J(HCHO) using published cross-sections and quantum yields49–52 were compared with values directly measured by chemical actinometry of NO2 and HCHO in the flow-reactor and showed good agreement (see Table 1).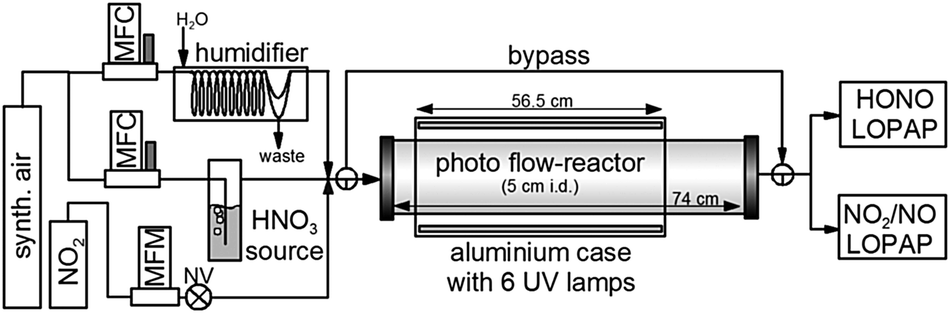 | ||
| Fig. 1 Experimental setup (MFC: mass flow controller, MFM: mass flow meter, NV: needle valve). | ||
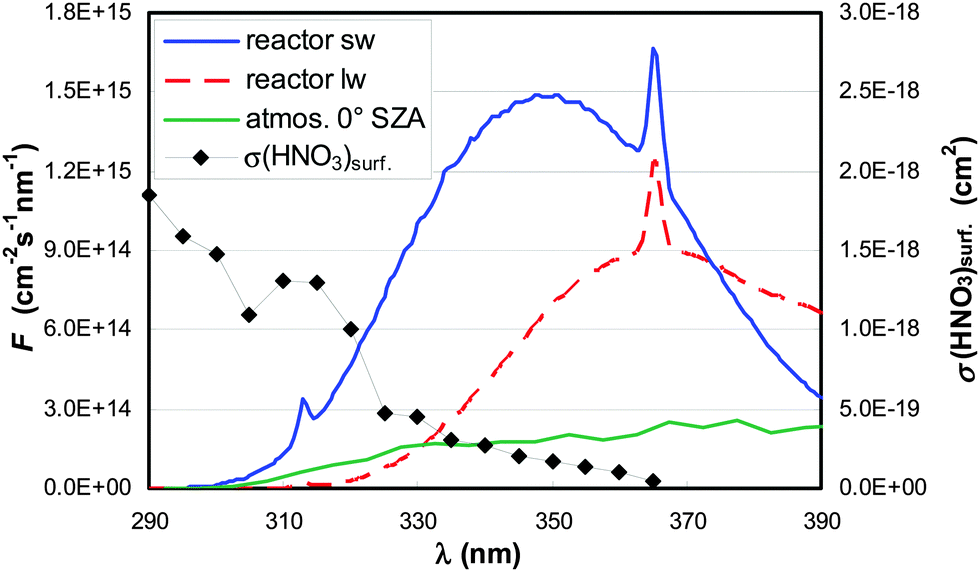 | ||
| Fig. 2 Actinic flux (F) spectra of the short-(sw) and long-(lw) wavelength lamps and from the atmosphere (0° SZA).50 The absorption spectrum σ of HNO3 adsorbed on fused silica is also shown.45,47 | ||
| lw: Philips, TL05, 40 W | sw: Wolff System, Helarium, 40 W | Atmosphere (0° SZA) | |
|---|---|---|---|
| J(NO2) (s−1) | 0.027 (0.029) | 0.036 | 0.010 |
| J(HONO) (s−1) | 5.1 × 10−3 | 8.7 × 10−3 | 2.2 × 10−3 |
| J(CH2O) (s−1) | 8.4 × 10−5 | 4.8 × 10−4 (4.2 × 10−4) | 8.9 × 10−5 |
Corrections of the observed HONO formation for its gas-phase photolysis were typically only ca. 5 and 8% for the two types of lamps used, respectively. Photolysis frequencies of HONO (J(HONO), see Table 1) were calculated using published cross-sections.53
2.2. Instruments
HONO was measured using the sensitive LOPAP technique10,55 with a detection limit and an accuracy of 2 pptV and 10%, respectively. NO2 and NO were detected by a recently developed NO2-LOPAP56 which was modified for the additional detection of NO, details of which will be published elsewhere. In short, NO passing the NO2 collection stripping coil was oxidized to NO2 by an acidic KMnO4 solution in a consecutive stripping coil and was detected downstream using the Griess–Saltzman reaction, similar to the NO2 channel of the instrument. Detection limits and accuracies for NO2/NO under the experimental conditions applied were 10/20 pptV and 7/10%, respectively. For the experimental determination of the photolysis frequency of HCHO an Aero Laser AL4001 Hantzsch instrument was used.57 Finally, adsorbed HNO3 was quantified by ion-chromatography (IC)58 after elution from the flow-reactor surface with ultra-pure water (MilliPore). IC detection was also used to measure gas-phase levels of HNO3 after bubbling through ultrapure water. The accuracy of the nitrate concentration determined by IC was 7% and those of HNO3 in the gas phase and on the surface were 10 and 12%, respectively.2.3. Experimental procedure
Prior to the experiments the reactor was cleaned with HF (5%) followed by ultra-pure water to minimize the contamination on the irradiated reactor surfaces. This was confirmed in blank experiments in which neither in the dark nor under irradiation any significant formation of HONO or NOx was observed. Different reactant mixtures were generated by mixing humidified synthetic air with a pure HNO3 gas-phase source58 and were flushed through the flow-reactor. For some experiments low levels of NO2 (Messer, 2 ppmV) were also added using a calibrated 10 mL min−1 flow meter and a needle valve. During one experiment a mixture of 29 VOCs (methane, ethane, propane, n-butane, iso-butane, n-pentane, n-hexane, n-heptane, n-octane, ethene, propene, iso-, trans-, and cis-butene, isoprene, β-pinene, limonene, ethine, benzene, toluene, o-, m-, and p-xylene, methanol, ethanol, n-propanol, n-butanol, acetone, and methacrolein) with individual mixing ratios in the range 48–69 ppbV was added using a 100 mL min−1 flow controller. In some experiments additional quartz glass plates were added to the flow-reactor to increase the irradiated surface area. The total gas flow rate was typically 2.7 L min−1, but was varied between 1.1 and 3.4 L min−1 in a few experiments leading to reaction times in the range 26–80 s.When constant reactant concentrations were reached, the lamps were switched on and the system was analysed for photochemical HONO and NOx formation. Immediately after each experiment and after disconnection from the HNO3 source, the reactor was eluted with ultra-pure water and the amount of adsorbed HNO3 was quantified. After saturation of the reactor surfaces with the continuously running HNO3 source (typically overnight), adsorbed HNO3 levels were assumed to be constant during each experiment. This assumption is considered plausible as the HNO3 source produced fairly stable gas-phase levels, as demonstrated also in a former laboratory study.58 In addition, constant gas phase HNO3 levels at the end of the flow tube confirmed equilibrium adsorption during the experiments. This observation also indicates that the surface concentration of HNO3 was homogeneously distributed on all the irradiated surfaces. For the calculation of surface concentrations and flux densities the geometric surface area of the flow tube was used.
