
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Three steps to gold: mechanism of protein adsorption revealed by Brownian and molecular dynamics simulations†
M.
Ozboyaci
ab,
D. B.
Kokh
a and
R. C.
Wade
*acd
aHeidelberg Institute for Theoretical Studies (HITS), Schloss-Wolfsbrunnenweg 35, 69118 Heidelberg, Germany. E-mail: rebecca.wade@h-its.org
bHeidelberg Graduate School of Mathematical and Computational Methods for the Sciences, Heidelberg University, INF 205, 69120 Heidelberg, Germany
cZentrum für Molekulare Biologie der Universität Heidelberg, DKFZ-ZMBH Allianz, INF 282, 69120 Heidelberg, Germany
dInterdisciplinary Center for Scientific Computing, Heidelberg University, INF 205, 69120 Heidelberg, Germany
First published on 21st March 2016
Abstract
The addition of three N-terminal histidines to β-lactamase inhibitor protein was shown experimentally to increase its binding potency to an Au(111) surface substantially but the binding mechanism was not resolved. Here, we propose a complete adsorption mechanism for this fusion protein by means of a multi-scale simulation approach and free energy calculations. We find that adsorption is a three-step process: (i) recognition of the surface predominantly by the histidine fusion peptide and formation of an encounter complex facilitated by a reduced dielectric screening of water in the interfacial region, (ii) adsorption of the protein on the surface and adoption of a specific binding orientation, and (iii) adaptation of the protein structure on the metal surface accompanied by induced fit. We anticipate that the mechanistic features of protein adsorption to an Au(111) surface revealed here can be extended to other inorganic surfaces and proteins and will therefore aid the design of specific protein–surface interactions.
1. Introduction
The interactions of biomolecules with solid surfaces have attracted considerable attention during the past few decades due to their wide range of application in the fields of physics, chemistry and biology.1–10 Protein–gold interactions are one of the most widely studied types of protein–surface interaction due to gold's unique optical, magnetic and chemical properties.9,11–16 Applications of protein–gold interactions include biological imaging,12,17 drug delivery18,19 and biosensors,20,21 see reviews.22–24Extensive effort has been put into experimental investigation of the adsorption properties of proteins and peptides on gold.25–29 Whereas cysteine–gold bond formation has been studied extensively, the effects of weaker interactions on protein–gold binding are still poorly understood due to experimental limitations in revealing the binding dynamics, and the adsorption conformation and orientation of a protein/peptide. Identification of the forces governing the binding of proteins to gold surfaces is necessary for understanding and predicting protein adsorption selectivity and for estimating protein binding propensities. To date, several promising attempts to model protein–gold interaction forces and to simulate the adsorption process have been made.30–38 Quantum mechanics and molecular dynamics (MD) approaches are among those widely used for obtaining interaction parameters and for simulation of adsorption dynamics, respectively. Complete sampling of the very large configurational space of binding orientations and conformations of a biomolecule is, however, beyond the scope of current MD methods because of the time and size limitations of these methods. Brownian dynamics (BD) simulations provide an alternative approach for simulating adsorption on more realistic timescales. However, they require a continuum solvent force-field optimized specifically for protein–surface interactions. An atomic-detail protein–metal continuum solvent force-field, ProMetCS, was developed by Kokh et al.39 It has been validated against MD39 and experimental data25 on the binding of amino acids to Au(111) surfaces, and has recently been used in BD simulations of ubiquitin adsorption to a gold surface.40 In the present study, ProMetCS was used in BD simulations of the adsorption of a protein, treated as a rigid body, to an Au(111) surface to provide starting configurations for MD simulations with full treatment of protein flexibility.
The interactions of an Au(111) surface with β-lactamase inhibitor protein (BLIP), as well as several other proteins, have been experimentally investigated25 using surface plasmon resonance. This study demonstrated that BLIP, when fused with three histidine residues at its N terminus (3H-BLIP), adsorbs both on a gold surface and on gold nanoparticles with almost no observable dissociation and with a notably higher binding propensity than that of wild-type BLIP (wtBLIP). Increasing the N-terminal extension up to 6 histidine residues improved binding, indicating that the tag residues play an important role in binding. Upon binding to the gold surface, 3H-BLIP was found to lose its inhibitory activity against TEM1 beta-lactamase, which is strongly inhibited by binding of BLIP to its active site.25 This loss of inhibitory activity was speculated to be due to unfolding of the protein upon adsorption. Moreover, experimental observations25 suggest that the footprint of adsorbed 3H-BLIP on gold nanoparticles is larger than that of the 3H peptide, suggesting a possible spreading or unfolding event during adsorption. Fusions of the N-terminus of BLIP with 3 Trp, Lys, Met or Tyr residues also resulted in strong binding to Au nanoparticles without observable dissociation, indicating a similar binding mechanism for these fusion proteins. From these experiments, however, the binding mechanism itself is unclear, in particular how histidine residues at the N-terminus help the protein bind much more strongly than wtBLIP, how the spreading of the protein occurs, and why BLIP loses its inhibitory activity against TEM1 beta-lactamase.
Here, we report a detailed analysis of the adsorption of 3H-BLIP and wtBLIP onto an Au(111) surface based on atomic-detail computer simulations. To efficiently sample binding orientations, we first performed rigid body BD simulations of wtBLIP and 3H-BLIP with an Au(111) surface. Then, to monitor the close inter-atomic interactions in the binding interface and the structural changes of the proteins upon binding, we carried out explicit solvent, all-atom MD simulations of the proteins on the Au(111) surface with a full treatment of protein flexibility. GolP,32 a modified OPLS force-field for protein–Au(111) surface interactions, in which the Lennard-Jones (LJ) terms are modified to account for chemisorption and physisorption to the surface and a charged rod model41 is used to account for the polarization of the metal surface, was employed in the MD simulations.
