
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Global optimization of small bimetallic Pd–Co binary nanoalloy clusters: a genetic algorithm approach at the DFT level†
Mikail
Aslan
a,
Jack B. A.
Davis
b and
Roy L.
Johnston
*b
aDepartment of Metallurgical and Materials Engineering, Gaziantep University, Gaziantep, Turkey
bSchool of Chemistry, University of Birmingham, Birmingham, UK. E-mail: r.l.johnston@bham.ac.uk
First published on 4th February 2016
Abstract
The global optimisation of small bimetallic PdCo binary nanoalloys are systematically investigated using the Birmingham Cluster Genetic Algorithm (BCGA). The effect of size and composition on the structures, stability, magnetic and electronic properties including the binding energies, second finite difference energies and mixing energies of Pd–Co binary nanoalloys are discussed. A detailed analysis of Pd–Co structural motifs and segregation effects is also presented. The maximal mixing energy corresponds to Pd atom compositions for which the number of mixed Pd–Co bonds is maximised. Global minimum clusters are distinguished from transition states by vibrational frequency analysis. HOMO–LUMO gap, electric dipole moment and vibrational frequency analyses are made to enable correlation with future experiments.
A. Introduction
The structural characterisation of clusters and nanoparticles is key in elucidating the size-dependent properties of nanoscale materials to facilitate many potential applications. Geometric structures of small nanoparticles (or subnanometre clusters) can be determined by coupling experimental measurements with theoretical calculations.3 The generation of possible geometric isomers that can be used to explain experimental findings can be made by intuition4–6 but this becomes impossible for larger systems and will also bias results. Another approach is to utilise an algorithm that explores configurational space to determine the global minimum (GM). Several computational methods that predict the globally stable structures of subnanometre clusters are available, such as statistical mechanical methods,7,8 basin hopping9 and genetic algorithms (GA).10 The choice of technique depends on how the potential energy surface is described and how complex it is.11,12 After the optimisation of the cluster structure using these methods, reoptimisation at the Density Functional Theory (DFT) level can be performed in order to correlate the predicted lowest energy structure with experimental results for free or supported clusters.13–15Experimental findings associated with theoretical investigations have revealed that the bonding situation for clusters and small nanoparticles is generally different from the corresponding bulk material16,17 and can also differ for two elements of the same group.18 For these reasons, electronic structure calculations are needed to predict the correct growth characteristics of small clusters. Thus, due to growing interest in the design of novel functional nanomaterials, the DFT analysis of clusters of metals such as cobalt, platinum, palladium, silver, and gold has become a hot research field for chemists, physicists, and materials scientists.19–33
Clusters become more complex when two or more metals are alloyed in order to tune the characteristics of the particles not only by size but also by composition and chemical ordering, possibly resulting in special synergistic effects for these “nanoalloys”.34 In recent years, bimetallic nanoparticles have been studied using many body empirical potentials, with the GM predicted using a GA, often yielding consistent results with experiments.35 However, an examination of the bonding in smaller (subnanometre) bimetallic clusters can only be achieved using electronic structure methods. For this reason, we have recently developed a program for the direct global optimisation of cluster geometries at the DFT level – the so-called GA-DFT approach.3,36 Another motivation is that nanoalloy particles often have superior chemical and physical properties compared to single element nanoparticles.37 Thus, nanoalloys are of great interest in the chemical industry: for example, one metal may adjust the catalytic properties of the other due to structural and/or electronic effects. In many nanoalloys, the lowest energy configurations have a core of one metal surrounded by a shell of the other metal, so that smaller volumes of a catalytically active element (thereby reducing the cost) might be sufficient to achieve similar effects as those of single element catalysts.38 Furthermore, in Fisher–Tropsch catalysis,39–41 PdCo nanoparticles have been shown to display higher selectivity than pure Co particles. Synthesis of hydrocarbon fuels by reacting carbon monoxide and hydrogen is appealing to many researchers in this area due to recent huge fluctuations in the prices of hydrocarbons.38
In this paper, we have used the Birmingham Cluster Genetic Algorithm (BCGA) with an interface to the PWscf DFT code within the Quantum Espresso (QE) package. This provides an unbiased search starting from entirely random coordinates for the search of GM of PdCo nanoalloys. Here we conduct the first GM search for 4–7 atom PdCo bimetallic nanoalloys, over the entire composition range, using the GA-DFT approach.
B. Methods
In the present study, the putative GM isomers are generated by the Birmingham Cluster Genetic Algorithm (BCGA)10 within the framework of Density Functional Theory, using an interface to the PWscf code of Quantum Espresso (QE) software.42 The BCGA is used for the structural characterisation of nanoparticles and nanoalloys. The interface with QE enables the energy landscape of a system to be searched for the GM at the DFT level.In the first step of the GA, a set of individuals is generated randomly to form an initial population of 10 members. Real valued Cartesian coordinates are chosen and each structure is relaxed by optimising the potential energy as a function of its coordinates. The BCGA is a “Lamarckian” type GA, which combines a GA step with a subsequent local minimisation of the energy. Each structure is assigned a fitness value such that the lowest energy structures correspond to highest fitness. In the GA-DFT approach, the energy of each member of the population is obtained from a PWscf DFT calculation. The crossover process (to generate a predetermined number of offspring) uses the roulette wheel selection criterion and the Deaven–Ho cut and splice method.43 Mutation is accomplished using a number of schemes in the BCGA, such as atom displacement, cluster twisting, cluster replacement (used here), and atom permutation and is performed to improve population diversity. The process of selection, crossover, and mutation is reiterated for a pre-determined maximum number of generations (here 200). However, if after a certain number of generations the lowest energy member of the population does not change, then the population is considered to have converged and the GA terminates.
Plane-wave PWscf calculations are conducted by applying ultrasoft type pseudopotentials44 for all metallic species, including scalar relativistic effects.45 We have adopted the Perdew–Burke–Ernzerhof (PBE) GGA exchange–correlation functional46 that has been widely used in the treatment of small, mixed clusters. For the primary screening of the structures with BCGA, the default density cutoff convergence criterion is applied with energy cutoff of 50 Ry. The Methfessel–Paxton smearing scheme47 with a value of 0.01 Ry is applied to aid metallic convergence.
Spin-polarised reminimisations of BCGA-DFT global minima were carried out using the orbital-based DFT package NWChem.48 PBE exchange–correlation functionals,46 LANL2DZ basis sets and relativistic effective core pseudopotentials (ECPs)49,50 were used for Pd and Co, where only the outer-most electrons are treated explicitly in the calculations. Default convergence criteria have been applied during calculations: 1 × 10−6 Hartree for energy and 5 × 10−4 Hartree a0−1 for the energy gradient. Geometry optimisations were performed without any symmetry constraints for a range of electron spin multiplicities.
