
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcentration dependent effects of urea binding to poly(N-isopropylacrylamide) brushes: a combined experimental and numerical study†
Samantha
Micciulla
a,
Julian
Michalowsky
b,
Martin A.
Schroer
cd,
Christian
Holm
b,
Regine
von Klitzing
a and
Jens
Smiatek
*b
aStranski-Laboratorium, Institut für Chemie, Technische Universität Berlin, D-10623 Berlin, Germany
bInstitut für Computerphysik, Universität Stuttgart, D-70569 Stuttgart, Germany. E-mail: smiatek@icp.uni-stuttgart.de
cDeutsches Elektronen-Synchrotron DESY, D-22607 Hamburg, Germany
dThe Hamburg Centre for Ultrafast Imaging (CUI), D-22761 Hamburg, Germany
First published on 21st January 2016
Abstract
The binding effects of osmolytes on the conformational behavior of grafted polymers are studied in this work. In particular, we focus on the interactions between urea and poly(N-isopropylacrylamide) (PNIPAM) brushes by monitoring the ellipsometric brush thickness for varying urea concentrations over a broad temperature range. The interpretation of the obtained data is supported by atomistic molecular dynamics simulations, which provide detailed insights into the experimentally observed concentration-dependent effects on PNIPAM–urea interaction. In particular, in the low concentration regime (cu ≤ 0.5 mol L−1) a preferential exclusion of urea from PNIPAM chains is observed, while in the high concentration regime (2 ≤ cu ≤ 7 mol L−1) a preferential binding of the osmolyte to the polymer surface is found. In both regimes, the volume phase transition temperature (Ttr) decreases with increasing urea concentration. This phenomenon derives from two different effects depending on urea concentration: (i) for cu ≤ 0.5 mol L−1, the decrease of Ttr is explained by a decrease of the chemical potential of bulk water in the surrounding aqueous phase; (ii) for cu ≥ 2 mol L−1, the lower Ttr is explained by the favorable replacement of water molecules by urea, which can be regarded as a cross-linker between adjacent PNIPAM chains. Significant effects of the concentration-dependent urea binding on the brush conformation are noticed: at cu = 0.5 mol L−1, although urea is loosely embedded between the hydrated polymer chains, it enhances the brush swelling by excluded volume effects. Beyond 0.5 mol L−1, the stronger interaction between PNIPAM and urea reduces the chain hydration, which in combination with cross-linking of monomer units induces the shrinkage of the polymer brush.
1 Introduction
Osmolytes are low-weight organic molecules which contribute to the regulation of the osmotic pressure inside the cells. Their presence allows biological cells to counteract a change of the external osmotic pressure in such a way that the isotonic balance is ensured. In addition, osmolytes have a strong influence on the structural conformation of proteins.1–4 It was reported that in high concentrated solutions of denaturants like guanidinium chloride or urea, protein unfolding occurs when a critical osmolyte concentration is reached.5–9 In contrast, the stabilization of protein structures was validated for protectants like trimethylamine-N-oxide (TMAO) and hydroxyectoine.10–13Experimental findings further evidenced that low concentrations of urea and guanidinium chloride can even stabilize protein native structures9 and lead to the fluidization of lipid bilayers and monolayers.12–14 The mechanisms and the different accumulation properties15–18 for protectants and denaturants around molecular surfaces can be brought into relation with the ‘law of matching water affinities’.15,19,20 As it was also recently demonstrated by computer simulations, the resulting binding behavior is a consequence of a subtle interplay between enthalpic and entropic effects, and the underlying charge of the solvated surface.21 In addition, further simulations of thermoresponsive polymers in presence of different salt concentration also reveal a concentration dependent change of binding properties for chaotropic salts.22 The possibility of investigating these effects is reasoned by the fact that the binding or the exclusion mechanism of the co-solute has a crucial influence on the stability of the macromolecule and therefore on the corresponding transition temperatures. Two distinct osmolyte binding mechanisms have been proposed: (1) direct interactions, related to the binding of the osmolyte to the surface of the macromolecule via hydrogen bonds, salt bridges or dispersion interactions,23–27 and (2) indirect interactions, mostly imposed by the influence of the osmolyte on the local water shell around the surface.28,29 However, it is still under debate which mechanism is predominant.