2. Methods
2.1 Preparation of initial coordinates
An atomic-detail model of an uncharged Au(111) surface was used in the BD and MD simulations as described by Kokh et al.32,38,39 The crystal structure of wtBLIP (PDB ID: 3GMU42), which has a resolution of 1.98 Å, was used. To model the missing 3H residues in the 3H-BLIP structure, the Modeller program43 was used. The Automodel class of Modeller was employed to build the initial coordinates of the missing residues with the default parameters. After building 1000 structures, each of them was minimized using the conjugate-gradient method implemented in Modeller with a maximum of 1000 steps. Amongst all the structures built, the one with the largest flat surface patch on the 3H was chosen. The protonation states of the residues at pH 7 were determined using the web server H++.44 The total charges of the wtBLIP and 3H-BLIP proteins were −1 and 0, respectively. No unusual protonation states were observed in the structures except for two histidine residues, one in the protein and another in the 3H tag. The H++ server determined the terminal histidine residue of the 3H tail in 3H-BLIP to be positively charged and the other two histidine residues in the 3H tag to be neutral. Both of the neutral histidine residues were protonated at their Nε atoms. The protein has three other histidine residues apart from the 3H tail and only one of them (at position 44) was determined to be charged by H++.2.2 BD simulations
SDA (Simulation of Diffusional Association) software45,46 (version 6) was used to perform BD simulations. The ProMetCS model39 was used to compute the protein–metal interaction forces. In this model, the energy function consists of two major terms: electrostatic interaction energy (due to metal polarization) and non-polar interaction energy. The electrostatic interaction energy is composed of Coulombic interaction and electrostatic protein and metal desolvation terms. The non-polar interaction energy is composed of LJ (van der Waals and weak chemical interactions32) and non-polar desolvation terms. Since the structures were treated as rigid bodies, individual energy and potential components were precomputed and saved on three dimensional grids centered on the protein and surface structures. The LJ terms were computed for two different surface atom types and stored in two energy grids each of which had dimensions of 350 × 350 × 350 points with a regular spacing of 0.2 Å. In the ProMetCS model, an image charge approximation is used to account for metal polarization effects. In this approximation, the protein is represented by a set of effective charges that electrostatically interacts with its image across the surface. To compute the electrostatic potential grid, the UHBD program47 was used. The size of the grid was set to 250 × 250 × 250 points with a regular spacing of 1 Å. The solvent and the protein dielectric constants were chosen as 78 and 4 respectively. The ionic strength was set to 10 mM to correspond to the experimental conditions. The effective charges on the protein were computed using the effective charges for macromolecules in solvent (ECM) method.48The structures of the protein and the surface were treated as rigid bodies in the BD simulations. BLIP was experimentally characterized as almost inert to the Au(111) surface,25 indicating that the protein structure does not change significantly upon contact with the gold surface, and hence justifying the rigid body treatment of the BLIP structures in the BD simulations. The coordinates of the surface were fixed and the protein moved in a box of size 100 Å × 100 Å in x and y directions with one boundary (z = 0) fixed at the Au surface. The protein center was placed on the z = 70 Å plane at the start of the simulations. When the separation between the protein center and the surface exceeded 140 Å, the simulations were terminated. To speed-up the simulations, a variable timestep was used. The minimum timestep was chosen as 0.2 ps and when the distance between the interacting bodies exceeded 30 Å, the timestep was gradually increased. In the simulations, the translational and rotational diffusion constants of 3H- and wtBLIP were set to 0.0123 Å2 ps−1, and 1.36 × 10−4 rad2 ps−1, respectively.
2.3 MD simulations
The MD simulations were performed using the GROMACS software package49 with GolP,32 a modified OPLS50 force-field, to account for the Au(111) surface. The GolP force-field includes parameters for water–gold, ion–gold and protein–gold interactions, as well as the rest of the OPLS parameters. The LJ parameters used in the MD simulation are the same as used in the BD simulations. In the MD simulations, the metal polarization is represented by a charged rod model.41 The three binding orientations that were obtained from BD simulations were used as starting positions in MD simulations. Each of the three orientations was translated away from the surface by 3 Å or by 5 Å. The six starting configurations were put in simulation boxes of size 80 Å × 80 Å × 140 Å, where the surface slab occupied the volume given by x = 0–80 Å, y = 0–80 Å and z from −70 to +70 Å (see Fig. S1, ESI†). The distance between the surface and its periodic image was large enough for the interactions to be negligible. The boxes were then solvated using the SPC water model51 and ionized with Na+ and Cl− ions at 15 mM concentration. The BD simulations were performed at 10 mM ionic strength but control calculations at 15 mM ionic strength did not produce significantly different results.The structures were energy minimized using the following procedure. Initially, the surface and protein atoms were fixed, and the water molecules and ions were subjected to energy minimization using the steepest descent method for 200 steps. Then, keeping the surface fixed, the rest of the system was minimized for 500 steps using the steepest descent method and then another 500 steps using the conjugate gradient method. During the minimizations, the bonds were constrained using the LINCS algorithm.52 After the minimization, the system was heated using a Nose–Hoover thermostat53,54 gradually to 300 K over a period of 1 ns in 60 K steps of 200 ps each.
The MD simulations were started with three different initial assignments of velocities for each of the six configurations of each protein–surface system. The temperature was kept constant at 300 K using a Nose–Hoover thermostat. Since the polarizable surface model was assessed for constant volume simulations in the original work, all the simulations in this study were performed under constant volume. Each of the different components (water, protein, the surface and the ions) in the system was coupled to the thermostat separately. The time constant for coupling was set to 0.1 ps. All bonds were constrained using the LINCS algorithm. The simulations were run for 25 ns with a timestep of 2 fs.
Further, three more simulations were performed starting with the same three binding orientations obtained by BD simulations. The structures were translated 12 Å away from the surface. The systems were built, minimized and run with the same parameters as described above. The simulations were run for 250, 250 and 55 ns, respectively, for the first three ranked orientations obtained by the single linkage clustering method.
Finally, to assess changes in protein conformation in solution, one simulation of 3H-BLIP without any Au(111) surface was carried out for 25 ns. The simulation procedure was the same as described above. RMS fluctuations were computed for the time interval 10–25 ns for this simulation as well as for the longer MD simulations with the surface. The fluctuations for the rest of the longer MD simulations (25–250 ns) were computed for 30 ns time periods.
3. Results and discussion
3.1 BD simulations
BD simulations were used to generate trajectories of the protein diffusion and to reveal the transient binding orientations (hereafter referred to as “encounter complexes”) of 3H-BLIP and wtBLIP on the Au(111) surface. To extract the most populated binding orientations, the protein orientations relative to the gold surface obtained from the BD trajectories were clustered using the single linkage algorithm. See ESI† for the details of the algorithm and the assessment of the results.Clustering analysis of 3H-BLIP encounter complexes resulted in three distinct binding orientations, see Fig. 1 and Table S1 (ESI†). The representative of the first, i.e. the most energetically favorable, cluster (orientation a) does not have a close interaction of the 3H residues with the surface. Instead, 3H-BLIP binds to the gold surface with a large number of residues on the opposite side of BLIP making favorable LJ interactions. Indeed, together with the nonpolar surface desolvation terms, which are roughly proportional to the protein–gold contact area, the non-polar interaction term (−126.3 kJ mol−1) provides the leading interaction component. The total electrostatic energy (−20.1 kJ mol−1) contributes rather little to binding (Table S1, ESI†). On the other hand, both the second and the third cluster representatives (orientations b and c) show strong interactions of the 3H residues with the surface and no direct contact of the BLIP protein with gold. The electrostatic interactions account for 38% and 66% of the total interaction energies for orientations b and c, respectively (see Table S1, ESI†). This result suggests that the binding of 3H-BLIP by its 3H tag is driven, not only by the LJ interactions, but also by favorable electrostatic interactions, consistent with one of the histidines in the 3H tag being doubly protonated. Noteworthy is that the second cluster constitutes 99% of all the encounter complexes obtained in the docking simulations, thus demonstrating that 3H-BLIP recognizes the gold surface by its 3H residues rather than by the globular part of 3H-BLIP. Even though the first ranked cluster is energetically more favorable, it is much less accessible than the second ranked cluster during the BD simulations.