DFT binding energies (Eb) are calculated from
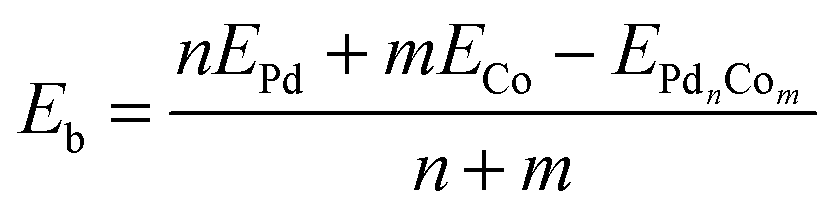 | (1) |
To further illustrate the stability of the nanoalloys and their size dependent behaviour, we have considered the second finite difference in energy, which is a sensitive quantity that is frequently used as a measure of the relative stability of a particular cluster with respect to neighbouring sizes or compositions and is often compared directly with the relative abundances determined in mass spectroscopy experiments. For a fixed size cluster, the second finite different energy (Dn,m) of the isomer PdnCom is calculated as
| Dn,m = En+1,m–1 + En–1,m+1 − 2En,m | (2) |
The mixing energies (Em) are listed in Table 1 to provide a measure of the stability of the bimetallic clusters with respect to the monometallic ones or the energy associated with alloying. Em is defined here as the (positive) quantity
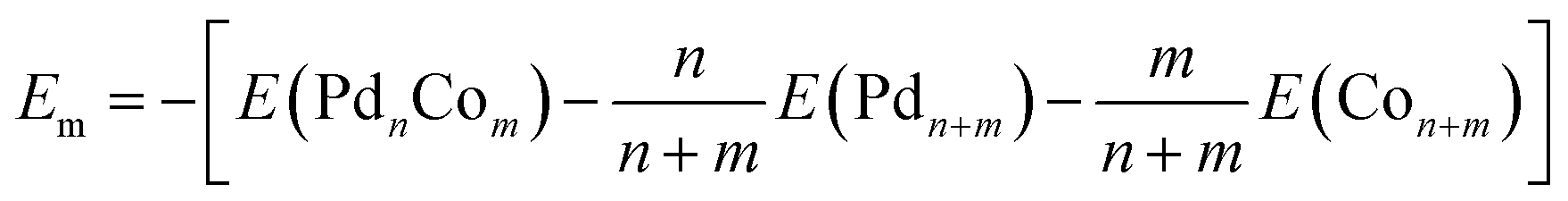 | (3) |
| Clusters | SM | SYM | E b | E m | Clusters | SM | SYM | E b | E m |
|---|---|---|---|---|---|---|---|---|---|
| a SM, spin moment in μB; SYM, point group symmetry; Eb, binding energy in eV per atom; Em, mixing energy in eV. b Ref. 1. c Ref. 2. | |||||||||
| Co3 | 7 | C 2v | 1.45 | — | Co6 | 14 | O h |
2.41
2.47c |
— |
| PdCo2 | 4 | C 2v | 1.62 | 0.60 | PdCo5 | 13 | C 4v | 2.38 | 0.16 |
| Pd2Co | 3 | C 2v | 1.70 | 0.93 | Pd2Co4 | 10 | C 2v | 2.35 | 0.32 |
| Pd3 | 2 | D 3h |
1.37
1.24b |
— | Pd3Co3 | 7 | C 1 | 2.30 | 0.38 |
| Co4 | 10 | C 2v | 1.79 | — | Pd4Co2 | 6 | C 2v | 2.27 | 0.55 |
| PdCo3 | 7 | C s | 1.86 | 0.29 | Pd5Co | 3 | C 4v | 2.21 | 0.51 |
| Pd2Co2 | 4 | C 2v | 1.93 | 0.57 | Pd6 | 2 | D 4h |
2.06
1.88b |
— |
| Pd3Co | 3 | C 3v | 1.97 | 0.73 | Co7 | 15 | C 1 |
2.51
2.45c |
0.00 |
| Pd4 | 2 | D 2d |
1.78
1.63b |
— | Pd1Co6 | 14 | C 3 | 2.54 | 0.64 |
| Co5 | 11 | C s |
2.10
2.29c |
— | Pd2Co5 | 13 | C S | 2.52 | 0.90 |
| PdCo4 | 8 | C 3v | 2.14 | 0.36 | Pd3Co4 | 10 | C 2v | 2.50 | 1.13 |
| Pd2Co3 | 7 | C 2v | 2.17 | 0.70 | Pd4Co3 | 7 | C 1 | 2.42 | 0.98 |
| Pd3Co2 | 4 | C 2v | 2.15 | 0.82 | Pd5Co2 | 6 | C S | 2.36 | 1.00 |
| Pd4Co | 3 | C 2v | 2.10 | 0.76 | Pd6Co1 | 3 | C 5 | 2.25 | 0.68 |
| Pd5 | 2 | C 4v |
1.91
1.74b |
— | Pd7 | 2 | D 5 |
2.10
1.90b |
0.00 |
C. Results and discussion