Due to the chemical variety and structural complexity, simplified homopolymers are more preferred than proteins in fundamental studies. Among them, poly(N-isopropylacrylamide) (PNIPAM) received a lot of scientific attention, because the mechanism underlying its thermoresponsive behavior, namely a coil-to-globule transition above the lower critical solution temperature (LCST), is closely related to the loss of protein structure upon addition of denaturating agents and resembles the behavior found for the cold denaturation of proteins.30 It has been validated that the predominant effects displayed above the LCST rely on the low number of hydrating water molecules bound to the macromolecule.31–35 Hence, the analysis of hydration properties, molecular size and transition temperatures represents a valid tool to study the influence of added solutes on the temperature-induced conformational changes of PNIPAM.36,37
In particular for PNIPAM, recent studies found that highly concentrated urea solutions lead to a significant decrease of the LCST.38 From the results of Fourier transformed infrared spectroscopy experiments, it was concluded that mainly direct interactions between urea and the carbonyl group of NIPAM promote the collapse into a globular state via a stabilizing bivalent hydrogen bond. In addition, more recent numerical studies also indicated that entropic effects arising from the accumulation of urea clouds around the PNIPAM molecule might have a crucial influence on the thermal behavior.27,39,40
Typical studies of PNIPAM–urea interactions were performed in bulk41–44 and very rarely for osmolyte concentrations as low as 0.5 mol L−1. Indeed, grafted chains are the base of a large number of smart coatings,45–47 therefore their conformational behavior in presence of osmolytes is of high interest. In this work, for the first time a systematic investigation of the swelling and collapse properties of PNIPAM brushes over a broad range of urea concentration from 0.1 to 7 mol L−1 is presented. The experimental approach consisted in monitoring the optical thickness of PNIPAM brushes by ellipsometry as a function of temperature for urea concentrations between 0.1 and 7 mol L−1. From the obtained data, the transition temperature (Ttr) and the swelling degree (ϕsw) of PNIPAM below (288 K) and above (328 K) the critical solution temperature were calculated. The hydration properties with regard to the underlying polymer–urea interactions were deduced from the observed swelling and collapse behavior. The interpretation of the experimental data is validated by atomistic MD simulation of PNIPAM chains in two different concentration regimes (low and high molar concentration) and for temperatures below and above the phase transition. The numerical studies further allow us to elucidate the balance between solvent (water) and co-solute (urea) binding on the conformational equilibrium of the polymer chain for the different combinations of urea concentration and temperature. In particular, at low molar urea concentrations, the observed shift of the transition temperature was governed by the dilution effect of urea on bulk water, which changes its chemical potential from pure solvent to dilute solution, and therefore favors brush dehydration across the phase transition. This effect was due to a preferential exclusion of urea from the macromolecule. In contrast, at high molar urea concentrations, the preferential binding of urea to PNIPAM was responsible for the pronounced decrease of Ttr, which was reasoned by the weakening of PNIPAM–water interactions, and the cross-linking of the osmolyte between adjacent PNIPAM chains.
Our study offers a deeper understanding on the impact of direct urea binding and indirect effects on the conformational equilibrium of the polymer. It contributes to widen the previous knowledge in this field and to understand the behavior of proteins in presence of osmolytes.