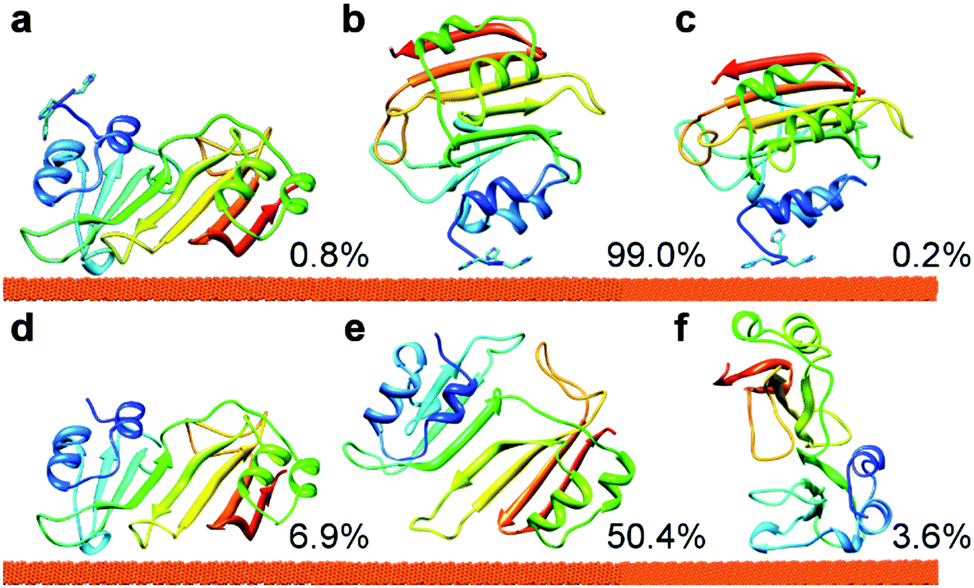 | ||
| Fig. 1 The binding orientations of 3H-BLIP (a–c) and wtBLIP (d–f) to the Au(111) surface obtained after clustering of the encounter complexes from BD simulations. Clustering of the 3H-BLIP encounter complexes yielded only three distinct protein binding orientations, whereas clustering of wtBLIP encounter complexes resulted in 10 distinct clusters. The clusters were, then, ranked by their mean total interaction energies, see Table S1 (ESI†). Along with their populations (given in %), the representatives of the three energetically most favorable clusters (1, 2 and 3) are shown for 3H-BLIP, in panels a, b and c, respectively and for wtBLIP, in panels d, e and f respectively. The protein is shown in cartoon representation, colored from N (blue) to C (red) terminus, with the N-terminal 3H tag shown with side-chains. | ||
In contrast to 3H-BLIP, wtBLIP binds to the gold surface in many different orientations, most of which have a small population, except for the second cluster (see Fig. S2 and Table S1 in ESI,† for the three energetically most favorable clusters). Orientation d (see Fig. 1) shows a much larger favorable mean interaction energy (−158.3 kJ mol−1) than the other clusters (−60.2 kJ mol−1 and −52.4 kJ mol−1 for orientations e and f, respectively). As seen in Fig. 1, orientation d corresponds to orientation a of 3H-BLIP obtained for the most energetically favorable cluster for 3H-BLIP. The remaining clusters have relatively smaller interaction energies, suggesting that wtBLIP has no pronounced specific binding site for the gold surface except that used in orientation d.
3.2 Potential of mean force (PMF) calculations
We calculated the PMFs of 3H-BLIP and wtBLIP along the reaction coordinate defined by the distance between the center of the protein and the center of atoms on the top layer of the gold slab (see Fig. 2). The PMF analysis not only supports the results from clustering analysis of the BD simulations, but also reveals two important points about the adsorption mechanism. First, strong adsorption of wtBLIP is hindered by a high energy barrier (60 kJ mol−1) caused mainly by the cost of replacing the water on the surface. Secondly, the energetically very favorable orientation achieved at a distance of 26 Å away from the surface by 3H-BLIP is the first step on the path towards the final adsorption of 3H-BLIP. The PMF was computed with a rigid body treatment but, in fact, the flexibility of 3H-BLIP could allow reorientation while the 3H-tail remains anchored on the surface. We therefore, next performed MD simulations with 3H-BLIP and wtBLIP treated as fully flexible. | ||
| Fig. 2 Potential of mean force (PMF) profiles of 3H-BLIP (red) and wtBLIP (blue). The orientations corresponding to the energy minima are shown for 3H-BLIP and wtBLIP below and above the graph, respectively. For 3H-BLIP, there are two energy wells with values of −112 kJ mol−1 and −68 kJ mol−1 and they correspond to the clusters obtained from BD simulations. For wtBLIP, on the other hand, there is one deep well with a minimum at −63 kJ mol−1 and several other shallow energy wells with minima above −26 kJ mol−1. The deepest energy well corresponds to the cluster with the representative orientation d and it is energetically and geometrically close to that of 3H-BLIP orientation a. The uncertainty due to the discretization along the z axis perpendicular to the gold surface, and the different definitions of the bound states is approximately 2.5 kJ mol−1. | ||
3.3 Dynamics of BLIP binding to the gold surface in 25 ns MD simulations
For both 3H-BLIP and wtBLIP, we performed 18 MD simulations starting from three different orientations of the protein relative to the gold surface. The initial orientations correspond to the representatives of the six clusters from the BD simulations: orientations a, b and c for 3H-BLIP and d, e and f for wtBLIP (see Fig. 1 and Table S1, ESI†). For each of the six binding orientations, two different sets of initial protein coordinates were generated by translating the protein by 3 Å and by 5 Å away from the surface to enable protein reorientation and rearrangements of the side chains near the surface. For each starting configuration, 3 independent simulations were performed with different starting velocities assigned. Each of the 36 systems was simulated for 25 ns.In the simulations started with orientations a, b, and c, the 3H-BLIP structures adsorbed to the gold surface during the first few ns of the simulations and no unbinding was observed except in one of the six simulations started with orientation c in which no adsorption occurred. In simulations started with orientation a, the structures were bound through BLIP and the interaction energies were dominated by the LJ term (Fig. S3, ESI†). Noteworthy is that in one of these simulations that started with orientation a, the 3H tag also adsorbed to the gold surface along with BLIP. In the simulations started with orientations b and c, on the other hand, adsorption was driven by the 3H tag and the electrostatic terms were more significant in the interaction energies owing to the +2e charge on the 3H tag. Contributions from the LJ term dominated the overall interaction energies later as the 3H tag adsorbed along with neighboring residues to the surface. Importantly, once adsorbed, the 3H tag stayed bound throughout the rest of the simulation even though the BLIP structure adopted different binding orientations in different simulations.
In all of the simulations started with orientations d and f, adsorption of wtBLIP structures took place and no unbinding occurred until the end of the simulations. On the other hand, in only two of the six simulations started with orientation e did wtBLIP adsorb to the surface. Simulations starting from orientation d, which is geometrically the closest to orientation a, resulted in the most favorable interaction energies with a dominant LJ term compared to those of orientations e and f (see Fig. S3, ESI†).
These relatively short MD simulations give useful insights into the early stages of adsorption. The most common binding orientation observed in BD simulations of wtBLIP does not form close contacts and moves away from the surface in most of the simulations. This suggests a role for the 3H tag in stabilizing the encounter complex in the early binding stage. Further, clustering of the 3H-BLIP and wtBLIP simulations shows that the protein adopts many different binding orientations (see Fig. 3). Importantly, orientations a and d kept their positions throughout the simulations regardless of whether they had a 3H tag to contact the surface. We conclude that the main contribution to the strength of binding of 3H-BLIP comes from the interactions of BLIP with the gold surface. To obtain a more complete picture of the mechanism of adsorption of 3H-BLIP on the gold surface, longer simulations were performed.