3. Results and discussion
In Fig. 3, a typical experiment is shown in which adsorbed HNO3 was irradiated without (first and last step in Fig. 3) and with different levels of added NO2. In the absence of additional NO2, clear positive steps in the HONO, NO and NO2 concentrations were observed, pointing to their photochemical production.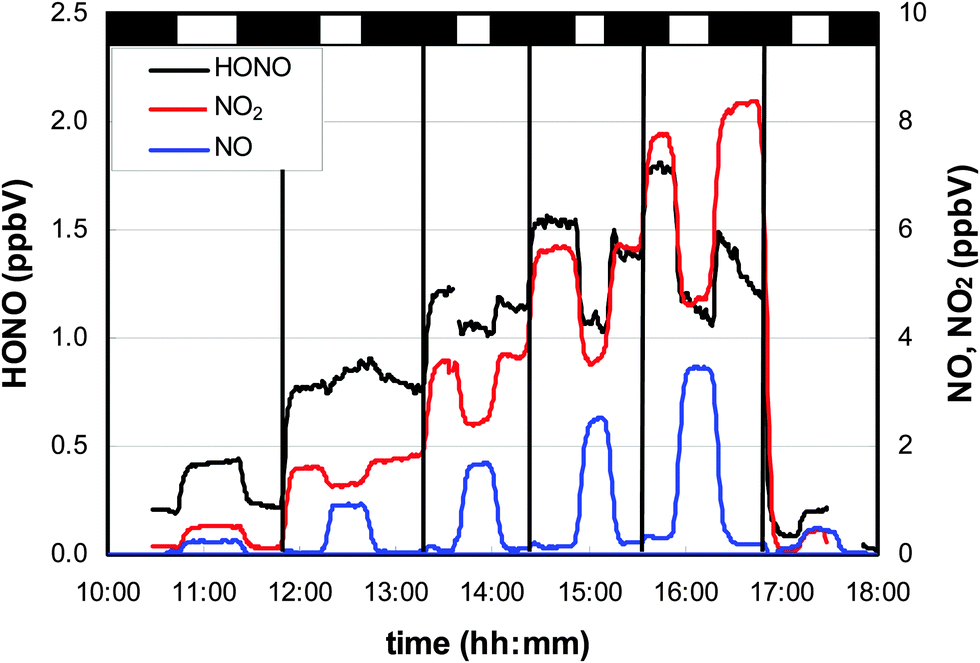 | ||
| Fig. 3 HONO and NOx formation from photolysis of adsorbed HNO3 (7.8 × 1013 cm−2) in the flow-reactor continuously flushed with humid (54% r.h.) gas-phase HNO3. After first irradiation, NO2 was added stepwise to the HNO3 mixture (marked with vertical black lines). For the last step, the NO2 addition was set to zero again. The vertical bars above the plot represent dark (black) and irradiated (white) periods. | ||
3.1. Surface dependency
To verify whether HONO formation was caused by the photolysis of adsorbed HNO3, reaction (R1), and not by its gas-phase photolysis, the surface area of the reactor was varied. For these experiments, additional quartz glass plates of different sizes were added to the flow-reactor. The HONO production during irradiation increased linearly with the surface area (Fig. 4), which demonstrates the heterogeneous nature of the process involved.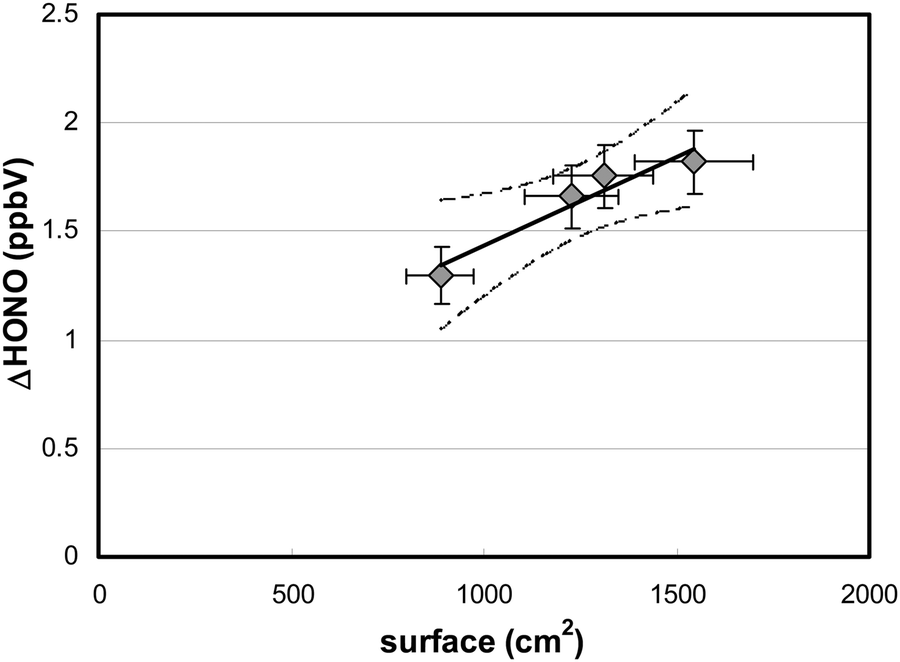 | ||
| Fig. 4 HONO formation in the photolysis of adsorbed HNO3 as a function of the reactor surface area ([HNO3]surf = 1.6 × 1014 cm−2, [HNO3]g = 86 ppbV, 59% r.h.). The straight line represents the linear regression of the data (r2: 0.93) with its 95% confidence limits (dashed lines). | ||
This result is in agreement with theoretical calculations performed using known gas-phase absorption cross-sections of HNO3,51 measured actinic flux, HNO3 gas-phase concentration, residence time in the flow-reactor and assuming an upper limit quantum yield for HONO formation of one (for calculations, see eqn (3)). The calculated maximum HONO formation by HNO3 photolysis in the gas phase of only 6 pptV is much smaller compared to the experimental results shown in Fig. 4 and thus, a heterogeneous reaction is proposed here.
3.2. HNO3 dependency
By varying the gas-phase concentration of HNO3 in the range 2–120 ppbV, different quantities of surface adsorbed HNO3 in the range 0.15–3.3 × 1014 molecules cm−2 were investigated. Recently, a surface monolayer concentration of HNO3 of 1.1 × 1014 molecules cm−2 was estimated based on its van der Waals radius,45 which is in fair agreement with the measured value for SiO2 particles (7 × 1013 molecules cm−2).59 Thus, the investigated surface levels varied from sub-monolayer to multilayer coverage, which were obtained at atmospherically relevant low ppbV mixing ratios of the sticky HNO3.Photochemical HONO formation was linearly correlated with the concentration of adsorbed HNO3, both, for the long and short wavelength lamps used (see Fig. 5). This observation is in line with a first-order surface photolysis process, reaction (R1), described by a photolysis frequency of adsorbed HNO3 for HONO formation, J(HNO3 → HONO), which is defined by:
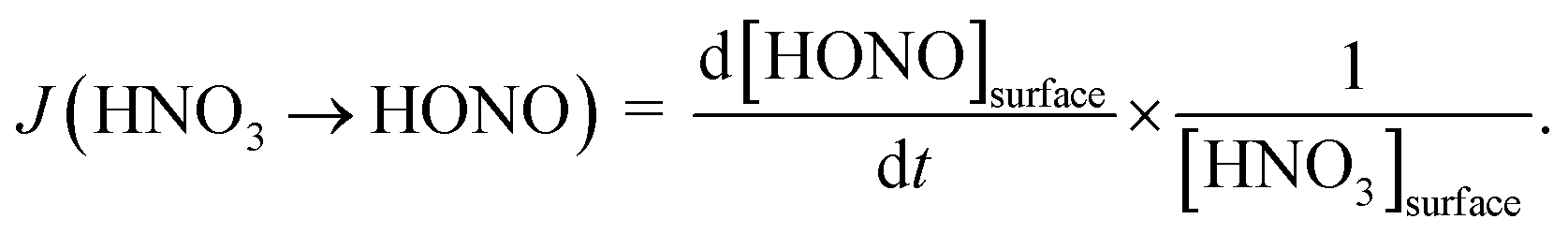 | (1) |
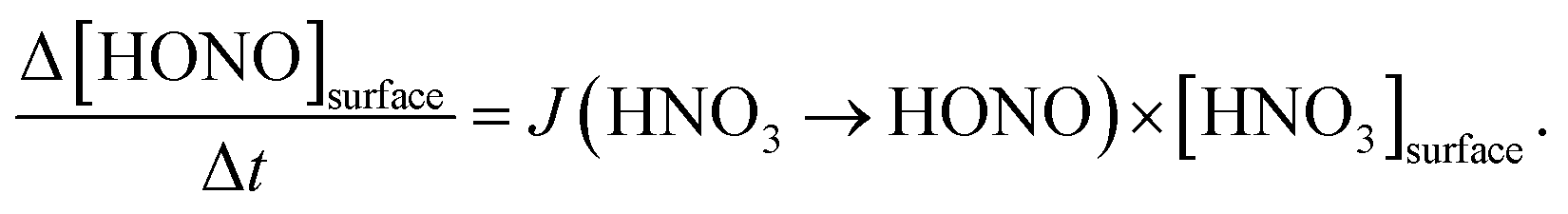 | (2) |
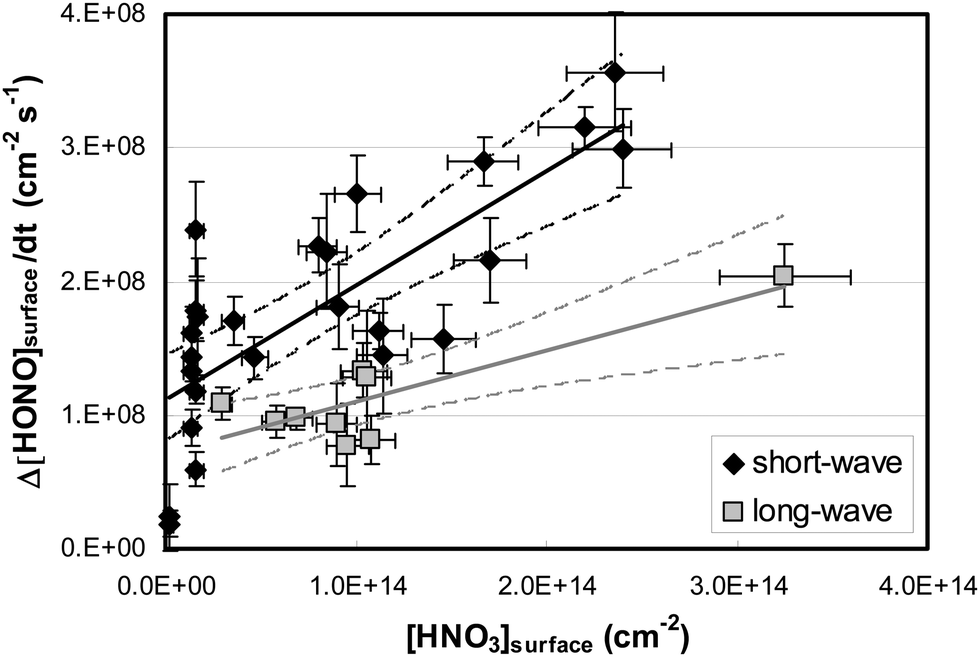 | ||
| Fig. 5 HONO flux density as a function of the measured surface concentration of HNO3 using two different sets of UV lamps (∼42–57% r.h.). The straight lines represent linear regressions of the data with their 95% confidence limits (dashed lines). | ||
3.3. Wavelength dependency