2 Experimental section
2.1 Materials
Silicon wafers (Siltronic AG, Germany) were used as solid substrates for the polymer brushes. N-Isopropylacrylamide (NIPAM) (98%, stabilized by methylhydroquinone) was purchased from TCI Tokyo Chemical Industry; N,N,N′,N′′,N′′-pentamethyldiethylene-triamine (PMDETA), Cu(I)Cl, methanol and urea were all purchased from Sigma Aldrich (Germany). All reagents were used as received without any further purification.2.2 Synthesis of polymer brushes
Polymer brushes were grown from silicon wafers by Atom Transfer Radical Polymerization (ATRP).48 First of all, a self-assembled monolayer of the ATRP initiator 2-bromo-2-methyl-N-[3-(triethoxysilyl)-propyl]-propanamide (BTPAm)49 in dry toluene (4 μL/10 mL) was adsorbed on the substrates for 24 h. A polymerization mixture of 2.83 g NIPAM in 50 mL of methanol and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), 183 μL N,N,N′,N′′,N′′-pentamethyldiethylene-triamine (PMDETA) and 25 mg copper(I) chloride was prepared and degassed by N2 bubbling for 40 minutes. The initiator-modified substrates were soaked in the reaction mixture and the polymerization was run for 8 minutes, followed by termination in water/methanol mixture and thorough rinsing in Milli-Q water. The grafting density σ was calculated considering the molecular structure of the initiator and the measured thickness of the self-assembled monolayer (details are reported in the ESI†). A value of σ ∼ 0.4 nm−2 was estimated, which corresponds to a moderate brush regime50 and allows urea to access the full solvent accessible surface area of PNIPAM. The brush thickness of the dry state (1.9 ± 0.1% r.h.) was measured by ellipsometry after drying the samples in a sealed cell flushed with nitrogen stream for 20 minutes. The measured value was ddry = (40.5 ± 0.9) nm and it remained constant for at least 10 minutes.
:1 v/v), 183 μL N,N,N′,N′′,N′′-pentamethyldiethylene-triamine (PMDETA) and 25 mg copper(I) chloride was prepared and degassed by N2 bubbling for 40 minutes. The initiator-modified substrates were soaked in the reaction mixture and the polymerization was run for 8 minutes, followed by termination in water/methanol mixture and thorough rinsing in Milli-Q water. The grafting density σ was calculated considering the molecular structure of the initiator and the measured thickness of the self-assembled monolayer (details are reported in the ESI†). A value of σ ∼ 0.4 nm−2 was estimated, which corresponds to a moderate brush regime50 and allows urea to access the full solvent accessible surface area of PNIPAM. The brush thickness of the dry state (1.9 ± 0.1% r.h.) was measured by ellipsometry after drying the samples in a sealed cell flushed with nitrogen stream for 20 minutes. The measured value was ddry = (40.5 ± 0.9) nm and it remained constant for at least 10 minutes.
The molecular weight (MW) of PNIPAM chains was estimated from the grafting density σ and the dry thickness ddry, according to the relation49,51,52
 | (1) |
000 g mol−1 (ca. 590 monomers per chain). Reported polydispersity indices (PDI) for ATRP polymerization are mostly between 1.2 and 1.5.53,54 A low PDI is also expected for our system, given the low standard deviation of the brush thickness (ca. 0.8% of the brush thickness). The swollen thicknesses of PNIPAM brushes in solutions of urea at different concentrations varied between 100 and 150 nm at 288 K, and between 47 and 80 nm at 328 K. The values are reported in Table S1 of the ESI.†