To check the validity of the computational method for the study of the bimetallic PdCo nanoalloys, the binding energies (BEs) of Pd and Co dimers were calculated as 0.76 and 1.03 eV per atom, respectively. The obtained results are in agreement with experimental51–55 and theoretical results.22,56–59 The BE of the PdCo dimer (1.21 eV) does not lie between those of Pd2 and Co2. However, PdnCom binary nanoalloys properties are expected to lie in between those of pure palladium and cobalt clusters (Fig. 1–4).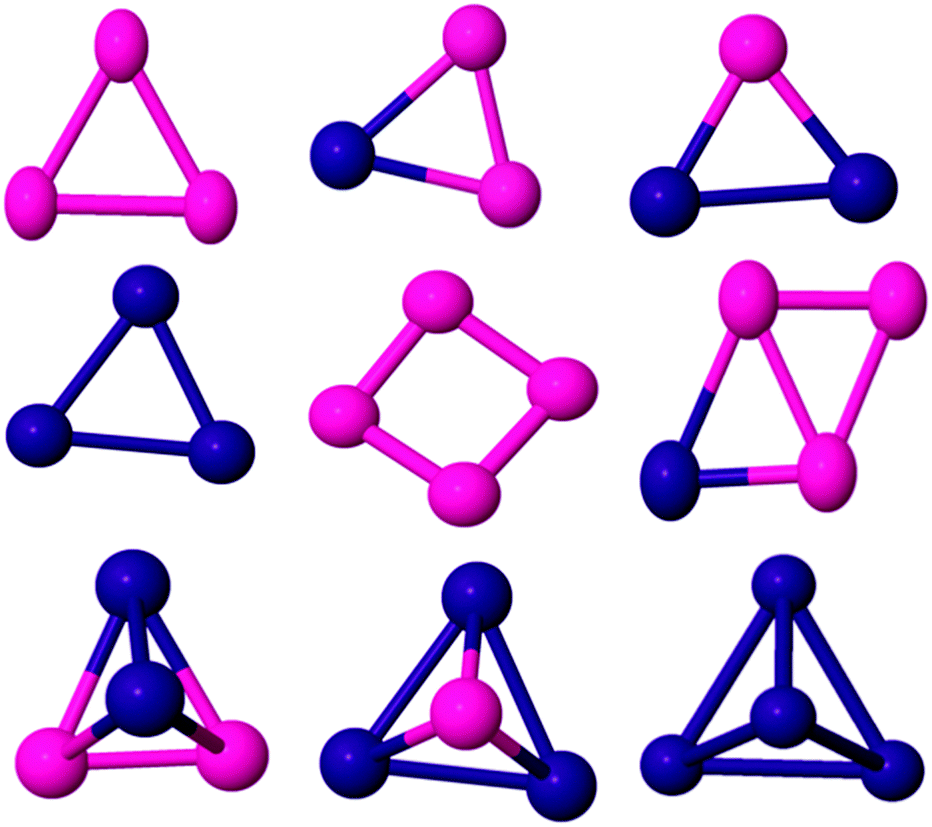 | ||
| Fig. 1 Global minima for three and four atom PdCo nanoalloys. Pd and Co are shown in purple and pink, respectively. | ||
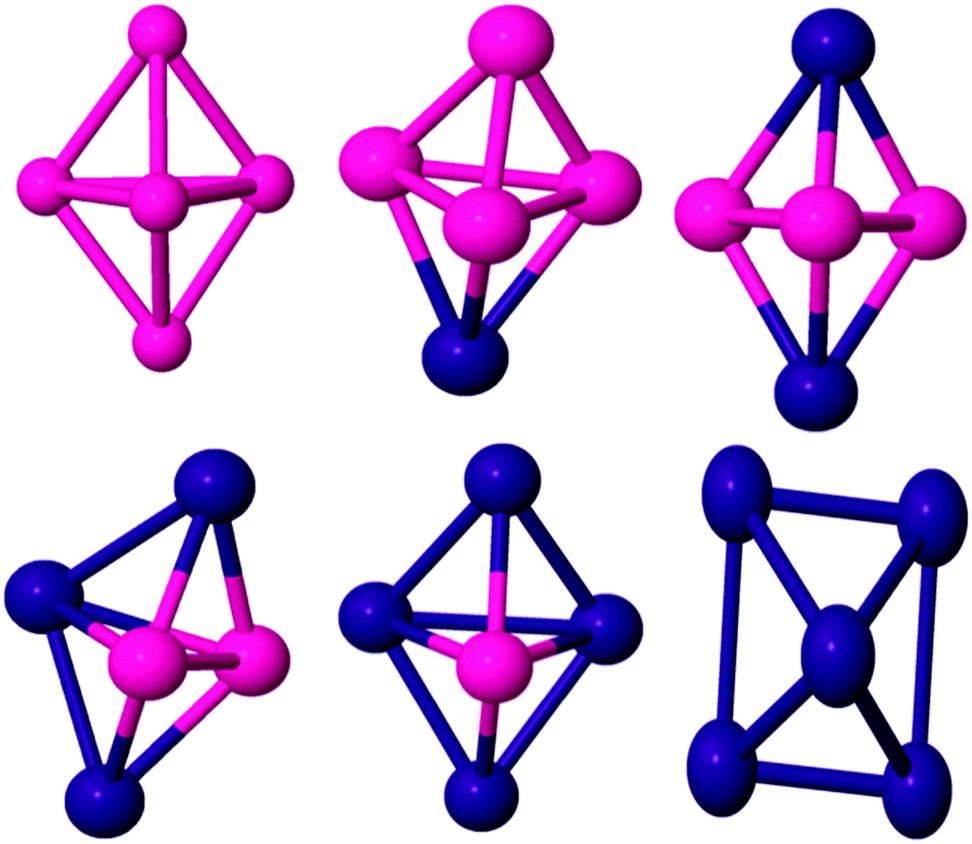 | ||
| Fig. 2 Global minima for five atom PdCo nanoalloys. | ||
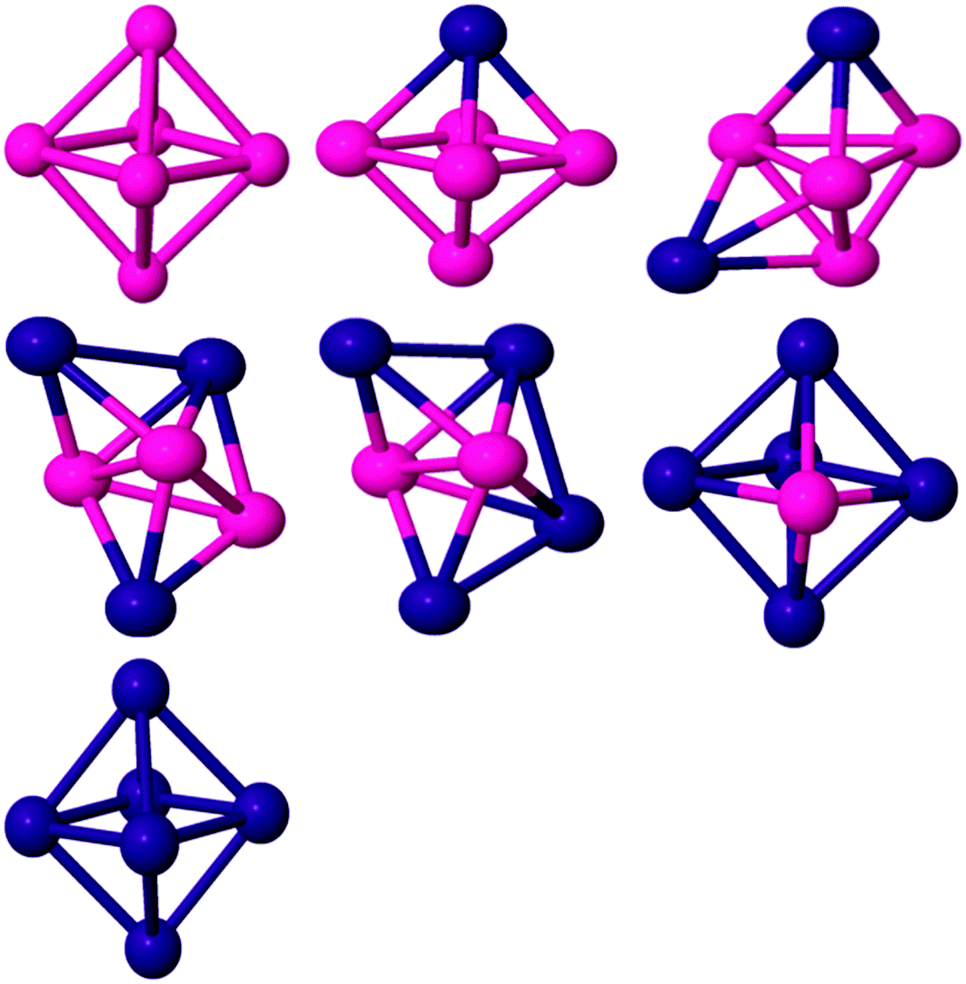 | ||
| Fig. 3 Global minima for six atom PdCo nanoalloys. | ||
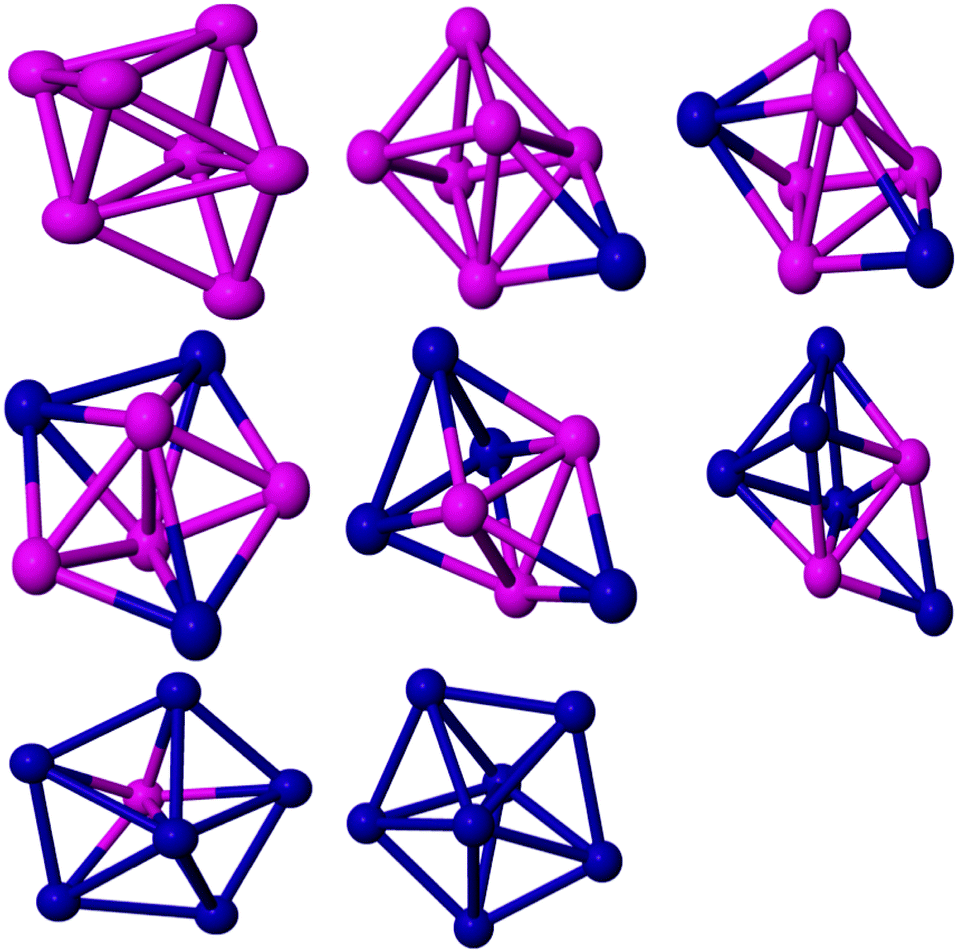 | ||
| Fig. 4 Global minima for seven atom PdCo nanoalloys. | ||
The ground state structure of the Co trimer is still controversial. In our calculation, the GM structure of Co3 has been identified as a C2v isosceles triangle, as previously reported.57,60,61 Experimentally, it was reported62 that the electron spin resonance spectrum of the cobalt trimer in an Ar/Kr matrix shows a triangular structure with a spin moment of 5 or 7 μB. However, Co3 was found to adopt a linear geometry in ref. 59 and 63. Datta et al.61 found the BE and spin moment as 1.78 eV per atom and 5 μB respectively while we found corresponding values of 1.45 eV per atom and 7 μB respectively (see Table 1). By introducing up to two Pd dopant atoms into the cluster, the overall structural motif is not changed significantly (see Fig. 1). For the PdCo2 cluster, an isosceles triangle with total magnetic moment 5 μB is found to be the GM with BE 1.62 eV per atom. The BE value is consistent with the result in ref. 38. When doped with two Pd atoms, the isosceles triangle remains the GM while the pure Pd trimer has a D3h equilateral triangle structure, though the calculated BE of this structure (1.37 eV per atom) is inconsistent with DFT results reported by other researchers.64,65