To estimate the atmospheric implication of the HNO3 photolysis, the results obtained in laboratory experiments cannot be used directly, since the actinic flux spectra of laboratory systems are typically very different from those of the atmosphere (cf.Fig. 2). To calculate atmospherically relevant HNO3 photolysis frequencies for HONO formation according to: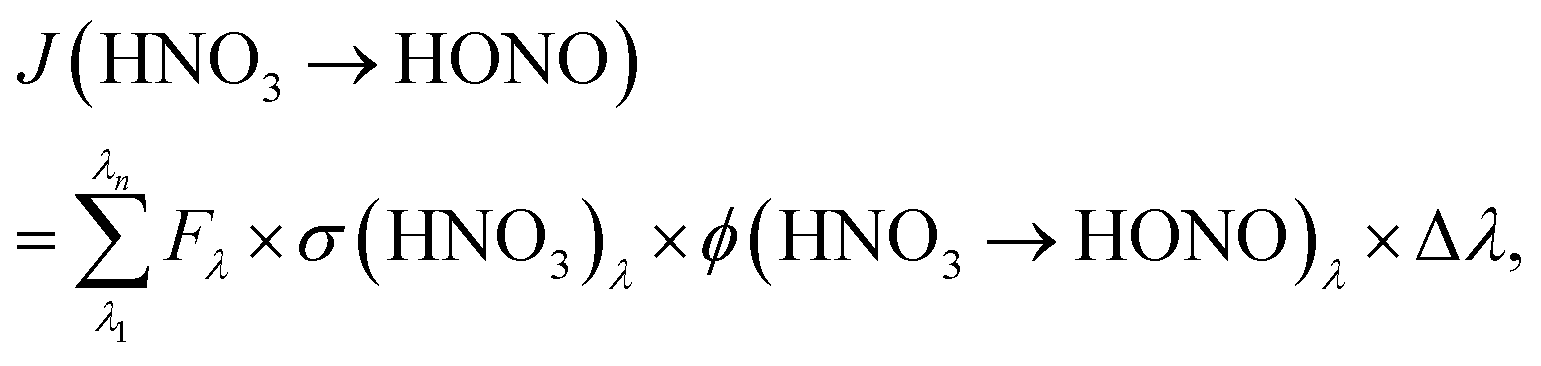 | (3) |
3.4. Influence of relative humidity
Similar to the study by Zhou et al.,24 a strong influence of the relative humidity on the HONO formation inside the flow-reactor was observed. HONO immediately decreased to very low levels after stopping the humidifier, indicating a direct influence of water on the HONO production. There are different potential explanations for this observation: (i) photochemical HONO formation is active only for H2O/HNO3 clusters26 and/or (ii) is caused by secondary chemistry via the additional reaction product NO2, for which strong humidity dependence of the heterogeneous HONO formation is known.613.5. NO2 dependency
To distinguish between these two explanations, the NO2 dependence of the HONO formation was studied. Besides HONO, NO2 is a main product of the HNO3 photolysis (see Fig. 3):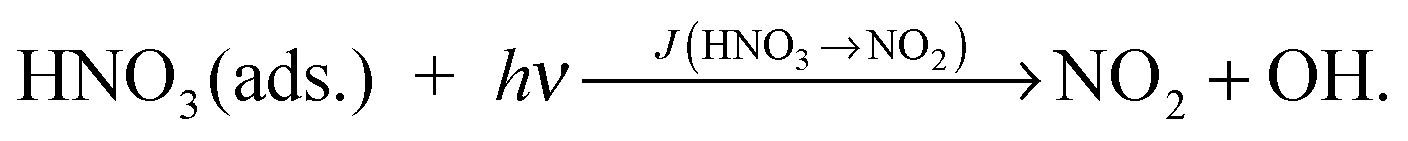 | (R2) |
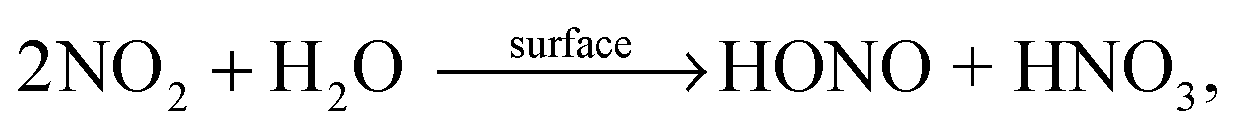 | (R3) |
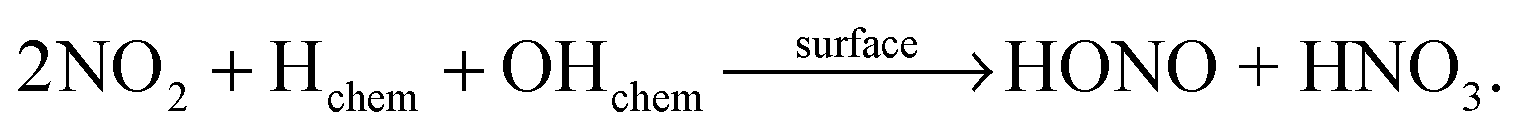 | (R4) |
 | (R5) |
To elucidate the contribution of NO2 to the observed HONO formation, NO2 was added to the reaction mixtures (see Fig. 3). From this and other similar experiments total HONO production rates were calculated from the concentration differences between the flow-reactor bypass and the dark or light steps, respectively, and plotted against the final NOx level at the flow-reactor exit (see Fig. 6). NOx was used here instead of the proposed precursor NO2 to take into account the significant NO2 photolysis in the flow-reactor (see Table 1). Thus, NO measured at the exit of the reactor mainly originated from the fast gas-phase photolysis of NO2:
| NO2 + hν → NO + O(3P), | (R6) |
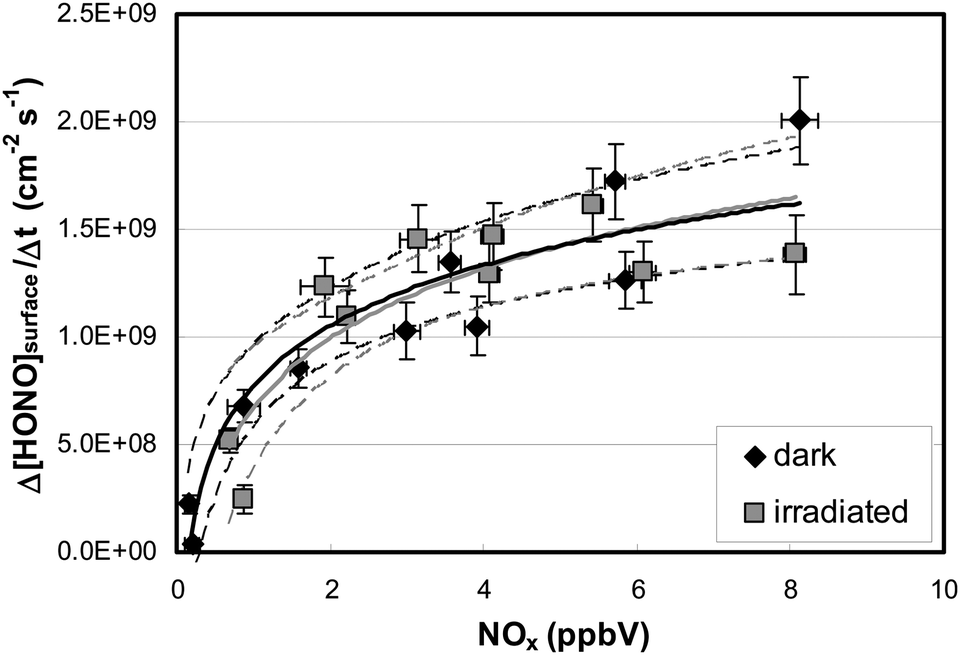 | ||
| Fig. 6 HONO flux as a function of the NOx concentration calculated from the differences (dark-bypass) and (illuminated-bypass), ([HNO3]surf ≤ 0.8 × 1014 cm−2, 51–54% r.h.). Within the experimental errors (95% confidence intervals, dashed lines), logarithmic fits (solid lines) show no difference between HONO formation in the dark (black) and under illumination (grey). | ||
Two important conclusions can be drawn from the experimental data:
(a) HONO formation depends strongly on the NO2 concentration and within the error of the intercept (±2 × 108 molecules cm−2 s−1) extrapolation to 0 ppbV NOx indicates no additional HONO formation by HNO3 photolysis alone (R1) under the experimental conditions applied. Thus, it is proposed that the observed HONO formation is caused by the heterogeneous conversion of NO2 on the reactor surfaces, reactions (R3) or (R4).
(b) HONO formation by heterogeneous NO2 conversion in the dark and under irradiation is similar, confirming a proposed dark formation of HONO, e.g. by reaction (R3) on clean quartz glass surfaces. This result is in excellent agreement with other studies,26,44 in which no photoenhancement of reaction (R3) was observed. Thus, the experimental results of the present study do not confirm photochemical HONO formation via excited NO2, reaction (R5), as proposed in another study by Abida et al.48 when using excimer laser photolysis. Very recently, the same group64 also excluded the direct formation of excited NO2 at least for the gas-phase photolysis of HNO3 and proposed secondary HONO formation by the heterogeneous reaction (R3). This conclusion is in agreement with studies of reaction (R5) in the gas phase,35,36 in which HONO formation via excited NO2 was excluded at atmospherically relevant actinic fluxes. Finally, the missing light-enhancement also excludes photolysis of complexes of HNO3 with nitrogen oxides as sources of HONO.63