2.3 Ellipsometry
Ellipsometry is a non-destructive, optical technique which is based on the detection of the change of polarization of light upon reflection from a substrate. Such a change is described by two parameters, ψ and Δ, which are related to the amplitude (E) and the phase (δ) of light by the following relations: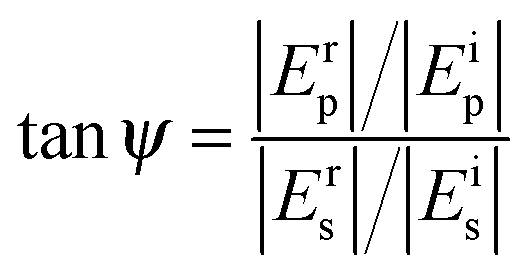 | (2) |
| Δ = (δrp − δrs) − (δip − δis) | (3) |
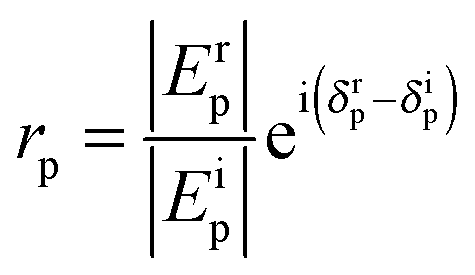 | (4) |
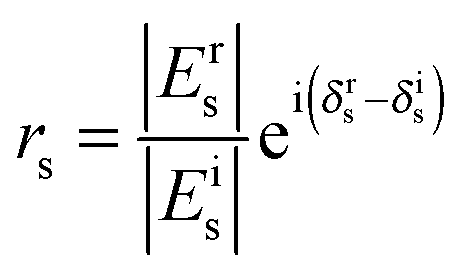 | (5) |
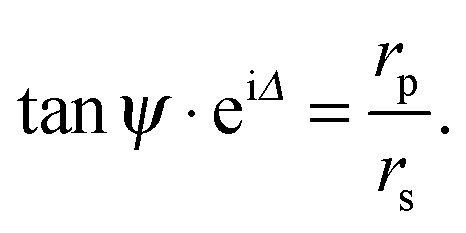 | (6) |
The use of a proper layer model, which describes the system under measurement (substrate-film-medium) in terms of its optical properties (thickness d and refractive index n) can be obtained from the measured parameters Δ and ψ, which in turn describe the interaction between polarized light and sample. Specific literature is available on the topic to which the reader is addressed for a more detailed description of the technique.55,56
For the data analysis, the measured values of Δ and Ψ were fitted with a five box model consisting, from the bottom to the top, of (i) silicon substrate (n = 3.8850, d = ∞), (ii) silicon oxide (n = 1.46, d = 1.5 nm), (iii) initiator BTPAm monolayer (n = 1.50, d = 0.7 nm), (iv) polymer brushes (n = nbrush, d = dbrush), (v) liquid medium (ncalc, d = ∞). From the measured values of Δ and ψ, the corresponding combination of n = nbrush, d = dbrush for the polymer brushes is obtained. The refractive index of solutions containing different urea concentrations was estimated by the empirical relations elaborated by Warren and Gordon57 from experimental data on urea solutions. The corresponding values are reported in Table S2 of the ESI.† The degree of swelling ϕsw for PNIPAM brushes was calculated from the thickness ddry of the dry brush and the thickness dsw(T) of the swollen brush at a given temperature T by
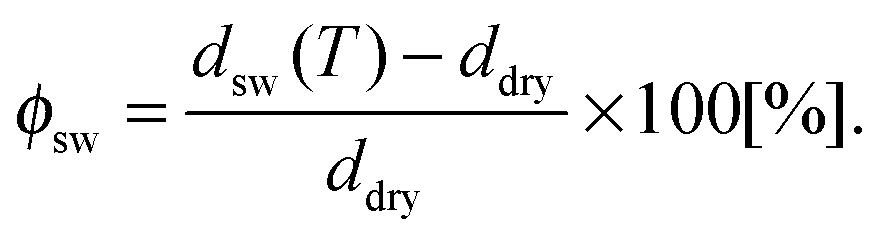 | (7) |
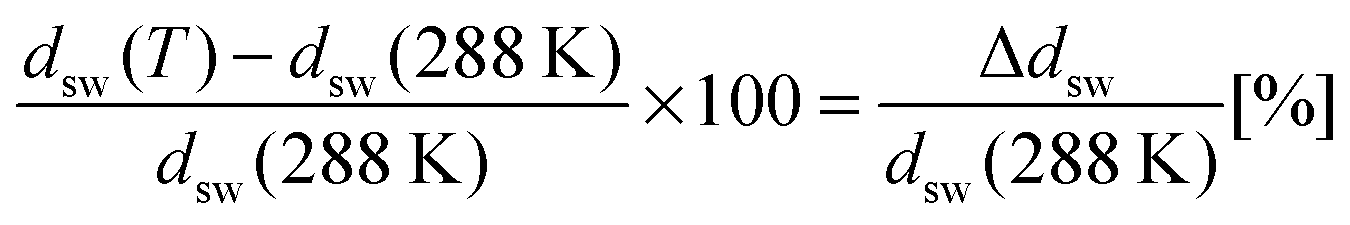 | (8) |
Ellipsometry measurements were carried out using a Multiscope Null-Ellipsometer from Optrel GbR (Germany). The instrument is equipped with a red laser (λ = 632.8 nm) and a PCSA (polarizer–compensator–sample-analyzer) setup. For the measurements in dry condition, a home-built humidity cell was used, whose inside is flushed by nitrogen stream, reaching a value of (1.9 ± 0.1)% relative humidity. The angle of incidence was set to 70° and the sample was left drying for 20 minutes before measurements. For the measurements in liquid, the sample was soaked in a stainless steel cell filled with the urea solution. The instrument was equipped with light guides to drive the incident beam directly at the substrate/water interface and avoid the reflection at the liquid/air interface. Prior to the measurements, the sample was swollen in urea solution at 288 K for at least 2 h. A thermal cycle was applied by heating the liquid by means of a copper plate underneath the sample holder. The temperature of the liquid environment was measured continuously with a precision of ΔT = ±0.01 K by means of a temperature sensor immersed in the sample cell.