The most stable isomer of Co4 is a non-planar butterfly-like structure with C2v symmetry (as the two diagonals are unequal in length). It has a BE of 1.79 eV per atom and a spin moment of 10 μB, as reported by Sebetci,59 Fan et al.,2 and Ma et al.66 but in contradiction to the findings of Datta et al.67 Experimentally, Jalink et al.68 identified Co4 as having a planar rhombus structure based on a gas phase vibrational spectroscopy. Replacing one of the Co atoms by Pd, results in a non-planar butterfly as the lowest energy morphology (see Fig. 1) and the symmetry is reduced to Cs, which removes orbital degeneracy, increases the d electron bandwidth and reduces the local magnetic moment of Co. As Pd doping increases, the BE of Pd2Co2 increases to 1.93 eV per atom in the quintet magnetic state and the structure changes to a C2v tetrahedron, where all faces are isosceles triangles. The structure of Pd3Co is a C3v tetrahedron. A triplet spin multiplicity with D2d symmetry is found to be the GM structure of Pd4, which has a D2d distorted-tetrahedral structure. This agrees with the results of Zanti et al.69 and Begum et al.70
For the Co pentamer, a distorted trigonal bipyramidal (TBP) structure has been found as the GM energy structure with a spin magnetic moment of 11 μB. There is no consensus on the structure of Co5 in the literature. Pereiro et al.,60 Datta et al.61 and Castro et al.58 have identified the ground state structure as the TBP configuration while a C4v pyramidal structure has been proposed by Fan et al.2 and Ma et al.66 The Pd2Co3, Pd3Co2 and Pd4Co clusters (see Fig. 2) all have C2v TBP geometries, with spin moments of 7, 4 and 3 μB, respectively, while the PdCo4 cluster has a C3v TBP structure and a spin moment of 8 μB. In Pd4Co and Pd3Co2, Co atoms preferably occupy the higher connectivity equatorial positions in the TBP, which maximise the number of Co–Co and Co–Pd bonds (both of which are stronger than Pd–Pd bonds). The preferential doping of Pd atoms into the lower-connected apical sites in PdCo4 and Pd2Co3 is consistent with this. In the case of the pure Pd pentamer, the lowest energy morphology changes to square a pyramid with C4v symmetry and a triplet magnetic state. This structure agrees with the work of Cantera and coworkers,38 and Zanti and coworkers69 while Jerzy and coworkers71 identified the GM as a TBP.