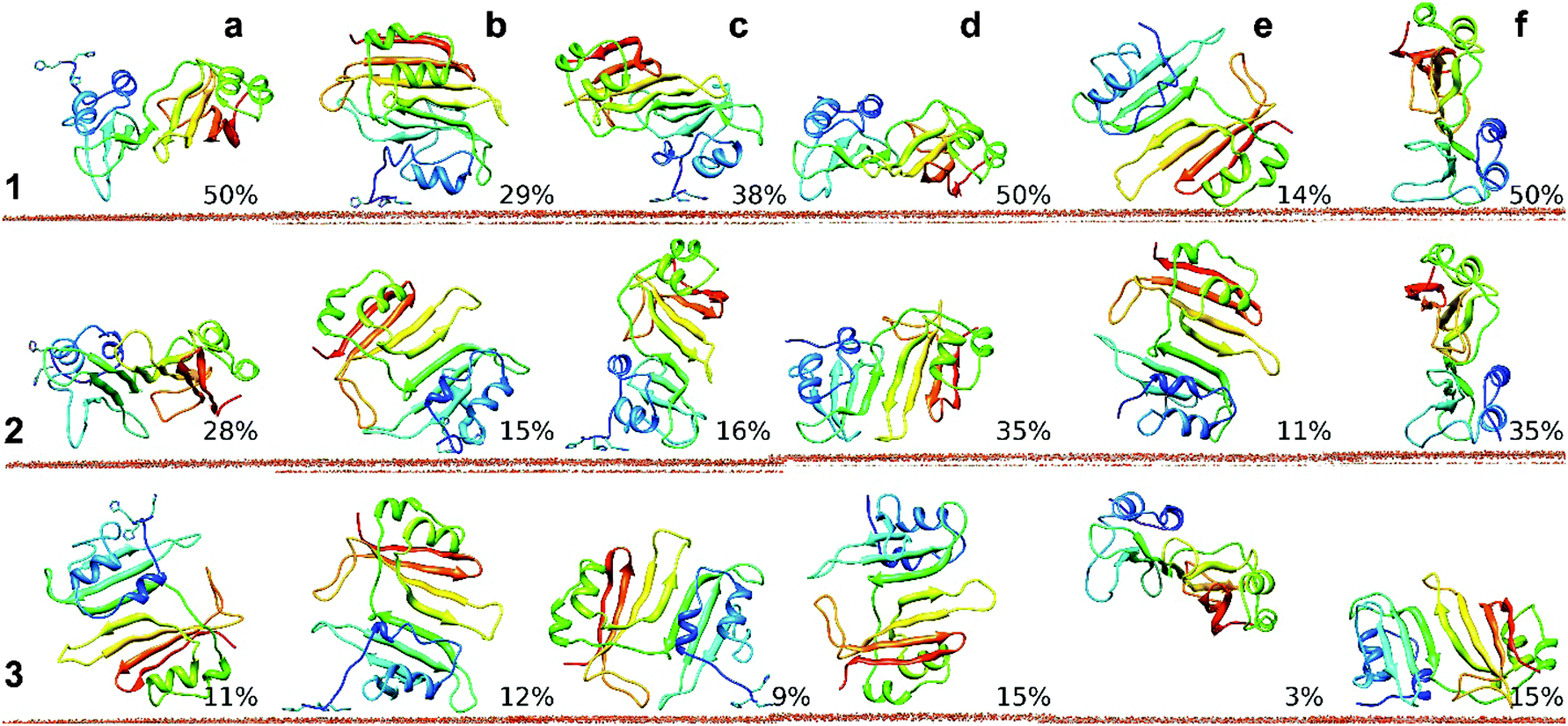 | ||
| Fig. 3 Representatives of the most populated clusters from analysis of the MD simulations (each of 25 ns duration) for 3H-BLIP and wtBLIP binding to the Au(111) surface. Simulations of 3H-BLIP started with orientations a, b and c (a, b, c) (see Table S1, ESI†) and of wtBLIP started with orientations d, e and f (d, e, f) were clustered and ranked by their population sizes. Representatives of the three most populated clusters (1, 2 and 3) along with their populations are depicted for each of the six starting orientations. Clustering of the trajectories for each of the 6 simulation sets showed that many distinct binding orientations were achieved. Amongst all orientations that simulations started with, a and d are the most well preserved ones. The representatives of the largest clusters from the simulations started with orientations a and d showed very close orientations to their corresponding starting orientations with RMSD values of 3.9 and 5.3 Å respectively. | ||
3.4 Longer time scale dynamics of 3H-BLIP on the gold surface
To investigate the binding mechanism in more detail, we performed several longer MD simulations of 3H-BLIP with the Au(111) surface, starting with the orientations shown in Fig. 1. The structures were translated 12 Å away from the surface, thereby allowing the protein and the surface to interact only through long range electrostatic interactions at the beginning of the simulations. The simulations were carried out for 250 ns, 250 ns and 55 ns for orientations a, b and c respectively. Since the simulation that started with orientation c resulted in diffusion of 3H-BLIP away from the surface and no binding was observed during the simulation time, we did not extend the simulation beyond 55 ns. This simulation will not be discussed further.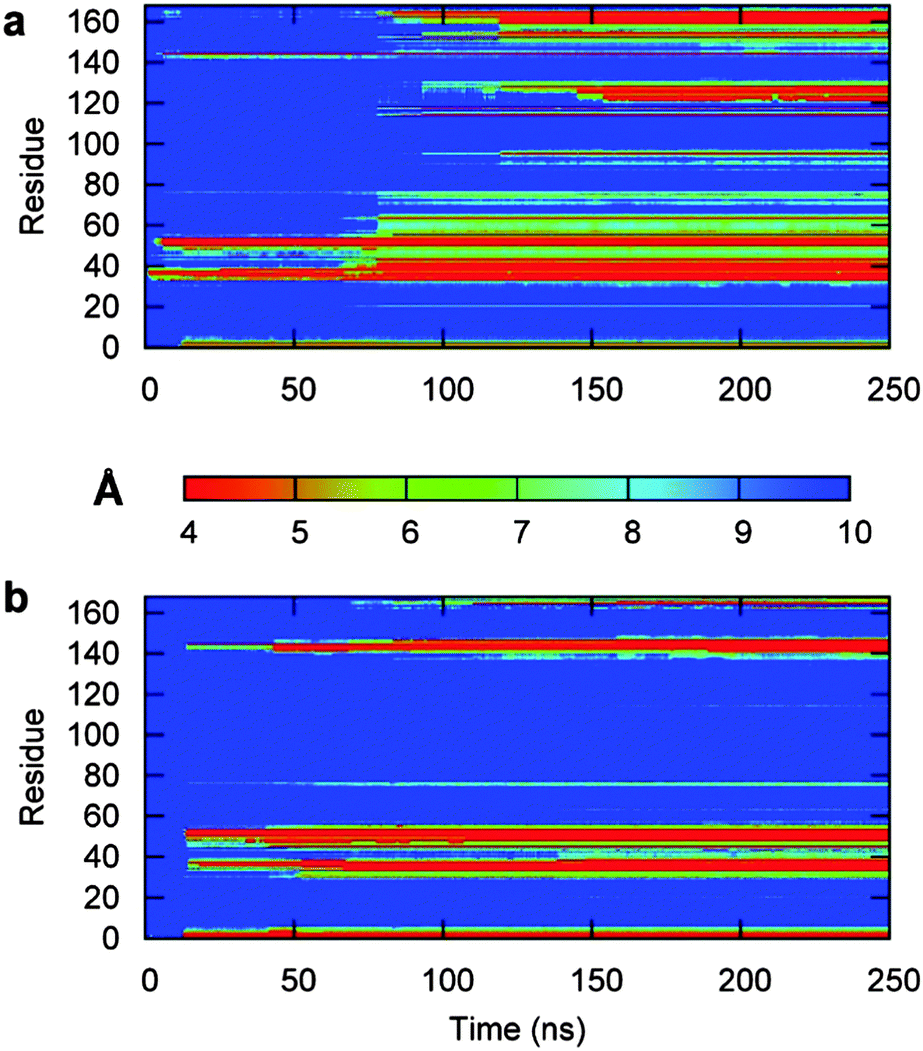 | ||
| Fig. 4 3H-BLIP residue–gold surface distance maps obtained from the two 250 ns long MD simulations started with orientation a (a) and b (b). Red regions indicate close interactions (4 Å), whereas the blue regions indicate no contacts (separation of 10 Å or more). (a) Starting with orientation a, the protein forms initial contacts with the metal surface within the first two ns through the side chains of only two BLIP residues. During the following 4 ns, the number of BLIP residues involved in tight binding slightly increases to 6–8. No unbinding occurs during this stage and the contacts are kept throughout the simulation. Between the 11th and 12th ns of the simulation, the 3H tag binds tightly to the surface. The protein maintains the interactions already formed but makes no further stable interactions with the surface until about the 80th ns. (b) Starting with orientation b, the protein moves to within 6 Å of the surface in the first ns. It approaches with its 3H tag closest to the surface during the first 2 ns. However, the protein then drifts away from the surface after 2.5 ns and, during the next 10 ns, the globular part of the protein does not form any close interactions with the surface. From 4.5 to 8.5 ns, the N-terminus is extended towards the surface and the residues appear to interact strongly with the surface. The N-terminus escapes from the surface during the next 2 ns and the protein changes its orientation by an RMSD of 12 Å. At about 10.5 ns, the protein starts to approach towards the surface again and at 12.5 ns, very close contacts are formed by the 3H tag. It takes another ns for the rest of the structure to start to contact the surface. After the first 13.5 ns, no unbinding occurs. | ||
As the simulation proceeded, 3H-BLIP started to undergo an induced fit on the surface (see Fig. 4a and 5a), mostly through side chains on its beta-strands without any significant changes in its secondary structure (see Fig. S4a, ESI†). Examination of the binding interface of 3H-BLIP within a distance of 6 Å of the surface (see Fig. 6a) shows that many of the side chains lie parallel to the surface. The loss of energetically favorable water–metal interactions due to the large binding area is compensated by the favorable non-polar desolvation term for the protein surface and the large number of aromatic side chains that form favorable pi-metal interactions with the gold surface. The aromatic residues at the binding interface undergo spreading on the surface, showing the need for reorientation of the side chains to adsorb fully on the surface.