2.4 Simulation details
A PNIPAM molecule with 24 monomers was studied by atomistic molecular dynamics (MD) simulations with the GROMACS 4.6.2 software package.58–60 The force field parameters for PNIPAM presented in ref. 35 have been used to guarantee the occurrence of the coil-to-globule transition at temperatures below 328 K34,35 while generalized AMBER force fields were used for urea.61,62 Typical swollen and collapsed PNIPAM configurations have been obtained by using the original Metadynamics approach63–65 as implemented within the PLUMED package.66 We extracted the most swollen (radius of gyration Rg = 1.4 nm) and the collapsed state of PNIPAM (radius of gyration Rg = 0.8 nm) as found during the Metadynamics simulation and inserted them into cubic simulation boxes to study the influence of pure water, low and high aqueous urea concentrations, respectively. The position of the backbone Cα carbon atoms have been kept fixed by using position restraints. We randomly inserted the corresponding number of urea and TIP3P water molecules67 to model the required urea concentrations. In addition, we also simulated a PNIPAM molecule in pure water as a reference system. Electrostatic interactions for all systems have been calculated by the Particle Mesh Ewald method68 and all bonds have been constrained by the LINCS algorithm.69 First, we performed an energy minimization followed by a 2 ns warm-up run in a NPT ensemble at 288 K and 328 K with the Berendsen barostat and thermostat,70 followed by NPT simulations of 20 ns for the production run with the same parameter sets. The minimum and maximum values for the length of the cubic simulation box are given by 〈l〉min = 4.7 nm and 〈l〉max = 6.49 nm in the NPT simulation depending on the system and the temperature. The corresponding effective urea concentrations for the different systems can be found in Table 1.| T [K] | R g [nm] | c u [mol L−1] | Conformation | Label |
|---|---|---|---|---|
| 288 | 0.8 | 0.5 | Collapsed | lc288 |
| 328 | 0.8 | 0.5 | Collapsed | lc328 |
| 288 | 1.4 | 0.5 | Swollen | ls288 |
| 328 | 1.4 | 0.5 | Swollen | ls328 |
| 288 | 0.8 | 6.1 | Collapsed | hc288 |
| 328 | 0.8 | 4.8 | Collapsed | hc328 |
| 288 | 1.4 | 6.1 | Swollen | hs288 |
| 328 | 1.4 | 4.8 | Swollen | hs328 |
Hydrogen bonds were defined by a maximum distance criterion of 0.35 nm between acceptor and donor pairs with a maximum angle of 35°.