Based on these two conclusions a two-step formation mechanism of HONO is proposed here. First, NO2 is formed in the photolysis of adsorbed HNO3, reaction (R2), at longer wavelengths compared to the gas and liquid phases. Second, NO2 is consecutively converted into HONO via the heterogeneous dark reactions (R3) or (R4) on the pure quartz glass surfaces. This consecutive formation of HONO via the intermediate NO2 was already proposed for the photolysis of nitrate in snow22,65 and in the aqueous phase.66 However, for snow the significant red-shift of the nitrate absorption compared to the liquid nitrate spectra was not observed leading to negligible NOx formation at wavelengths >345 nm.67 Finally, the observed NO formation (see Fig. 3) is explained by the gas-phase photolysis of NO2, reaction (R6).
To determine HNO3 photolysis frequencies for the primary reaction product NO2:
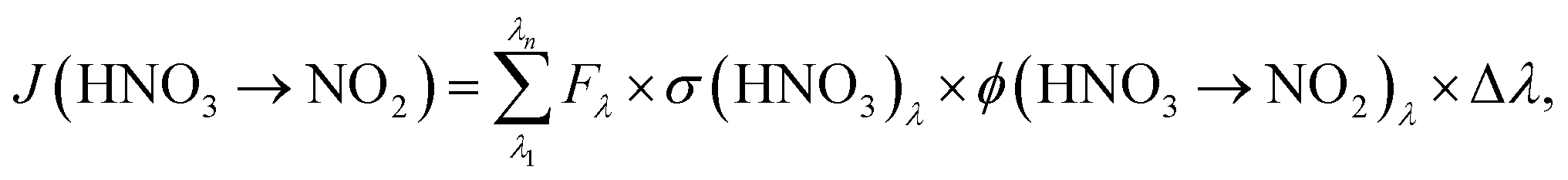 | (4) |
Using the absorption cross-sections of adsorbed HNO3 from the studies of Zhu et al.45 and Du and Zhu,47 measured actinic fluxes of both setups and measured values of J(HNO3 → NO2) from the present study, similar quantum yields for NO2 formation, ϕ(HNO3 → NO2), of (3.6 ± 0.8) × 10−4 and (3.2 ± 2.5) × 10−4 were determined for the short and the long wavelength setups, respectively. The averaged quantum yield for both setups of (3.4 ± 2.6) × 10−4 is orders of magnitude lower compared to the studies of Zhu et al.46 and Abida et al.,48 in which quantum yields near unity were observed in excimer laser photolysis experiments at 308 and 351 nm. Reasons for the strong discrepancy are still unclear and may be explained by:
(a) Overestimation of the HNO3 absorption cross-sections in the studies of Zhu et al.45 and Du and Zhu.47 In these studies, the concentrations of adsorbed HNO3 were only indirectly determined by adsorption isotherms based on measured light absorption and by using an estimated van der Waals radius for HNO3 resulting in a monolayer coverage only at very high gas-phase levels (15–20 ppmV) of the highly sticky HNO3. In contrast, in the present study three orders of magnitude lower gas-phase concentrations (∼10 ppbV) were necessary to obtain similar levels of adsorbed HNO3, which were directly measured by ion chromatography. If surface levels of HNO3 were much higher than proposed in the studies of Zhu et al.45 and Du and Zhu,47 absorption cross-sections may be overestimated, which was also recently proposed by Tadić;68
(b) Overestimation of the quantum yields ϕ(HNO3 → NO2) in the studies of Zhu et al.46 and Abida et al.48 caused by higher photon flux densities of the excimer laser used for the photolysis experiments compared to atmospheric conditions. High photon flux densities may have caused stronger NO2 formation, e.g. by a potential multi-photon excitation, which was also recently observed for the gas-phase reaction of NO2* + H2O.36
Independent of which explanation is correct, photolysis of pure HNO3 will be of much lower importance for renoxification than previously assumed, since the presented NO2 flux densities were determined under realistic atmospheric conditions (HNO3 level, humidity, actinic flux). A strong indication for an overestimation of either the absorption cross-sections or the quantum yield of adsorbed HNO3 in the former studies45–47 is the resulting unrealistic short photolytic lifetime of adsorbed HNO3 of only ∼5 min (see Section 4). If published absorption cross-sections are overestimated, our calculated quantum yields, which are based on these values, will be strongly underestimated.