 | ||
| Fig. 5 Changes in protein–surface contacts and interaction energy components during the two 250 ns-long MD simulations of 3H-BLIP and the Au(111) surface started with orientations a (a) and b (b). The time evolution of the number of the gold atoms within 6 Å of the protein atoms (blue), as well as the LJ (red) and the Coulomb (green) interaction terms, are shown. The number of surface contacts increased steeply (step-wise) after the 80th ns in the simulation that started with orientation a, corresponding to formation of an encounter complex, and continued to increase slowly up to about 200 ns. In the simulation that started with orientation b, on the other hand, the number of surface contacts increased rather steeply for about 45 ns to formation of an encounter complex and then gradually with convergence only starting after 200 ns. The change can be seen in the interaction energies, as both the increase in the number of contacts were accompanied by favorable changes in the LJ terms and, to a lesser extent, the Coulombic interaction terms. Upon binding of the 3H-BLIP structures, the electrostatic and LJ interactions with the surface become more favorable. However, it is the LJ term that dominates the adsorption energies in the simulations, once the protein structures are bound to the surface. | ||
 | ||
| Fig. 6 The Au(111) binding surface of 3H-BLIP obtained from the two 250 ns long MD simulations started with orientation a (a) and b (b). The residues within 7 Å of the surface are shown in licorice representation and colored according to their types: polar (green), nonpolar (yellow), positively (blue) and negatively (red) charged. Side-on views of the corresponding binding orientations of 3H-BLIP colored from N (blue) to C (red) terminus with the 3H tag in licorice representation (pink) are shown in the insets. The orientations (shown in the insets of panels a and b) differ by an angle of about 30° computed between the two planes obtained by least square plane fitting of the backbone atoms of the two orientations from MD simulations. In the simulation started with orientation a, His, Phe, Tyr and Trp residues at positions 1, 3 and 44; 39 and 145; 53, 54, 56, 118 and 122; and 115, 153 and 165, respectively, formed stable interactions upon adsorption and retained their binding positions until the end of the simulation. In the simulation started with orientation b, the His1, His2, His3, Phe39, Tyr53, Tyr54, Phe145, Tyr146 and Trp165 residues formed stable interactions with the surface upon adsorption. Tyr56 and His44, on the other hand, in the same simulation, formed looser interactions with fewer atoms involved. Apart from the aromatic residues, there are charged residues in both of the binding interfaces. In the simulation started with orientation a, the residues Arg46 and Glu34 (panel a), and in the simulation started with orientation b, the residues Arg147 and Asp166, as well as Arg46 and Glu34 (panel b) form salt bridges and interact with the surface. Many other relatively less stable interactions are also formed during the simulations. | ||
The interaction energy decomposition for the simulation shown in Table S2 (ESI†) suggests that the binding of 3H-BLIP is initiated by both LJ and electrostatic terms. The residues involved in close interactions with the surface are depicted in Fig. 6b. The binding interface shows that many of the adsorbed 3H-BLIP residues are the same as those adsorbed in the simulation starting from orientation a and that the aromatic residues in the protein structure contribute to the interaction energy significantly (see Fig. S5b, ESI†).
As shown in Fig. 6a and b, both of the 250 ns-long simulations result in the protein binding to the surface with approximately the same side of the protein. In the simulation that started with orientation b, the initial interactions with the metal surface are formed through the 3H peptide and then the protein adopts its final orientation with the globular part of the protein binding the surface. The order of events is reversed in the simulation started with orientation a: first, interactions were formed through the globular BLIP part of the protein and then the 3H terminus reoriented and adsorbed to the surface during the first 13 ns of the simulation. In both simulations, the 3H tag demonstrates an anchoring role.