For pure Co6 the GM structure is a regular octahedron with Oh symmetry and a magnetic moment of 14 μB, which is entirely consistent with previous results.61,66,72 On replacing one Co by Pd (see Fig. 3), the overall octahedral structure is retained. The GM structure has C4v symmetry and a total magnetic moment of 13 μB. When Pd atom doping continues, a capped TBP (or bicapped tetrahedron) is found to be GM for Pd2Co4 (C2v), Pd3Co3 (C1) and Pd4Co2 (C2v). As for the pentamers, the Co atoms preferentially occupy the higher coordinate sites and the Pd atoms occupy the low coordinate sites, leading to more Co–Co (and Co–Pd) and fewer Pd–Pd bonds, again correlating with bond strengths and bulk cohesive energies.73 The overall effect is for core segregation of Co and surface segregation of Pd, with this core–shell type of segregation (which is related to the lower surface energy of Pd compared to Co, since the element having smaller surface energy and cohesive energy favours occupying the surface to minimise the total energy74) also predicted for larger PdCo clusters.75 Janssens et al.76 found that with fewer than 50 atoms, for AgnCom with n ≫ m the cobalt dopants occupy highly coordinated sites and are strongly bound, for clusters with m ≫ n the silver atoms are poorly coordinated surface atoms and are loosely bound. The GM structure of Pd5Co is an octahedron (C4v symmetry) in the quartet magnetic state. The BE of Pd5Co has been calculated as 2.21 eV per atom. It should be noted that the dominant growth patterns for the studied bimetallic nanoalloys generally keep similar frameworks to those of the pure Co clusters. Pd6 has a D4h tetragonally distorted octahedral geometry. This is consistent with previous DFT studies.65,77,78
The most stable isomer of Co7 is a capped octahedron-like structure. It has a BE of 2.51 eV per atom while Datta et al.67 calculated the BE as 2.97 eV per atom. The structure has a spin moment of 15, which is consistent with the experimental result (2.36 ± 0.25 μB per atom79). For PdCo6, the overall capped octahedron-like structure is retained (see Fig. 4). The GM structure has C3 symmetry and a spin moment of 14 μB. A low symmetry Cs isomer, with a spin moment of 13 μB, is found to be the GM for Pd2Co5. On further Pd doping, the structural motif changes to a pentagonal pyramid for Pd3Co4. Pd4Co3 has C1 symmetry and a spin moment of 7 μB. The BE of Pd4Co3 has been calculated as 2.42 eV. For Pd5Co2, this structural motif is retained. The spin moment of Pd5Co2 is calculated as 6 μB and it has Cs symmetry. On further Pd doping, the structural motif changes to the pentagonal bipyramid. The pentagonal bipyramid GM structure for Pd7 is consistent with the findings of Kalita and Deka.80 The BE of this structure in the doublet state is calculated as 2.10 eV per atom, which is consistent with the result of ref. 38.
The magnetic spin moments of the GM structures of PdCo clusters within the studied size range are shown in Fig. 5. The magnetic moments of bimetallic PdCo clusters exhibit a zigzag pattern upon successive addition of Pd atoms. It should be noted that all pure Pd clusters in this study have triplet magnetic ground states, which is consistent with the results of ref. 69. As the Pd atom has a closed shell electronic configuration (4d10), to make a stable a metal–metal bond, some 4d electronic density needs to be promoted into the lowest unoccupied orbitals, in this case 5s. Occupying this orbital leads to a triplet state. As expected, Pd atoms have a quenching effect on the magnetism of bimetallic PdCo nanoalloys, so successive Pd doping of a Co cluster leads to a decrease of the spin moment of the ground state structure. We could not find any clear size dependence, but for CoMn clusters, based on Stern–Gerlach (SG) experiments, Yin et al.81 have concluded that the magnetic enhancement of ConMnm (n ≤ 60, m ≤ n/3) is independent of the cluster size and composition. However, the study conducted by Zanti and coworkers showed that clusters enriched in palladium atoms have spin multiplicities that increase with the cluster size while clusters enriched in gold atoms maintain the lowest possible spin multiplicity for the structure.69
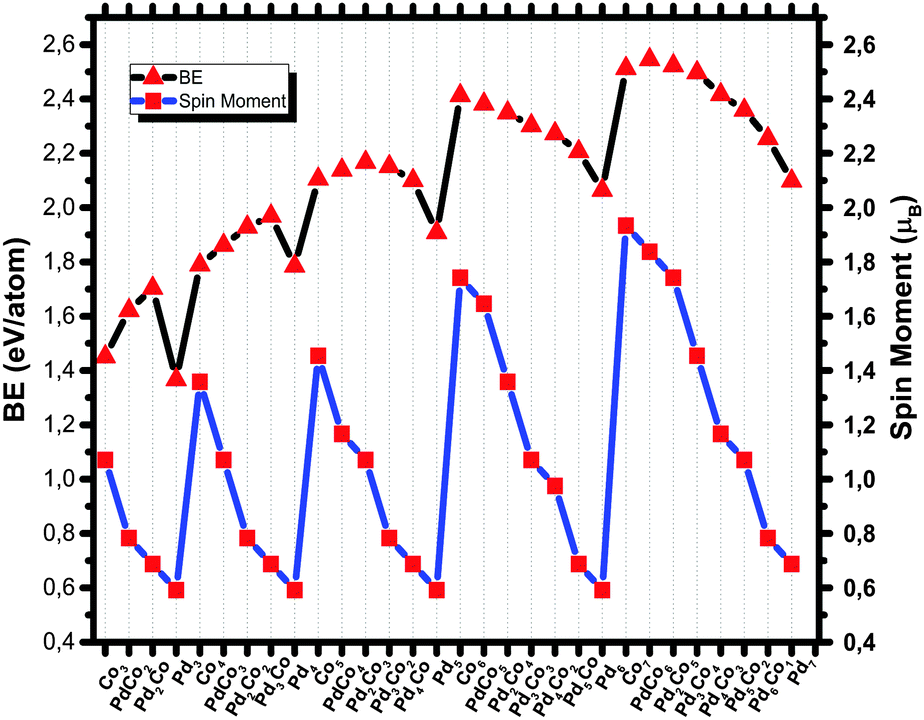 | ||
| Fig. 5 Binding energies and spin moments of PdCo nanoalloys. | ||
Nanoparticle stability can be analyzed in various ways, the most common being the computation of the energy released during the growth of metal nanoparticles starting from isolated atoms, corresponding to the BE. The dependence of the BE on the composition of PdCo nanoalloys within the studied range is shown in Fig. 5. As expected, increasing nuclearity makes the BE larger. This is because during the growth process the number of nearest neighbors increases, leading to a larger number of interactions per atom but the proportional gain gets smaller as the cluster gets larger, eventually approaching the bulk cohesive energy asymptotically. For bimetallic PdCo clusters, the BE is expected to lie between those of pure Pd and Co clusters. However, up to size 5, the BEs are (as seen in Table 1) larger than both the pure Pd and Co clusters, while for size 6 and 7, the BE values usually lie between those of the pure Pd and Co nanoparticles, as expected. Bakken and Swang82 have found that for small cobalt clusters, substituting rhenium stabilizes the clusters.