3.6. Influence of VOCs
To explain enhanced HONO formation, photosensitized conversion of HNO3/nitrate by adsorbed organic compounds was recently proposed.69–72 In addition, increased HONO formation by scavenging of OH radicals formed viareaction (R2) by organic compounds was proposed for the liquid phase photolysis of nitrate.66 To test the influence of organic compounds on the reaction system, in one experiment a GC calibration mixture of 29 VOCs (≤C10, see Section 2.3. for details; each with a final mixing ratio of 3–4 ppbV) was continuously added to the humidified HNO3 mixture, both, during surface saturation overnight and during the photolysis. While some of the used VOCs clearly do not adsorb onto surfaces (e.g. C1–C4), others are expected to be surface active (e.g. β-pinene, limonene, xylenes, n-butanol, methacrolein) and substituted aromatic compounds are even proposed to participate in redox reactions of NOy.18 However, within the experimental errors photochemical HONO formation was not enhanced compared to the absence of the VOCs. A similar result was obtained when the flow tube was not cleaned after flushing with laboratory air for one day before the photolysis experiment. Thus, at least for the smaller VOCs and at atmospherically relevant concentrations, HONO formation by photosensitized conversion of HNO3 could not be confirmed. However, further experiments using more complex VOCs, known as photosensitizers (e.g. aromatic carbonyls, hydroxy-PAHs, humic acid etc.), are recommended in the future.4. Atmospheric implications
Based on the experimental results HONO formation from the photolysis of adsorbed nitric acid was estimated under typical atmospheric conditions. The present study indicates no direct photochemical HONO formation viareaction (R1), but secondary formation by the heterogeneous dark conversion of NO2 on humid surfaces, reaction (R3) or (R4). At high NOx levels, reaction (R3) can be a significant night-time ground surface source of HONO in the atmosphere,12,61 but is too slow to explain daytime HONO levels.41 Here, other NO2 reactions, e.g. the photosensitized conversion on humic acid surfaces have been proposed.19,20Although HONO is not the primary HNO3 photolysis product, reaction (R1), the formal quantum yield for HONO formation of ϕ(HNO3 → HONO) = (7.6 ± 1.5) × 10−5, as determined in Section 3.3., was considered here. Using this wavelength independent quantum yield, published absorption cross-sections of adsorbed HNO345,47 and the upper limit actinic flux in the atmosphere at 0° SZA50 a formal photolysis frequency J(HNO3 → HONO) of only 2.4 × 10−7 s−1 was derived using eqn (3). This value is almost two orders of magnitude lower compared to another laboratory study by Zhou and coworkers,24 in which atmospheric HONO formation in a rural environment could be well explained by reaction (R1). In contrast, using the results of the present study and assuming a high surface coverage of HNO3 on atmospheric surfaces of 1014 cm−2 (∼one monolayer45), an upper limit HONO flux density of only 2.4 × 107 cm−2 s−1 can be calculated using eqn (1) at 0° SZA. This upper limit is 2–3 orders of magnitude lower compared to HONO flux densities typically observed over irradiated surfaces in the atmosphere.25,42,43 Thus, HONO formation by photolysis of pure HNO3 is too slow to explain field observations, even in rural environments. Reasons for the strong discrepancy between the present study and the one by Zhou et al.24 are yet unclear but may possibly be explained by:
(a) the indirect extrapolation of the laboratory results to the atmosphere via the chemical actinometry used by Zhou et al.,24 by which OH radicals formed in the liquid phase photolysis of nitrate were used to quantify J(HNO3) for the red-shifted (compared to the liquid phase) photolysis of adsorbed HNO3.45,47 In contrast, in the present study a well calibrated spectro-radiometer was used to quantify actinic fluxes and to calculate J(HNO3) using eqn (3). Here, the wavelength dependent absorption cross-sections of adsorbed HNO345,47 were considered;
(b) different surface properties of the reactors and the different NO2 levels used. While in the present experiments, the flow-reactor was chemically cleaned by HF prior to each experiment, impurities on irradiated surfaces and higher NO2 levels in the experiments by Zhou et al.24 may have caused increased HONO yields viareaction (R3) and (R4), or by photosensitized conversion of NO2 on adsorbed organic impurities.18–21 Alternatively, direct photosensitized conversion of HNO3/nitrate by organic impurities69–72 may also be another explanation for the higher HONO yields observed by Zhou et al.24
However, in the present study the latter mechanism could not be confirmed at least for a mixture of 29 smaller VOCs (≤C10) or when the reactor was not cleaned after flushing with laboratory air for one day before the experiment. Thus, in the future, HONO formation from the photolysis of adsorbed HNO3 should be studied on more realistic organic surfaces, for example, on leaves, urban grime or humic acid. However, for the photolysis of pure HNO3 adsorbed on non-reactive surfaces, HONO formation is of minor importance.
Similar conclusions can be drawn for a possible renoxification by photolysis of pure adsorbed HNO3 in the absence of photosensitizers. Extrapolation of the experimental results from the present study to atmospheric conditions (see above) yields an upper photolysis frequency limit of J(HNO3 → NO2) = 1.1 × 10−6 s−1 (0° SZA). This value translates into photolytic lifetimes of HNO3 on surfaces of ∼30 days when the diurnal variability of the actinic flux in the atmosphere is considered. This contrasts starkly with an atmospheric photolytic lifetime of adsorbed HNO3 of only ∼5 min (0° SZA), which is derived when published cross-sections and quantum yields are used.48 Such a short lifetime is highly unreasonable and would result in negligible amounts of nitrates on atmospheric surfaces during daytime, which is not in agreement with direct field observations.25
Summary
In the present study heterogeneous formation of nitrous acid (HONO) from photolysis of adsorbed HNO3 was studied in a photo flow-reactor, from which a HNO3 photolysis frequency for HONO formation, J(HNO3 → HONO), of only 2.4 × 10−7 s−1 is extrapolated for atmospheric noontime conditions (0° SZA). The slow HONO formation is explained by secondary heterogeneous formation via the primary HNO3 photolysis product NO2. On pure quartz glass surfaces, the heterogeneous conversion of NO2 into HONO shows no photoenhancement, in good agreement with other studies in simulation chambers.For the primary photolysis product NO2 a low photolysis frequency of J(HNO3 → NO2) = 1.1 × 10−6 s−1 (0° SZA) is calculated. From the wavelength dependence of the HONO formation a red-shift of the UV absorption of adsorbed HNO3 is inferred in agreement with other recent studies. However, in contrast to these studies, it is concluded that photolysis of pure adsorbed HNO3 is only a minor source of HONO and NO2 in the atmosphere, at least on non-reactive substrates. For the future, HONO formation by the photolysis of adsorbed HNO3 should also be studied on more complex surfaces, for example, on leaves, urban grime or humic acid.