We hypothesize that the 3H tag helps to decrease the dissociation of the complex by prolonging the residence time of the encounter complex because of its high affinity for the gold surface and the high flexibility of the poly-histidine chain. Further, the results show that, upon adsorption, the protein undergoes an induced fit but that this does not significantly perturb the secondary or tertiary structure. Binding is stabilized by the interactions of hydrophobic and aromatic side chains with the protein surface. The contribution of the aromatic residues with a value of −600 kJ mol−1 to the total LJ interaction term constitutes almost half of this term (see Fig. 5 and Fig. S5a, ESI†) at the end of the simulation started with orientation a. The total accessible surface area of these aromatic residues (see Fig. S6, ESI†) increases from about 1000 Å2 in the crystal structure to 1500 Å2 for the final structure from the MD simulation, demonstrating that the aromatic residues play a significant role in adsorption of BLIP. The final orientations of the adsorbed proteins also reveal that the gold binding site of the protein overlaps with the TEM1 binding site (see Fig. S7, ESI†), thereby explaining the experimentally observed loss of activity of 3H-BLIP towards TEM1 upon adsorption.25
3.5 Role of 3H tag in the recognition and binding mechanism
The 250 ns-long simulation of 3H-BLIP that started with orientation b shows that the orientations of the 3H residues before and after its binding promote the recognition and adsorption of the protein. Shown in Fig. 7, the orientation of the 3H may help to keep a close interaction of the peptide with the surface while perturbing the hydration shell on the metal surface minimally. The fluctuations of the angles that the 3H side chain rings make with the gold surface during this period decreases significantly once the 3H peptide is bound on the surface. After binding, two of the 3H histidines undergo parallel adsorption with the ring facing the gold surface, hence strengthening the interactions of the 3H side chains with the gold surface atoms. Noteworthy is that the 3H peptide does not unbind from the surface even though the rest of the protein is not adsorbed for a further 1 ns.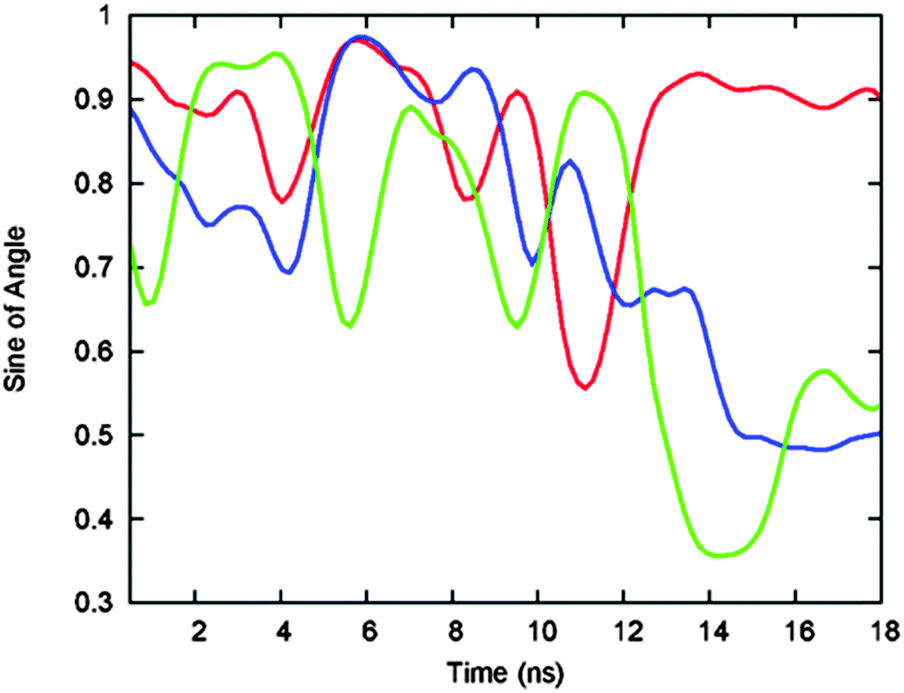 | ||
| Fig. 7 Time evolution of the sine of the planar angle between each of the side chain rings of His1 (red), His2 (blue) and His3 (green) of the 3H tag of 3H-BLIP and the surface during the beginning of the 250 ns-long MD simulation started with orientation b. The orientation of these residues during the first 10 ns before binding is such that at least one of the imidazole groups tends to stay perpendicular to the surface. Upon binding of the 3H tag to the surface, the fluctuations of the angles tend to decrease and the rings of two of the histidines lay parallel to the surface. | ||
The 250 ns-long simulation that started with orientation a, on the other hand, shows weak binding of BLIP to the surface before its 3H tag. Although the 3H tag is, in the beginning of the simulations, on the opposite side of the 3H-BLIP structure from the surface, it binds to the surface during the first 15 ns and stays bound throughout the rest of the simulation. Due to the flexibility of the N-terminus, the binding of the 3H tag does not require the protein to undergo a significant change in its orientation. The 3H peptide thereby plays an important role in the recognition (orientation b), and anchoring (orientations a and b) of the protein.
3.6 Role of the interfacial dielectric permittivity
Water has a high polarizability that shields electrostatic interactions between solutes. A recent study55 showed that the dielectric constant of water is anisotropic between two associating proteins. The dielectric constant was found to be lower in the direction of association in the interface region, resulting in enhanced electrostatic interactions compared to bulk solvent. This behavior was explained by the presence of an adhesive hydrogen bond network in the interfacial gap.In our protein–metal surface MD simulations, water forms three layers on the metal surface that are more structured than bulk water (see Fig. S8, ESI†). As discussed by Penna et al.56 the orientational ordering of water in the interface causes formation of charged layers which we also observe in our simulations. They hypothesized that the dipole alignment arising from the charged layers is responsible for the biased diffusion that was observed in their study and by Hoefling et al.34 To investigate the role of the water as a mediator in the adsorption process further, we computed the tensor of the dielectric permittivity in the interfacial region between the surface and the protein as described by Ahmad et al.55 (see Methods S5 in ESI†). As shown in Fig. S9 (ESI†), the permittivity of the interfacial water with at least two structured layers in the z-direction is approximately 4% lower than that of bulk water. Therefore, the effect of an electrostatic field in the direction of association is slightly enhanced near to the surface, strengthening initial protein–surface electrostatic interactions and facilitating the recognition of the surface. In summary, our results suggest that, in addition to the charged layers from the water on the surface, the reduced permittivity of the interfacial water in the direction of the adsorption contributes to the biased diffusion of a protein/peptide from the bulk water to the adsorption interface.
3.7 Internal protein motions upon binding the Au(111) surface
Relatively small fluctuations of the free 3H-BLIP suggest that the tagged protein is not very flexible except for two short loop regions around the 38th and 122nd residues. In the simulation that started with orientation a, BLIP–surface interactions upon binding, although strong, allow the protein to search for an energetically better conformation with good LJ interactions between the globular part of the protein and the surface suggested by its RMS fluctuations decreasing from 2.4 Å to 0.5 Å, and from 4.5 Å to 2.3 Å around each of the two flexible loop regions, respectively (see Fig. S10, ESI†). The fluctuations overall dampened towards the end of the simulation as the number of contacts with the surface grew, supporting the observation of induced fit to a stable structure in this simulation.Surprisingly, the 3H tag fluctuates less (∼1 Å) in the simulation without the surface than in the other two simulations during the corresponding time intervals (∼3 Å). This observation suggests that the 3H tag preferentially maintains a stable position next to the rest of the protein structure and that external forces, e.g. from a surface, are required for it to extend outwards.