For the pentamer PdCo nanoalloys, Pd4Co has a much higher Dn,m energy than Pd3Co2. This shows that it is more stable than the latter species but the result is not consistent with the mixing energy result. For the hexamer structures, the dip is seen at Pd3Co3 in Fig. 6. This species is expected to be less abundant in mass spectra than the other corresponding clusters.
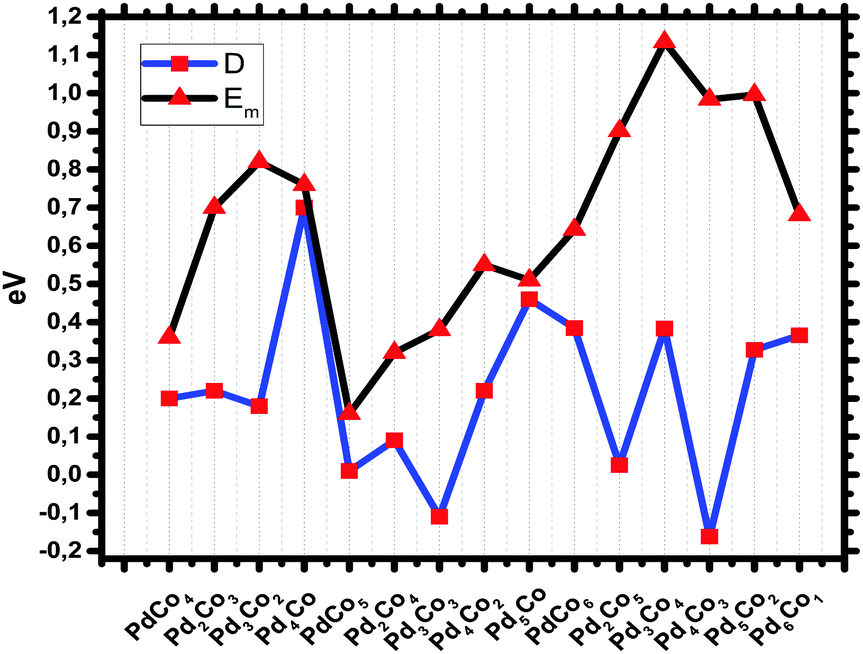 | ||
| Fig. 6 Second finite difference energies (D) and mixing energies (Em) of 5–7-atom PdCo nanoalloys. | ||
The mixing energy Em can be used to evaluate the effect of mixing in a system. The evolution of the mixing energy with composition is shown for n + m = 5, 6 and 7 in Fig. 6. The more positive values of Em show a stronger mixing tendency. The maximum value is seen for Pd2Co in Table 1. This is consistent with the BE result. Comparing the different nuclearities, it should be noted that the mixing energy of bimetallic PdnCom nanoparticles is independent of the cluster size. However, up to 6 atoms, the values of mixing energy of the studied nanoalloys are proportional to the proportion of Pd atoms in the cluster, with the exception of Pd4Co and Pd5Co.
A large HOMO–LUMO gap (HLG) is generally considered as a significant requirement for chemical stability since HLG indicates the ability of electrons to hop from HOMO to LUMO and indicates the tendency of a molecule to be involved in chemical reactions to some degree. Pd3Co2 (0.81 eV) has the highest HLG among the species studied in this work. Thus, one can expect high chemical stability for this cluster. It can be noted that pure Pd clusters generally have higher chemical activity than pure cobalt clusters. Furthermore, among the studied species, Pd-rich clusters are expected to have high chemical activity due to their low HLG values (see Table 2).
| Clusters | HLG | DM | FRQ | Clusters | HLG | DM | FRQ |
|---|---|---|---|---|---|---|---|
| a HLG, HOMO–LUMO gap in eV, DM; electric dipole moment in Debyes; FRQ, lowest, weighted, highest vibrational frequencies, respectively, in cm−1. | |||||||
| Co3 | 0.68 | 0.40 | 96, 231, 319 | Co6 | 0.32 | 0.00 | 120, 269, 344 |
| PdCo2 | 0.71 | 1.16 | 170, 170, 296 | PdCo5 | 0.29 | 0.52 | 81, 261, 318 |
| Pd2Co | 0.76 | 0.75 | 148, 148, 286 | Pd2Co4 | 0.50 | 0.25 | 78, 154, 301 |
| Pd3 | 0.01 | 0.13 | 159, 166, 237 | Pd3Co3 | 0.37 | 0.72 | 66, 151, 287 |
| Co4 | 0.61 | 0.05 | 66, 233, 267 | Pd4Co2 | 0.32 | 0.05 | 54, 257, 290 |
| PdCo3 | 0.59 | 1.32 | 46, 111, 290 | Pd5Co | 0.54 | 0.74 | 83, 256, 256 |
| Pd2Co2 | 0.51 | 1.35 | 111, 176, 289 | Pd6 | 0.08 | 0.00 | 99, 184, 220 |
| Pd3Co | 0.48 | 0.84 | 95, 156, 285 | Co7 | 0.18 | 0.31 | 66, 253, 304 |
| Pd4 | 0.01 | 0.00 | 111, 184, 239 | Pd1Co6 | 0.37 | 0.85 | 60, 254, 295 |
| Co5 | 0.52 | 0.53 | 74, 191, 327 | Pd2Co5 | 0.33 | 0.64 | 85, 248, 299 |
| PdCo4 | 0.43 | 0.87 | 115, 192, 328 | Pd3Co4 | 0.58 | 0.61 | 64, 151, 278 |
| Pd2Co3 | 0.59 | 0.15 | 82, 256, 303 | Pd4Co3 | 0.33 | 0.34 | 75, 257, 282 |
| Pd3Co2 | 0.81 | 0.89 | 83, 173, 288 | Pd5Co2 | 0.21 | 0.75 | 60, 122, 272 |
| Pd4Co | 0.55 | 0.67 | 50, 238, 261 | Pd6Co1 | 0.33 | 0.39 | 29, 226, 234 |
| Pd5 | 0.20 | 0.31 | 53, 195, 221 | Pd7 | 0.14 | 0.00 | 56, 167, 207 |
Electric dipole moment calculations have been performed to enable predictions to be made for future electron beam deflection experiments, which is one of few experimental methods for the investigation of small neutral clusters. The dipole moments (see Table 2) are small due to the low charge distribution within the cluster with the exception of PdCo2, PdCo3 and Pd2Co2, which have clearly distinguishable dipole moments. These clusters should be easily discriminated by experiment.