Acknowledgements
The financial support for this work by the Deutsche Forschungsgemeinschaft (DFG) under contract number (KL 1392/3-1) is gratefully acknowledged.References
- X. Zhou, K. Civerolo, H. Dai, G. Huang, J. Schwab and K. L. Demerjian, Summertime Nitrous Acid Chemistry in the Atmospheric Boundary Layer at a Rural Site in New York State, J. Geophys. Res., 2002, 107, 4590 Search PubMed.
- K. Acker, D. Möller, W. Wieprecht, F. X. Meixner, B. Bohn, S. Gilge, C. Plass-Dülmer and H. Berresheim, Strong Daytime Production of OH from HNO2 at a Rural Mountain Site, Geophys. Res. Lett., 2006, 33, L02809 CrossRef.
- M. Sörgel, I. Trebs, A. Serafimovich, A. Moravek, A. Held and C. Zetzsch, Simultaneous HONO Measurements in and above a Forest Canopy: Influence of Turbulent Exchange on Mixing Ratio Differences, Atmos. Chem. Phys., 2011, 11, 841–855 CrossRef.
- G. Villena, P. Wiesen, C. A. Cantrell, F. Flocke, A. Fried, S. R. Hall, R. S. Hornbrook, D. Knapp, E. Kosciuch, R. L. Mauldin III, J. A. McGrath, D. Montzka, D. Richter, K. Ullmann, J. Walega, P. Weibring, A. Weinheimer, R. M. Staebler, J. Liao, L. G. Huey and J. Kleffmann, Nitrous Acid (HONO) during Polar Spring in Barrow, Alaska: A net Source of OH Radicals?, J. Geophys. Res.: Atmos., 2011, 116, D00R07 CrossRef.
- R. Oswald, M. Ermel, K. Hens, A. Novelli, H. G. Ouwersloot, P. Paasonen, T. Petäjä, M. Sipilä, P. Keronen, J. Bäck, R. Königstedt, Z. Hosaynali Beygi, H. Fischer, B. Bohn, D. Kubistin, H. Harder, M. Martinez, J. Williams, T. Hoffmann, I. Trebs and M. Sörgel, Comparison of HONO Budgets for two Measurement Heights at a Field Station within the Boreal Forest in Finland, Atmos. Chem. Phys., 2015, 15, 799–813 CrossRef.
- A. Neftel, A. Blatter, R. Hesterberg and T. Staffelbach, Measurements of Concentration Gradients of HNO2 and HNO3 over a Semi-Natural Ecosystem, Atmos. Environ., 1996, 30, 3017–3025 CrossRef CAS.
- T. Staffelbach, A. Neftel, A. Blatter, A. Gut, M. Fahrni, J. Stähelin, A. Prévôt, A. Hering, M. Lehning, B. Neininger, M. Bäumle, G. L. Kok, J. Dommen, M. Hutterli and M. Anklin, Photochemical Oxidant Formation Over Southern Switzerland 1. Results from Summer 1994, J. Geophys. Res., 1997, 102, 23345–23362 CrossRef CAS.
- J. Kleffmann, T. Gavriloaiei, A. Hofzumahaus, F. Holland, R. Koppmann, L. Rupp, E. Schlosser, M. Siese and A. Wahner, Daytime Formation of Nitrous Acid: A Major Source of OH-Radicals in a Forest, Geophys. Res. Lett., 2005, 32, L05818 CrossRef.
- V. Michoud, A. Colomb, A. Borbon, K. Miet, M. Beekmann, M. Camredon, B. Aumont, S. Perrier, P. Zapf, G. Siour, W. Ait-Helal, C. Afif, A. Kukui, M. Furger, J. C. Dupont, M. Haeffelin and J. F. Doussin, Study of the Unknown HONO Daytime Source at a European Suburban Site during the MEGAPOLI Summer and Winter Field Campaigns, Atmos. Chem. Phys., 2014, 14, 2805–2822 Search PubMed.
- J. Kleffmann, J. Heland, R. Kurtenbach, J. C. Lörzer and P. Wiesen, A new Instrument (LOPAP) for the Detection of Nitrous acid (HONO), Environ. Sci. Pollut. Res., 2002, 9(special issue 4), 48–54 Search PubMed.
- X. Ren, H. Harder, M. Martinez, R. L. Lesher, A. Oliger, J. B. Simpas, W. H. Brune, J. J. Schwab, K. L. Demerjian, Y. He, X. Zhou and H. Gao, OH and HO2 Chemistry in the Urban Atmosphere of New York City, Atmos. Environ., 2003, 37, 3639–3651 CrossRef CAS.
- B. Vogel, H. Vogel, J. Kleffmann and R. Kurtenbach, Measured and Simulated Vertical Profiles of Nitrous Acid – Part II, Model Simulations and Indications for a Photolytic Source, Atmos. Environ., 2003, 37, 2957–2966 CrossRef CAS.
- K. Acker, A. Febo, S. Trick, C. Perrino, P. Bruno, P. Wiesen, D. Möller, W. Wieprecht, R. Auel, M. Guisto, A. Geyer, U. Platt and I. Allegrini, Nitrous Acid in the Urban Area of Rome, Atmos. Environ., 2006, 40, 3123–3133 CrossRef CAS.
- X. Ren, W. H. Brune, J. Mao, M. J. Mitchell, R. L. Lesher, J. B. Simpas, A. R. Metcalf, J. J. Schwab, C. Cai, Y. Li, K. L. Demerjian, H. D. Felton, G. Boynton, A. Adams, J. Perry, Y. He, X. Zhou and J. Hou, Behavior of OH and HO2 in the Winter Atmosphere in New York City, Atmos. Environ., 2006, 40(supplement 2), 252–263 CrossRef.
- Y. F. Elshorbany, R. Kurtenbach, P. Wiesen, E. Lissi, M. Rubio, G. Villena, E. Gramsch, A. R. Rickard, M. J. Pilling and J. Kleffmann, Oxidation Capacity of the City Air of Santiago, Chile, Atmos. Chem. Phys., 2009, 9, 2257–2273 CrossRef CAS.
- Y. F. Elshorbany, J. Kleffmann, R. Kurtenbach, E. Lissi, M. Rubio, G. Villena, E. Gramsch, A. R. Rickard, M. J. Pilling and P. Wiesen, Seasonal Dependence of the Oxidation Capacity of the City of Santiago de Chile, Atmos. Environ., 2010, 44, 5383–5394 CrossRef CAS.
- F. Hendrick, J.-F. Müller, K. Clémer, P. Wang, M. De Mazière, C. Fayt, C. Gielen, C. Hermans, J. Z. Ma, G. Pinardi, T. Stavrakou, T. Vlemmix and M. Van Roozendael, Four Years of Ground-based MAX-DOAS Observations of HONO and NO2 in the Beijing Area, Atmos. Chem. Phys., 2014, 14, 765–781 CrossRef CAS.
- C. George, R. S. Strekowski, J. Kleffmann, K. Stemmler and M. Ammann, Photoenhanced Uptake of Gaseous NO2 on Solid Organic Compounds: A Photochemical Source of HONO?, Faraday Discuss., 2005, 130, 195–210 RSC.
- K. Stemmler, M. Ammann, C. Dondors, J. Kleffmann and C. George, Photosensitized Reduction of Nitrogen Dioxide on Humic Acid as a Source of Nitrous Acid, Nature, 2006, 440, 195–198 CrossRef CAS PubMed.
- K. Stemmler, M. Ndour, Y. Elshorbany, J. Kleffmann, B. D'Anna, C. George, B. Bohn and M. Ammann, Light Induced Conversion of Nitrogen Dioxide into Nitrous Acid on Submicron Humic Acid Aerosol, Atmos. Chem. Phys., 2007, 7, 4237–4248 CrossRef CAS.
- Y. Sosedova, A. Rouvière, T. Bartels-Rausch and M. Ammann, UVA/Vis-induced Nitrous Acid Formation on Polyphenolic Films Exposed to Gaseous NO2, Photochem. Photobiol. Sci., 2011, 10, 1680–1690 CAS.