4. Summary and concluding discussion
We have studied the adsorption of 3H-BLIP and wtBLIP on an Au(111) surface using continuum solvent BD and explicit solvent MD simulations. Our results imply that the adsorption of 3H-BLIP is a three-step process: (i) recognition of the surface mostly by the 3H tag through electrostatic interactions and formation of an encounter complex with the 3H tag; (ii) weak adsorption of 3H-BLIP onto the gold surface and achievement of a single, well-defined orientation; and (iii) induced fit of 3H-BLIP on the gold surface by spreading of its sidechains and loop regions on the surface. In the first step, the BD simulations show that the protein is more likely to form interactions through the 3H tag, but binding of BLIP itself or both BLIP and 3H is possible. When BLIP binds first, the binding mode can be stabilized by subsequent anchoring by the 3H tag, as seen in the MD simulations. Our results show a directionally reduced relative permittivity in addition to the ordered charged layers caused by the orientational ordering of the solvent in the binding region, which, together, enhance the recognition due to electrostatic interactions, particularly of the 3H tag. The first step of adsorption therefore shares similarities with protein–protein binding events57,58 that are driven by long-range electrostatics to form diffusional encounter complexes, even though the electrostatic interactions are rather short-range in the case of BLIP. However, long-range electrostatics will definitely play a role in the adsorption of proteins that are highly charged on to polarizable surfaces and this will be mediated by the interfacial water. Wang and colleagues59 proposed a three-step model for protein adsorption on Au nanoparticles. The “hardening” of the proteins via cysteine chemisorption was, however, described as the third step in their model with the first and second being recognition and reorientation steps, respectively. Thus, it is reasonable to expect that the mechanism we have observed here for BLIP is also relevant for the initial recognition process in the binding of proteins that undergo irreversible chemisorption to gold.The results presented here show that the 3H tag also plays a critical role by increasing the lifetime of the encounter complex in the second step and this helps the protein adopt a defined orientation on the surface. This, in turn is achieved by flexibility of the N-terminus of the protein including the 3H that allows the rest of the protein to change its orientation easily to a more energetically favorable arrangement. In the last step of the adsorption mechanism, the protein undergoes an induced fit, entailing rearrangements of the loop regions and of the side chains on the binding interface. Upon full adsorption, 3H-BLIP exhibits a very large interaction energy with the surface and its binding site mainly consists of aromatic and hydrophobic residues. The interactions are thus predominantly due to weak chemical and van der Waals interactions and not due to favorable electrostatic interactions as speculated by Cohavi et al.25 No unfolding was observed in any of the simulations, although the simulations revealed an induced fit of the protein upon adsorption that might explain the almost irreversible nature of the binding. Further, our results show that the binding site of BLIP to the TEM1 beta-lactamase overlaps with the BLIP binding site to the gold surface, thereby explaining the experimental observation25 that the activity of BLIP towards TEM1 beta-lactamase is lost after adsorption.
Adsorption experiments for wtBLIP and a 3H peptide to Au(111) surfaces have shown that wtBLIP has weak binding potency to the gold surface, in contrast to the binding of 3H-BLIP25 although binding of both proteins is almost irreversible. On the other hand, the binding of the 3H peptide to the surface is reversible. Therefore, the strong irreversible binding of 3H-BLIP to the gold surface cannot only be explained by the high gold binding propensity of histidines. It is clear that the 3H peptide facilitates the adsorption to the gold surface. As the wtBLIP does not have the 3H peptide to facilitate recognition, the relative propensity of binding of BLIP is much smaller. However, it is also partially the second and the third steps in the binding process that cause weaker binding of wtBLIP compared to 3H-BLIP. To adsorb strongly, as discussed above, BLIP requires a single orientation to form strong interactions and many side chain reorientations to adsorb by induced fit. We surmise that wtBLIP, due to the lack of an anchor peptide, is not able to sufficiently expose its strong binding residues to the surface, hence prohibiting a strong adsorption.
Since no equilibrium constant or adsorption free energy could be obtained from experimental studies,25 we cannot directly compare the binding free energy calculations we performed in this study. However, to compare to the association constants obtained in that same study, we inspected the times required to form an encounter complex with the surface in BD simulations. Our results (data not shown) did not show a significant difference between the time required for formation of an initial encounter of the 3H-BLIP and wtBLIP proteins on the surface. This suggests that a simple two stage Langmuir model, as used previously25 may be inadequate in accounting for the kinetics of adsorption of wtBLIP and 3H-BLIP. Thus, these results support our notion of a more complicated binding mechanism.
Simulations of ubiquitin40 and fibronectin domains34 binding to gold surfaces also showed the tendency for planar protein side chains to reorient to lie flat on the protein surface. The ability of flexible tails and loops in proteins to bind to gold surfaces seems to require that they contain residues with high gold-binding propensities, such as His. In agreement with this, experimental and computational studies40,60 of ubiquitin suggest that its N-terminal Met residue adsorbs to gold nanoparticles, whereas the long flexible C-terminus does not play an important role in the bound state.
Of note is that the role of the water and a flexible peptide sequence in the structure of the protein is similar to that of the peptide/surface adsorption described by Penna et al.56 Terminal residues with higher affinities towards the surface of interest play anchor roles in both cases and provide the rest of the peptide/protein sufficient time to adsorb with a lockdown mechanism.56 Hence, using the results presented here, a roadmap can be drawn for designing proteins with desired binding selectivity and propensity for an Au(111) surface. Adding a flexible peptide tag that consists of a specific number and type of amino-acid residues to any protein can be a first step to obtain surface binding. A second step is to design proteins that can undergo the desired extent of structural change upon binding with the required number of residues that can make flat sidechain interactions (His, Arg, Phe) at the interface.
Here, we have focused on the adsorption mechanisms of 3H-BLIP and wtBLIP to an Au(111) surface. However, many of the determinants of BLIP–surface binding, such as the dynamics of the water in the interfacial region, the crystalline structure of the surface, the accessibility of strongly surface-binding residues, and the flexibility of the protein structure, can be expected to be of importance for similar, non-covalently interacting, protein–surface systems. The determinants emerge from the properties of the major partners (i.e. the surface, the solvent and the protein) in the binding process and the interactions between them. To derive a complete set of rules governing the binding of proteins to gold or other metal surfaces, studies of further proteins will be necessary but we anticipate that many of the features of protein–surface adsorption observed here will be general.
Acknowledgements
The authors thank Bingding Huang for the script for calculating the flat surface patches on proteins in the SDA 6 software package. Financial support for this work was provided by HGS MathComp (University of Heidelberg Graduate School of Mathematical and Computational Methods for the Sciences), the Klaus Tschira Foundation, and the European Union (FP6 NEST project no. 028331-Prosurf). We acknowledge the support of Heidelberg University for open access publication of this article.References
- J.-W. Choi, J. S. Kim, S.-U. Kim and J. Min, BioChip J., 2009, 3, 157–163 Search PubMed.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS PubMed.
- D. R. Jackson, S. Omanovic and S. G. Roscoe, Langmuir, 2000, 16, 5449–5457 CrossRef CAS.
- S. Laera, G. Ceccone, F. Rossi, D. Gilliland, R. Hussain, G. Siligardi and L. Calzolai, Nano Lett., 2011, 11, 4480–4484 CrossRef CAS PubMed.
- L. Qin, M. J. Banholzer, J. E. Millstone and C. A. Mirkin, Nano Lett., 2007, 7, 3849–3853 CrossRef CAS PubMed.
- Y. Hu, A. Das, M. H. Hecht and G. Scoles, Langmuir, 2005, 21, 9103–9109 CrossRef CAS PubMed.
- Y. Xu, Q. Cao, F. Svec and J. M. J. Fréchet, Anal. Chem., 2010, 82, 3352–3358 CrossRef CAS PubMed.
- H. D. Hill, R. A. Vega and C. A. Mirkin, Anal. Chem., 2007, 79, 9218–9223 CrossRef PubMed.
- J. M. Slocik, A. O. Govorov and R. R. Naik, Nano Lett., 2011, 11, 701–705 CrossRef CAS PubMed.
- D. Millo, M.-E. Pandelia, T. Utesch, N. Wisitruangsakul, M. A. Mroginski, W. Lubitz, P. Hildebrandt and I. Zebger, J. Phys. Chem. B, 2009, 113, 15344–15351 CrossRef CAS PubMed.
- Q. Wei, H.-M. Song, A. P. Leonov, J. A. Hale, D. Oh, Q. K. Ong, K. Ritchie and A. Wei, J. Am. Chem. Soc., 2009, 131, 9728–9734 CrossRef CAS PubMed.
- J. Yguerabide and E. E. Yguerabide, J. Cell. Biochem., 2001, 84, 71–81 CrossRef PubMed.