In fact, the study of polymer brushes is a challenging task for atomistic simulations. For the minimization of finite-size effects, a large number of grafted PNIPAM chains have to be considered which limits the applicability of atomistic approaches. In contrast, insights into polymer brush behavior can be provided by computationally efficient coarse-grained (CG) simulations71 in which specific chemical details are usually neglected. Due to these reasons, CG approaches are not suitable for the detailed study of the PNIPAM–urea interactions such that we decided to simulate single PNIPAM molecules in an atomistic approach. The usage of restrained PNIPAM configurations was motivated by recent publications.27,40 In fact, the simulation of PNIPAM coil-to-globule changes is a challenging task which might result in an insufficient sampling accuracy with regard to the interpretation of conformational transitions as rare events whose timescale usually exceeds the simulated time interval. Furthermore, the direct evaluation of free energy landscapes by free energy methods like metadynamics or umbrella sampling might also lead to wrong results due to inappropriate reaction coordinates and the presence of hysteresis effects.72 Thus, the usage of restrained PNIPAM conformations as reference states avoids these drawbacks and provides a meaningful interpretation of the observed effects.27,40
2.5 Implications of the Kirkwood–Buff theory and local/bulk partition coefficients
The Kirkwood–Buff (KB) integral is given by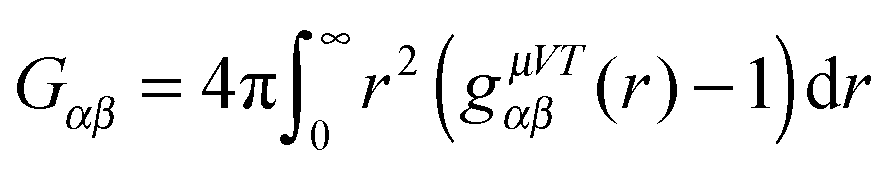 | (9) |
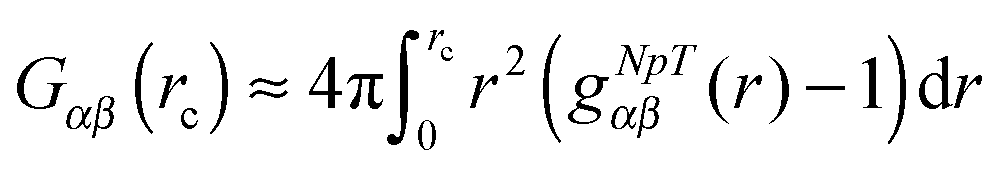 | (10) |
The preferential binding coefficient ν23 between PNIPAM and urea can be obtained from the difference of the KB integrals describing the distribution of water and urea around a PNIPAM chain, according to