- X. Zhou, H. J. Beine, R. E. Honrath, J. D. Fuentes, W. Simpson, P. B. Shepson and J. W. Bottenheim, Snowpack Photochemical Production of HONO: A major Source of OH in the Arctic Boundary Layer in Springtime, Geophys. Res. Lett., 2001, 28, 4087–4090 CrossRef CAS.
- X. Zhou, Y. He, G. Huang, T. D. Thornberry, M. A. Carroll and S. B. Bertman, Photochemical Production of Nitrous Acid on Glass Sample Manifold Surface, Geophys. Res. Lett., 2002, 29(14), 1681 CrossRef.
- X. Zhou, H. Gao, Y. He, G. Huang, S. B. Bertman, K. Civerolo and J. Schwab, Nitric Acid Photolysis on Surfaces in Low-NOx Environments: Significant Atmospheric Implications, Geophys. Res. Lett., 2003, 30, 2217 Search PubMed.
- X. Zhou, N. Zhang, M. TerAvest, D. Tang, J. Hou, S. Bertman, M. Alaghmand, P. B. Shepson, M. A. Caroll, S. Griffith, S. Dusanter and P. S. Stevens, Nitric Acid Photolysis on Forest Canopy Surface as a Source for Tropospheric Nitrous Acid, Nat. Geosci., 2011, 4, 440–443 CrossRef CAS.
- K. A. Ramazan, D. Syomin and B. J. Finlayson-Pitts, The Photochemical Production of HONO during the Heterogeneous Hydrolysis of NO2, Phys. Chem. Chem. Phys., 2004, 6, 3836–3843 RSC.
- H. Su, Y. Cheng, R. Oswald, T. Behrendt, I. Trebs, F. X. Meixner, M. O. Andreae, P. Cheng, Y. Zhang and U. Pöschl, Soil Nitrite as a Source of Atmospheric HONO and OH Radicals, Science, 2011, 333, 1616–1618 CrossRef CAS PubMed.
- R. Oswald, T. Behrendt, M. Ermel, D. Wu, H. Su, Y. Cheng, C. Breuninger, A. Moravek, E. Mougin, C. Delon, B. Loubet, A. Pommerening-Röser, M. Sörgel, U. Pöschl, T. Hoffmann, M. O. Andreae, F. X. Meixner and I. Trebs, HONO Emissions from Soil Bacteria as a Major Source of Atmospheric Reactive Nitrogen, Science, 2013, 341, 1233–1235 CrossRef CAS PubMed.
- M. Maljanen, P. Yli-Pirilä, J. Hytönen and J. Joutsensaari, Acidic Northern Soils as a Source of Atmospheric Nitrous Acid (HONO), Soil Biol. Biochem., 2013, 67, 94–97 CrossRef CAS.
- D. Wu, C. J. Kampf, U. Pöschl, U. Oswald, J. Cui, M. Ermel, C. Hu, I. Trebs and M. Sörgel, Novel Tracer Method to Measure Isotopic Labeled Gas-phase Nitrous Acid (HO15NO) in Biogeochemical Studies, Environ. Sci. Technol., 2014, 48, 8021–8027 CrossRef CAS PubMed.
- M. A. Donaldson, A. E. Berke and J. D. Raff, Uptake of Gas Phase Nitrous Acid onto Boundary Layer Soil Surfaces, Environ. Sci. Technol., 2013, 48, 375–383 CrossRef PubMed.
- T. C. VandenBoer, S. S. Brown, J. G. Murphy, W. C. Keene, C. J. Young, A. A. P. Pszenny, S. Kim, C. Warneke, J. A. de Gouw, J. R. Maben, N. L. Wagner, T. P. Riedel, J. A. Thornton, D. E. Wolfe, W. P. Dubé, F. Öztürk, C. A. Brock, N. Grossberg, B. Lefer, B. Lerner, A. M. Middlebrook and J. M. Roberts, Understanding the Role of the Ground Surface in HONO Vertical Structure: High Resolution Vertical Profiles during NACHTT-11, J. Geophys. Res.: Atmos., 2013, 118, 10155–10171 Search PubMed.
- T. C. VandenBoer, M. Z. Markovic, J. E. Sanders, X. Ren, S. E. Pusede, E. C. Browne, R. C. Cohen, L. Zhang, J. Thomas, W. H. Brune and J. G. Murphy, Evidence for a Nitrous Acid (HONO) Reservoir at the Ground Surface in Bakersfield, CA, during CALNex 2010, J. Geophys. Res.: Atmos., 2014, 119, 9093–9106 CAS.
- S. Li, J. Matthews and A. Sinha, Atmospheric Hydroxyl Radical Production from Electronically Excited NO2 and H2O, Science, 2008, 319, 1657–1660 CrossRef CAS PubMed.
- J. N. Crowley and S. A. Carl, OH Formation in the Photoexcitation of NO2 Beyond the Dissociation Threshold in the Presence of Water Vapor, J. Phys. Chem. A, 1997, 101, 4178–4184 CrossRef CAS.
- D. Amedro, A. E. Parker, C. Schoemaecker and C. Fittschen, Direct Observation of OH Radicals after 565 nm Multi-photon Excitation of NO2 in the Presence of H2O, Chem. Phys. Lett., 2011, 513, 12–16 CrossRef CAS.
- I. Bejan, Y. Abd El Aal, I. Barnes, T. Benter, B. Bohn, P. Wiesen and J. Kleffmann, The Photolysis of Ortho-Nitrophenols: A New Gas Phase Source of HONO, Phys. Chem. Chem. Phys., 2006, 8, 2028–2035 RSC.
- X. Li, F. Rohrer, A. Hofzumahaus, T. Brauers, R. Häseler, B. Bohn, S. Broch, H. Fuchs, S. Gomm, F. Holland, J. Jäger, J. Kaiser, F. N. Keutsch, I. Lohse, K. Lu, R. Tillmann, R. Wegener, G. M. Wolfe, T. F. Mentel, A. Kiendler-Scharr and A. Wahner, Missing Gas-phase Source of HONO Inferred from Zeppelin Measurements in the Troposphere, Science, 2014, 344, 292–296 CrossRef CAS PubMed.
- X. Li, F. Rohrer, A. Hofzumahaus, T. Brauers, R. Häseler, B. Bohn, S. Broch, H. Fuchs, S. Gomm, F. Holland, J. Jäger, J. Kaiser, F. N. Keutsch, I. Lohse, K. Lu, R. Tillmann, R. Wegener, G. M. Wolfe, T. F. Mentel, A. Kiendler-Scharr and A. Wahner, Response to Comment on “Missing Gas-Phase Source of HONO Inferred from Zeppelin Measurements in the Troposphere”, Science, 2015, 348, 1326e CrossRef PubMed.
- C. Ye, X. Zhou, D. Pu, J. Stutz, J. Festa, M. Spolaor, C. Cantrell, R. L. Mauldin, A. Weinheimer and J. Haggerty, Comment on “Missing Gas-phase Source of HONO Inferred from Zeppelin Measurements in the Troposphere”, Science, 2015, 348, 1326d CrossRef PubMed.
- J. Kleffmann, Daytime Sources of Nitrous Acid (HONO) in the Atmospheric Boundary Layer, ChemPhysChem, 2007, 8, 1137–1144 CrossRef CAS PubMed.
- X. Ren, J. E. Sanders, A. Rajendran, R. J. Weber, A. H. Goldstein, S. E. Pusede, E. C. Browne, K.-E. Min and R. C. Cohen, A Relaxed Eddy Accumulation System for Measuring Vertical Fluxes of Nitrous Acid, Atmos. Meas. Tech., 2011, 4, 2093–2103 CrossRef CAS.
- N. Zhang, X. Zhou, S. Bertman, D. Tang, M. Alaghmand, P. B. Shepson and M. A. Carroll, Measurements of Ambient HONO Concentrations and Vertical HONO Flux above a Northern Michigan Forest Canopy, Atmos. Chem. Phys., 2012, 12, 8285–8296 CrossRef CAS.