- K. C. Grabar, R. G. Freeman, M. B. Hommer and M. J. Natan, Anal. Chem., 1995, 67, 735–743 CrossRef CAS.
- S. H. D. P. Lacerda, J. J. Park, C. Meuse, D. Pristinski, M. L. Becker, A. Karim and J. F. Douglas, ACS Nano, 2010, 4, 365–379 CrossRef PubMed.
- R. Shukla, V. Bansal, M. Chaudhary, A. Basu, R. R. Bhonde and M. Sastry, Langmuir, 2005, 21, 10644–10654 CrossRef CAS PubMed.
- L. R. Ditzler, A. Sen, M. J. Gannon, A. Kohen and A. V. Tivanski, J. Am. Chem. Soc., 2011, 133, 13284–13287 CrossRef CAS PubMed.
- Y.-S. Chen, W. Frey, S. Kim, P. Kruizinga, K. Homan and S. Emelianov, Nano Lett., 2011, 11, 348–354 CrossRef CAS PubMed.
- P. Mukherjee, Clin. Cancer Res., 2005, 11, 3530–3534 CrossRef CAS PubMed.
- R. Bhattacharya, C. R. Patra, R. Verma, S. Kumar, P. R. Greipp and P. Mukherjee, Adv. Mater., 2007, 19, 711–716 CrossRef CAS.
- T. J. Park, S. Y. Lee, S. J. Lee, J. P. Park, K. S. Yang, K.-B. Lee, S. Ko, J. B. Park, T. Kim, S. K. Kim, Y. B. Shin, B. H. Chung, S.-J. Ku, D. H. Kim and I. S. Choi, Anal. Chem., 2006, 78, 7197–7205 CrossRef CAS PubMed.
- I. Maier, M. R. A. Morgan, W. Lindner and F. Pittner, Anal. Chem., 2008, 80, 2694–2703 CrossRef CAS PubMed.
- O. Salata, J. Nanobiotechnol., 2004, 2, 1–6 CrossRef PubMed.
- A. S. Thakor, J. Jokerst, C. Zavaleta, T. F. Massoud and S. S. Gambhir, Nano Lett., 2011, 11, 4029–4036 CrossRef CAS PubMed.
- Z. Wang and L. Ma, Coord. Chem. Rev., 2009, 253, 1607–1618 CrossRef CAS.
- O. Cohavi, D. Reichmann, R. Abramovich, A. B. Tesler, G. Bellapadrona, D. B. Kokh, R. C. Wade, A. Vaskevich, I. Rubinstein and G. Schreiber, Chem. – Eur. J., 2011, 17, 1327–1336 CrossRef CAS PubMed.
- B. N. Johnson and R. Mutharasan, Langmuir, 2012, 28, 6928–6934 CrossRef CAS PubMed.
- S. H. Brewer, W. R. Glomm, M. C. Johnson, M. K. Knag and S. Franzen, Langmuir, 2005, 21, 9303–9307 CrossRef CAS PubMed.
- Z. Deng, N. Thontasen, N. Malinowski, G. Rinke, L. Harnau, S. Rauschenbach and K. Kern, Nano Lett., 2012, 12, 2452–2458 CrossRef CAS PubMed.
- C. R. So, C. Tamerler and M. Sarikaya, Angew. Chem., 2009, 121, 5276–5279 CrossRef.
- A. Vila Verde, P. J. Beltramo and J. K. Maranas, Langmuir, 2011, 27, 5918–5926 CrossRef CAS PubMed.
- M. Hoefling, F. Iori, S. Corni and K.-E. Gottschalk, ChemPhysChem, 2010, 11, 1763–1767 CrossRef CAS PubMed.
- F. Iori, R. Di Felice, E. Molinari and S. Corni, J. Comput. Chem., 2009, 30, 1465–1476 CrossRef CAS PubMed.
- J. Yu, M. L. Becker and G. A. Carri, Langmuir, 2012, 28, 1408–1417 CrossRef CAS PubMed.
- M. Hoefling, S. Monti, S. Corni and K. E. Gottschalk, PLoS One, 2011, 6, e20925 CAS.
- R. Di Felice and S. Corni, J. Phys. Chem. Lett., 2011, 2, 1510–1519 CrossRef CAS.
- F. Iori, S. Corni and R. Di Felice, J. Phys. Chem. C, 2008, 112, 13540–13545 CAS.
- J. L. C. Fajín, J. R. B. Gomes and M. N. D. S. Cordeiro, Langmuir, 2013, 29, 8856–8864 CrossRef PubMed.
- M. Hoefling, F. Iori, S. Corni and K.-E. Gottschalk, Langmuir, 2010, 26, 8347–8351 CrossRef CAS PubMed.
- D. B. Kokh, S. Corni, P. J. Winn, M. Hoefling, K. E. Gottschalk and R. C. Wade, J. Chem. Theory Comput., 2010, 6, 1753–1768 CrossRef CAS PubMed.
- G. Brancolini, D. B. Kokh, L. Calzolai, R. C. Wade and S. Corni, ACS Nano, 2012, 6, 9863–9878 CrossRef CAS PubMed.
- F. Iori and S. Corni, J. Comput. Chem., 2008, 29, 1656–1666 CrossRef CAS PubMed.
- M. Gretes, D. C. Lim, L. de Castro, S. E. Jensen, S. G. Kang, K. J. Lee and N. C. J. Strynadka, J. Mol. Biol., 2009, 389, 289–305 CrossRef CAS PubMed.
- A. Šali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef PubMed.
- J. C. Gordon, J. B. Myers, T. Folta, V. Shoja, L. S. Heath and A. Onufriev, Nucleic Acids Res., 2005, 33, W368–W371 CrossRef CAS PubMed.
- R. R. Gabdoulline and R. C. Wade, Biophys. J., 1997, 72, 1917–1929 CrossRef CAS PubMed.
- R. R. Gabdoulline and R. C. Wade, Methods, 1998, 14, 329–341 CrossRef CAS PubMed.
- M. E. Davis, J. D. Madura, B. A. Luty and J. A. McCammon, Comput. Phys. Commun., 1991, 62, 187–197 CrossRef CAS.
- R. R. Gabdoulline and R. C. Wade, J. Phys. Chem., 1996, 100, 3868–3878 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren and J. Hermans, Intermol. Forces, 1981, 11, 331–338 Search PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef.
- M. Ahmad, W. Gu, T. Geyer and V. Helms, Nat. Commun., 2011, 2, 261 CrossRef PubMed.
- M. J. Penna, M. Mijajlovic and M. J. Biggs, J. Am. Chem. Soc., 2014, 136, 5323–5331 CrossRef CAS PubMed.
- G. Schreiber and A. R. Fersht, Nat. Struct. Biol., 1996, 3, 427–431 CrossRef CAS PubMed.
- T. Selzer and G. Schreiber, Proteins: Struct., Funct., Genet., 2001, 45, 190–198 CrossRef CAS PubMed.
- A. Wang, K. Vangala, T. Vo, D. Zhang and N. C. Fitzkee, J. Phys. Chem. C, 2014, 118, 8134–8142 CAS.
- L. Calzolai, F. Franchini, D. Gilliland and F. Rossi, Nano Lett., 2010, 10, 3101–3105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of PMF and relative permittivity calculations and clustering methods. Fig. S1–S10 and Tables S1–S5 show further details of the simulation set-up and results. See DOI: 10.1039/c6cp00201c |
| This journal is © the Owner Societies 2016 |