
An ab initio molecular dynamics study of D2 dissociation on CO-precovered Ru(0001)
Abstract
In dynamics calculations of H2 dissociating on metal surfaces often clean, high-symmetry surfaces are used. Few such dynamics studies have been performed on surfaces with pre-adsorbed molecules, especially when also the motion of the surface and the adsorbate are considered. In this study, the dissociation of H2 on a carbon monoxide-covered Ru(0001) surface is considered. Ab initio molecular dynamics (AIMD) calculations are performed on this system using the PBE-vdW-DF2 functional, which accurately describes the reaction probability for H2 on Ru(0001). Using this functional, the reaction probability of H2 on the CO-covered Ru(0001) surface is found to be too low when compared to experiments. This suggests that exchange–correlation functionals that can describe the reaction of H2 on a bare metal surface are not in general able to describe the reaction of H2 on a CO-precovered surface of the same metal, with the same accuracy. However, it cannot be ruled out that the discrepancy between theory and experiment is partly due to an inhomogeneous coverage of the surface by CO in the experiments. The incorporation of the motion of the surface has only a small effect on the reaction probability. It is found that when including surface motion for this system, the size of the simulation cell can be important. Upon collision, a considerable amount of energy is transferred to the surface, causing the adsorbed CO molecules to move apart, which opens the surface for reaction. In order to obtain converged reaction probabilities with respect to the size of the simulation cell, at least a 3 × 3 simulation cell is needed, because in the smaller 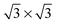 cell the CO molecules cannot be pushed apart as only a single independent CO molecule is present, also leading to less energy exchange with the surface.
cell the CO molecules cannot be pushed apart as only a single independent CO molecule is present, also leading to less energy exchange with the surface.

- This article is part of the themed collection: Developments in Density Functional Theory
 Please wait while we load your content...
Please wait while we load your content...