- F. Rohrer, B. Bohn, T. Brauers, D. Brüning, F.-J. Johnen, A. Wahner and J. Kleffmann, Characterisation of the Photolytic HONO-source in the Atmosphere Simulation Chamber SAPHIR, Atmos. Chem. Phys., 2005, 5, 2189–2201 CrossRef CAS.
- C. Zhu, B. Xiang, L. Zhu and R. Cole, Determination of Absorption Cross Sections of Surface-adsorbed HNO3 in the 290–330 nm Region by Brewster Angle Cavity Ring-down Spectroscopy, Chem. Phys. Lett., 2008, 458, 373–377 CrossRef CAS.
- C. Zhu, B. Xiang, L. T. Chu and L. Zhu, 308 nm Photolysis of Nitric Acid in the Gas Phase, on Aluminum Surfaces, and on Ice Films, J. Phys. Chem. A, 2010, 114, 2561–2568 CrossRef CAS PubMed.
- J. Du and L. Zhu, Quantification of the Absorption Cross Sections of Surface-adsorbed Nitric Acid in the 335–365 nm Region by Brewster Angle Cavity Ring-down Spectroscopy, Chem. Phys. Lett., 2011, 511, 213–218 CrossRef CAS.
- O. Abida, J. Du and L. Zhu, Investigation of the Photolysis of the Surface-adsorbed HNO3 by Combining Laser Photolysis with Brewster Angle Cavity Ring-down Spectroscopy, Chem. Phys. Lett., 2012, 534, 77–82 CrossRef CAS.
- E. P. Gardner, G. D. Sperry and J. G. Calvert, Primary Quantum Yields of NO2 Photodissociation, J. Geophys. Res., 1987, 92, 6642–6652 CrossRef CAS.
- B. J. Finlayson-Pitts and J. N. Pitts Jr., Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic Press, 2000 Search PubMed.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Volume I – Gas Phase Reactions of Ox, HOx, NOx and SOx Species, Atmos. Chem. Phys., 2004, 4, 1461–1738 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Volume II – Gas Phase Reactions of Organic Species, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS.
- J. Stutz, E. S. Kim, U. Platt, P. Bruno, C. Perrino and A. Febo, UV-visible Absorption Cross Sections of Nitrous Acid, J. Geophys. Res., 2000, 105, 14585–14592 CrossRef CAS.
- TUV, Quick TUV Calculator, NCAR Earth System Laboratory, Atmospheric chemistry Division, 2015, http://cprm.acd.ucar.edu/Models/TUV/Interactive_TUV/.
- J. Heland, J. Kleffmann, R. Kurtenbach and P. Wiesen, A New Instrument to Measure Gaseous Nitrous Acid (HONO) in the Atmosphere, Environ. Sci. Technol., 2001, 35, 3207–3212 CrossRef CAS PubMed.
- G. Villena, I. Bejan, R. Kurtenbach, P. Wiesen and J. Kleffmann, Development of a new Long Path Absorption Photometer (LOPAP) Instrument for the Sensitive Detection of NO2 in the Atmosphere, Atmos. Meas. Tech., 2011, 4, 1663–1676 CrossRef CAS.
- C. Hak, I. Pundt, S. Trick, C. Kern, U. Platt, J. Dommen, C. Ordóñez, A. S. H. Prévôt, W. Junkermann, C. Astorga-Lloréns, B. R. Larsen, J. Mellqvist, A. Strandberg, Y. Yu, B. Galle, J. Kleffmann, J. C. Lörzer, G. O. Braathen and R. Volkamer, Intercomparison of Four Different In situ Techniques For Ambient Formaldehyde Measurements in Urban Air, Atmos. Chem. Phys., 2005, 5, 2881–2900 CrossRef CAS.
- J. Kleffmann, T. Benter and P. Wiesen, Heterogeneous Reaction of Nitric Acid with Nitric Oxide on Glass Surfaces under Simulated Atmospheric Conditions, J. Phys. Chem. A, 2004, 108, 5793–5799 CrossRef CAS.
- A. L. Goodman, E. T. Bernard and V. H. Grassian, Spectroscopic Study of Nitric Acid and Water Adsorption on Oxide Particles: Enhanced Nitric Acid Uptake Kinetics in the Presence of Adsorbed Water, J. Phys. Chem. A, 2001, 105, 6443–6457 CrossRef CAS.
- J. J. Jankowski, D. J. Kieber and K. Mopper, Nitrate and Nitrite Ultraviolet Actinometers, Photochem. Photobiol., 1999, 70(3), 319–328 CrossRef CAS.
- B. J. Finlayson-Pitts, L. M. Wingen, A. L. Sumner, D. Syomin and K. A. Ramazan, The Heterogeneous Hydrolysis of NO2 in Laboratory Systems and in Outdoor and Indoor Atmospheres: An Integrated Mechanism, Phys. Chem. Chem. Phys., 2003, 5, 223–242 RSC.
- R. J. Gustafsson, G. Kyriakou and R. M. Lambert, The Molecular Mechanism of Tropospheric Nitrous Acid Production on Mineral Dust Surfaces, ChemPhysChem, 2008, 9, 1390–1393 CrossRef CAS PubMed.
- M. A. Kamboures, J. D. Raff, Y. Miller, L. F. Phillips, B. J. Finlayson-Pitts and R. B. Gerber, Complexes of HNO3 and NO3− with NO2 and N2O4, and their Potential Role in Atmospheric HONO formation, Phys. Chem. Chem. Phys., 2008, 10, 6019–6032 RSC.
- L. Zhu, M. Sangwan, L. Huang, J. Du and L. T. Chu, Photolysis of Nitric Acid at 308 nm in the Absence and in the Presence of Water Vapor, J. Phys. Chem. A, 2015, 119, 4907–4914 CrossRef CAS PubMed.
- H.-W. Jacobi and B. Hilker, A Mechanism for the Photochemical Transformation of Nitrate in Snow, J. Photochem. Photobiol., A, 2007, 185, 371–382 CrossRef CAS.
- N. K. Scharko, A. E. Berke and J. D. Raff, Release of Nitrous Acid and Nitrogen Dioxide from Nitrate Photolysis in Acidic Aqueous Solutions, Environ. Sci. Technol., 2014, 48, 11991–12001 CrossRef CAS PubMed.
- E. S. N. Cotter, A. E. Jones, E. W. Wolff and S. J.-B. Bauguitte, What Controls Photochemical NO and NO2 Production from Antarctic Snow? Laboratory Investigation Assessing the Wavelength and Temperature Dependence, J. Geophys. Res., 2003, 108(D4), 4147 CrossRef.
- J. M. Tadić, Comment on “308 nm Photolysis of Nitric Acid in the Gas Phase, on Aluminum Surfaces, and on Ice Films”, J. Phys. Chem. A, 2012, 116, 10463–10464 CrossRef PubMed.
- S. R. Handley, D. Clifford and D. J. Donaldson, Photochemical Loss of Nitric Acid on Organic Films: A Possible Recycling Mechanism for NOx, Environ. Sci. Technol., 2007, 41, 3898–3903 CrossRef CAS PubMed.
- R. Ammar, M. E. Monge, C. George and B. D'Anna, Photoenhanced NO2 Loss on Simulated Urban Grime, ChemPhysChem, 2010, 11, 3956–3961 CrossRef CAS PubMed.
- A. M. Baergen and D. J. Donaldson, Photochemical Renoxification of Nitric Acid on Real urban Grime, Environ. Sci. Technol., 2013, 47, 815–820 CrossRef CAS PubMed.
- D. Cazoir, M. Brigante, R. Ammar, B. D'Anna and C. George, Heterogeneous Photochemistry of Gaseous NO2 on Solid Fluoranthene Films: A Source of Gaseous Nitrous Acid (HONO) in the Urban Environment, J. Photochem. Photobiol., A, 2014, 273, 23–28 CrossRef CAS.
| This journal is © the Owner Societies 2016 |