| ν23 = ρ3(G23 − G21) | (11) |
| ΔF23 = −RTν23 | (12) |
The effects of osmolytes on macromolecular conformations can be evaluated in terms of the chemical equilibrium constant K = πs/πc where πs and πc denote the fraction of PNIPAM molecules in the swollen (s) and the collapsed state (c). From the relation
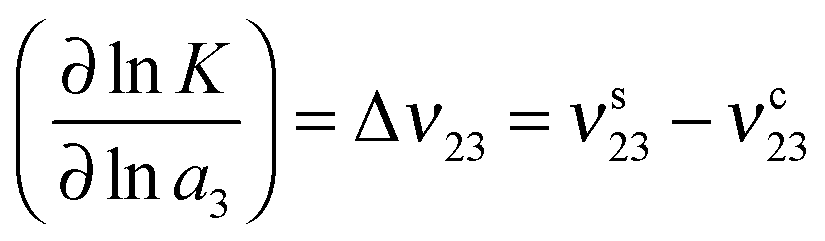 | (13) |
| ΔΔF23 = ΔFs23 − ΔFc23 | (14) |
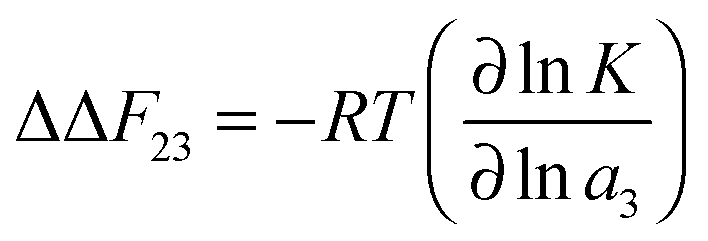 | (15) |
Another robust parameter to study the osmolyte accumulation behavior is given by the local/bulk partition coefficient40,81 according to
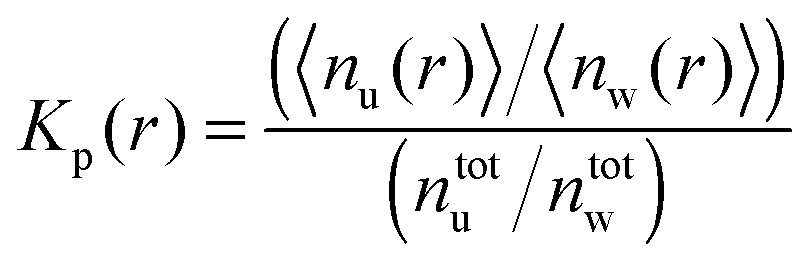 | (16) |
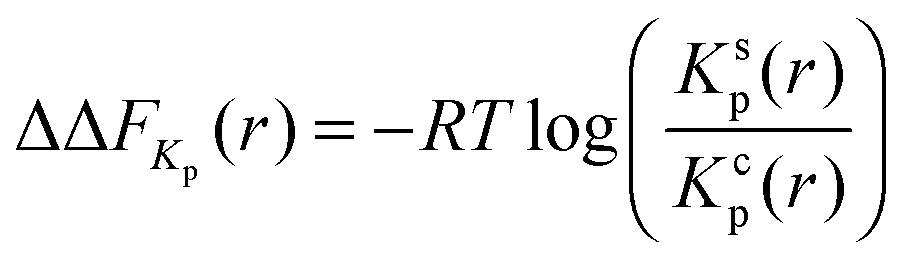 | (17) |
3 Results
3.1 Experimental results
The relative change of ellipsometric thickness Δdsw/dsw(288 K) calculated according to eqn (8) is reported in Fig. 1 as a function of temperature for different urea concentrations (cu). Two important features in the obtained trends can be noticed: a shift of Ttr to lower values for increasing urea concentration cu as denoted by the left arrow, and a weaker brush collapse in urea solutions compared to pure water as denoted by the right arrow. The weaker brush collapse above the transition temperature at high urea concentrations can be discussed with the arguments given by Cremer and co-workers in ref. 38, where increasing PNIPAM radii observed at increasing urea concentration were explained by bridging effects of the osmolyte in order to cross-link PNIPAM chains. An analogous effect in this case might be responsible for the reduced brush collapse at high cu. In order to analyze the effects of urea on the