The vibrational frequencies (see Table 2) lie between 29 and 344 cm−1 and show no clear dependence on cluster size and composition. All vibrational frequencies, projected infrared intensities, Mulliken charge analysis, electric field gradients, dipole, quadrupole and octupole moments are listed as ESI.†
D. Conclusions
We have performed a computational study of PdCo nanoalloys ranging from 3 to 7 atoms. The structural, electronic and magnetic properties of bimetallic PdCo nanoalloys have been studied within the framework of the GA-DFT approach that performs an unbiased global optimisation search for the lowest energy isomer, for each size and composition, at the DFT level.The calculations reveal that Pd atoms segregate to the peripheral positions of the clusters to enable maximisation of the stronger Co–Co and Co–Pd bonds and also the lower surface energy of Pd. Up to size 5, the doped nanoalloys possess higher binding energies indicating that the bimetallic species possess enhanced chemical stability than their pure counterparts. Pd atoms have a quenching effect on the magnetism of bimetallic PdCo nanoalloys, so successive Pd doping of a Co cluster leads to a decrease of the spin moment of the ground state structure. For the size dependence of magnetic moment we could not find any clear relation. It can be noted that pure Pd clusters generally have higher chemical activity than those pure cobalt clusters. Furthermore, among the studied species, Pd-rich clusters are expected to have high chemical activity due to their low HLG values. The dipole moments are small due to the low charge distribution within the cluster, with the exception of PdCo2, PdCo3 and Pd2Co2, which have clearly distinguishable dipole moments.
In the future, the study we have carried out will be extended to larger bimetallic PdCo nanoalloys, where the dependence of chemical ordering and structure on the composition will be analyzed by using the Pool-BCGA code,83,84 which is a new parallel implementation of the code, which reduces computational costs significantly for larger clusters.
Acknowledgements
The calculations reported here have been performed on the following HPC facilities: The University of Birmingham BlueBEAR facility (see http://www.bear.bham.ac.uk/bluebear for a description of the Blue-BEAR HPC facility), the MidPlus Regional Centre of Excellence for Computational Science, Engineering and Mathematics, funded under EPSRC grant EP/K000128/1 (RLJ); and via membership of the UK's HPC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202), this work made use of the facilities of ARCHER, the UK's national high-performance computing service, which is funded by the Office of Science and Technology through EPSRC's High End Computing Programme. MA acknowledges financial support by The Scientific and Technological Research Council of Turkey (TUBITAK) under the program 2214-A.References
- P. Nava, Density Functional Theory Calculations on Palladium Clusters and on an AgInS Semiconductor Compound, Cuvillier Verlag, 2005 Search PubMed.
- H.-J. Fan, C.-W. Liu and M.-S. Liao, Chem. Phys. Lett., 1997, 273, 353–359 CrossRef CAS.
- S. Heiles, A. J. Logsdail, R. Schafer and R. L. Johnston, Nanoscale, 2012, 4, 1109–1115 RSC.
- V. Bonačić-Koutecký, J. Burda, R. Mitrić, M. Ge, G. Zampella and P. Fantucci, J. Chem. Phys., 2002, 117, 3120 CrossRef.
- H. M. Lee, M. Ge, B. R. Sahu, P. Tarakeshwar and K. S. Kim, J. Phys. Chem. B, 2003, 107, 9994–10005 CrossRef CAS.
- G. F. Zhao and Z. Zeng, J. Chem. Phys., 2006, 125, 014303 CrossRef CAS PubMed.
- J. M. Montejano-Carrizales and J. L. Morán-López, Surf. Sci., 1990, 239, 169–177 CrossRef CAS.
- M. Polak and L. Rubinovich, Surf. Sci., 2005, 584, 41–48 CrossRef CAS.
- D. J. Wales and J. P. K. Doye, J. Phys. Chem. A, 1997, 101, 5111–5116 CrossRef CAS.
- R. L. Johnston, Dalton Trans., 2003, 4193–4207 RSC.
- A. Sutton and J. Chen, Philos. Mag. Lett., 1990, 61, 139–146 CrossRef.
- J. N. Murrell and R. E. Mottram, Mol. Phys., 1990, 69, 571–585 CrossRef CAS.
- B. C. Curley, R. L. Johnston, N. P. Young, Z. Li, M. Di Vece, R. E. Palmer and A. L. Bleloch, J. Phys. Chem. C, 2007, 111, 17846–17851 CAS.
- R. Ferrando, A. Fortunelli and R. L. Johnston, Phys. Chem. Chem. Phys., 2008, 10, 640–649 RSC.
- O. Kostko, B. Huber, M. Moseler and B. von Issendorff, Phys. Rev. Lett., 2007, 98, 043401 CrossRef PubMed.
- F. Chen and R. L. Johnston, Acta Mater., 2008, 56, 2374–2380 CrossRef CAS.
- F. Y. Chen and R. L. Johnston, Appl. Phys. Lett., 2007, 90, 3123 Search PubMed.
- J. Jellinek, Faraday Discuss., 2008, 138, 11–35 RSC.
- L. B. Vilhelmsen and B. Hammer, J. Chem. Phys., 2014, 141, 044711 CrossRef PubMed.
- A. Sebetci, Comput. Mater. Sci., 2013, 78, 9–11 CrossRef CAS.
- A. Sebetci, Comput. Mater. Sci., 2012, 58, 77–86 CrossRef CAS.
- J. B. A. Davis, S. L. Horswell and R. L. Johnston, J. Phys. Chem. A, 2013, 118, 208–214 CrossRef PubMed.
- I. Demiroglu, D. Stradi, F. Illas and S. T. Bromley, J. Phys.: Condens. Matter, 2011, 23, 334215 CrossRef PubMed.
- M. Aslan, Z. Öztürk and A. Sebetci, J. Cluster Sci., 2014, 25, 1187–1201 CrossRef CAS.
- J. B. A. Davis, R. L. Johnston, L. Rubinovich and M. Polak, J. Chem. Phys., 2014, 141, 224307 CrossRef PubMed.
- C. Lueng, P. J. Metaxas and M. Kostylev, Conference on Optoelectronic and Microelectronic Materials and Devices (COMMAD), IEEE, 2014, pp. 27–29.
- A. Prasad and P. J. Cascone, U. S. Pat. No. 8,623,272, U.S. Patent and Trademark Office, Washington, DC, 2014 Search PubMed.
- S. Ramaprabhu, B. A. R. O. Mridula, P. Nayak and T. T. Baby, U. S. Pat., No. 9,149,833, U.S. Patent and Trademark Office, Washington, DC, 2015 Search PubMed.
- S. Akbayrak, M. Kaya, M. Volkan and S. Özkar, Appl. Catal., B, 2014, 147, 387–393 CrossRef CAS.
- K. Mech, G. Boczkal, P. Pałka, P. Zabiński and R. Kowalik, J. Solid State Electrochem., 2014, 18, 3121–3127 CrossRef CAS.
- B. Li, X. Liu, M. Zhu, Z. Wang, A. Adeyeye and W. Choi, J. Nanosci. Nanotechnol., 2015, 15, 4332–4338 CrossRef CAS PubMed.
- H. Yildirim, A. Kara and T. S. Rahman, J. Phys. Chem. C, 2012, 116, 281–291 CAS.
- H. Arslan, Int. J. Mod. Phys. C, 2008, 19, 1243–1255 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- D. T. Tran and R. L. Johnston, Proc. R. Soc. A, 2011, 467, 2004–2019 CrossRef CAS.
- S. Heiles and R. L. Johnston, Int. J. Quantum Chem., 2013, 113, 2091–2109 CrossRef CAS.
- F. Pittaway, L. O. Paz-Borbón, R. L. Johnston, H. Arslan, R. Ferrando, C. Mottet, G. Barcaro and A. Fortunelli, J. Phys. Chem. C, 2009, 113, 9141–9152 CAS.
- H. Cantera-López, J. M. Montejano-Carrizales, F. Aguilera-Granja and J. L. Morán-López, Eur. Phys. J. D, 2010, 57, 61–69 CrossRef.
- S. Bischoff, A. Weigt, K. Fujimoto and B. Lücke, J. Mol. Catal. A: Chem., 1995, 95, 259–268 CrossRef CAS.
- M. Heemeier, A. F. Carlsson, M. Naschitzki, M. Schmal, M. Bäumer and H.-J. Freund, Angew. Chem., Int. Ed., 2002, 41, 4073–4076 CrossRef CAS.
- F. B. Noronha, M. Schmal, C. Nicot, B. Moraweck and R. Frety, J. Catal., 1997, 168, 42–50 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- D. Deaven and K. Ho, Phys. Rev. Lett., 1995, 75, 288 CrossRef CAS PubMed.
- A. M. Rappe, K. M. Rabe, E. Kaxiras and J. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 1227 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- M. Methfessel and A. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616 CrossRef CAS.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. Van Dam, D. Wang, J. Nieplocha, E. Apra and T. L. Windus, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- A. Kant and B. Strauss, J. Chem. Phys., 1964, 41, 3806–3808 CrossRef CAS.
- L. M. Russon, S. A. Heidecke, M. K. Birke, J. Conceicao, M. D. Morse and P. Armentrout, J. Chem. Phys., 1994, 100, 4747–4755 CrossRef CAS.
- K. A. Gingerich, Faraday Symp. Chem. Soc., 1980, 14, 109–125 RSC.
- S. Taylor, E. M. Spain and M. D. Morse, J. Chem. Phys., 1990, 92, 2698–2709 CrossRef CAS.
- K. Koyasu, M. Mitsui, A. Nakajima and K. Kaya, Chem. Phys. Lett., 2002, 358, 224–230 CrossRef CAS.
- B. Sahu, G. Maofa and L. Kleinman, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 115420 CrossRef.
- C. Jamorski, A. Martinez, M. Castro and D. R. Salahub, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10905 CrossRef CAS.
- M. Castro, C. Jamorski and D. R. Salahub, Chem. Phys. Lett., 1997, 271, 133–142 CrossRef CAS.
- A. Sebetci, Chem. Phys., 2008, 354, 196–201 CrossRef CAS.
- M. Pereiro, D. Baldomir, M. Iglesias, C. Rosales and M. Castro, Int. J. Quantum Chem., 2001, 81, 422–430 CrossRef CAS.
- S. Datta, M. Kabir, S. Ganguly, B. Sanyal, T. Saha-Dasgupta and A. Mookerjee, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 014429 CrossRef.
- R. Van Zee, Y. Hamrick, S. Li and W. Weltner, Chem. Phys. Lett., 1992, 195, 214–220 CrossRef CAS.
- Q. M. Ma, Z. Xie, J. Wang, Y. Liu and Y. C. Li, Phys. Lett. A, 2006, 358, 289–296 CrossRef CAS.
- G. Qiu, M. Wang, G. Wang, X. Diao, D. Zhao, Z. Du and Y. Li, THEOCHEM, 2008, 861, 131–136 CrossRef CAS.
- M. Moseler, H. Häkkinen, R. N. Barnett and U. Landman, Phys. Rev. Lett., 2001, 86, 2545–2548 CrossRef CAS PubMed.
- Q.-M. Ma, Z. Xie, J. Wang, Y. Liu and Y.-C. Li, Phys. Lett. A, 2006, 358, 289–296 CrossRef CAS.
- S. Datta, M. Kabir, T. Saha-Dasgupta and A. Mookerjee, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 085418 CrossRef.
- J. Jalink, Metal clusters: from geometric to electronic and magnetic properties, Doctoral Thesis, Radboud University Nijmegen, Netherlands, 2014 Search PubMed.
- G. Zanti and D. Peeters, Eur. J. Inorg. Chem., 2009, 3904–3911 CrossRef CAS.
- P. Begum, D. Bhattacharjee, B. K. Mishra and R. C. Deka, Theor. Chem. Acc., 2014, 133, 1–11 CrossRef CAS.
- J. Moc, D. G. Musaev and K. Morokuma, J. Phys. Chem. A, 2003, 107, 4929–4939 CrossRef CAS.
- J. Rodríguez-López, F. Aguilera-Granja, K. Michaelian and A. Vega, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 174413 CrossRef.
- C. Kittel, Introduction to solid state physics, Wiley, 2005 Search PubMed.
- Q. L. Lu, L. Z. Zhu, L. Ma and G. H. Wang, Phys. Lett. A, 2006, 350, 258–262 CrossRef CAS.
- H. Arslan, A. K. Garip and R. L. Johnston, Phys. Chem. Chem. Phys., 2015, 42, 28311–28321 RSC.
- E. Janssens, T. Van Hoof, N. Veldeman, S. Neukermans, M. Hou and P. Lievens, Int. J. Mass Spectrom., 2006, 252, 38–46 CrossRef CAS.
- P. Nava, M. Sierka and R. Ahlrichs, Phys. Chem. Chem. Phys., 2003, 5, 3372–3381 RSC.
- C. Luo, C. Zhou, J. Wu, T. Dhilip Kumar, N. Balakrishnan, R. C. Forrey and H. Cheng, Int. J. Quantum Chem., 2007, 107, 1632–1641 CrossRef CAS.
- M. B. Knickelbein, J. Chem. Phys., 2006, 125, 44308 CrossRef PubMed.
- B. Kalita and R. C. Deka, J. Chem. Phys., 2007, 127, 244306 CrossRef PubMed.
- S. Yin, R. Moro, X. Xu and W. A. de Heer, Phys. Rev. Lett., 2007, 98, 113401 CrossRef PubMed.
- V. Bakken and O. Swang, J. Chem. Phys., 2008, 128, 084712 CrossRef PubMed.
- A. Shayeghi, D. Götz, J. Davis, R. Schaefer and R. L. Johnston, Phys. Chem. Chem. Phys., 2015, 17, 2104–2112 RSC.
- J. B. Davis, A. Shayeghi, S. L. Horswell and R. L. Johnston, Nanoscale, 2015, 7, 14032–14038 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp00342g |
| This journal is © the Owner Societies 2016 |