thermal behavior of PNIPAM in more detail, Ttr and ϕsw were extracted from the ellipsometry curves. The values of Ttr for PNIPAM brushes in presence of different urea concentrations are presented in Fig. 2. A systematic decrease of Ttr for increasing urea concentrations was found, with the evidence of two distinct regimes: a low concentration regime between 0 and 0.5 mol L−1, with a linear decrease of Ttr as a function of cu (inset in Fig. 2), and a high urea concentration regime between 2 and 7 mol L−1 with a weaker, but constant decrease of Ttr. Two distinct regimes, although not so noticeable, were found also for single PNIPAM chains.38 Thus, it can be concluded that this effect is not exclusively characteristic for PNIPAM brushes and it demonstrates that molecular PNIPAM–urea interactions dominate the chain behavior, independent of the system geometry. This allows us to apply the knowledge of single chain–urea interactions to more complex and crowded molecular systems, like polymer brushes, which might be of paramount importance for the application of grafted biomolecular (e.g. proteins) systems.71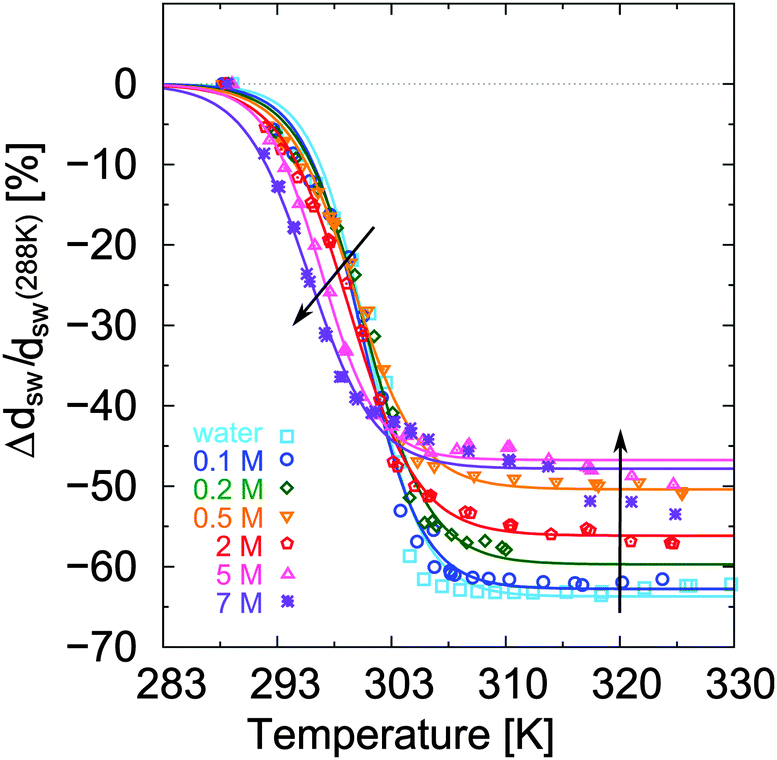 | ||
| Fig. 1 Relative thickness change, Δdsw/dsw(288 K), as a function of temperature measured in urea solution at different molar concentration. The lines are the fit of a sigmoid on the experimental data and are traced as a guide for the eye. Arrows indicate the effect of added urea. | ||
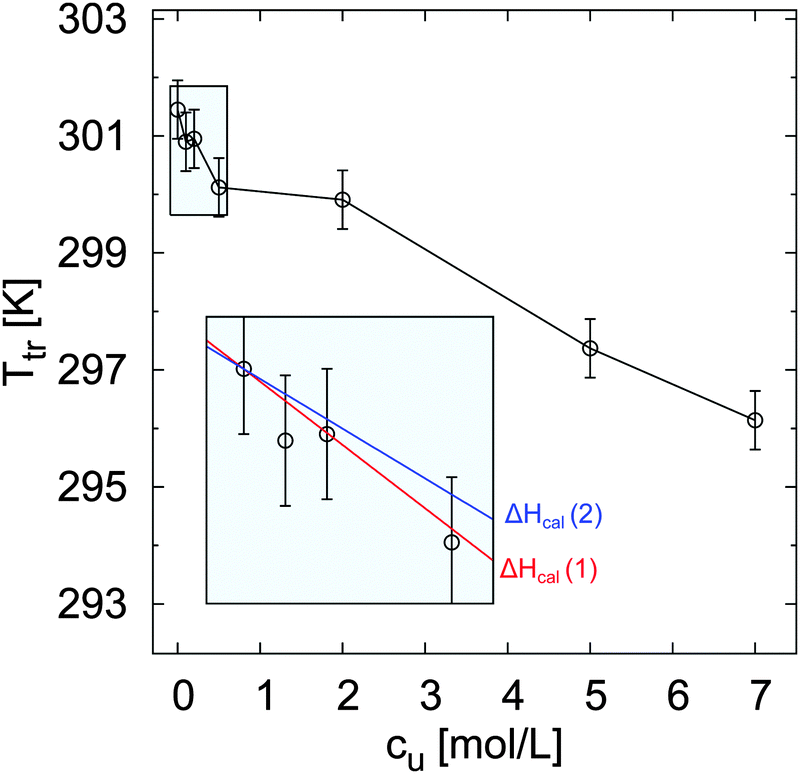 | ||
| Fig. 2 Transition temperature Ttr of PNIPAM brushes as a function of urea concentration. The samples were subjected to a heating cycle from 288 K to 328 K. The values Ttr can be related to the temperature where half of the brush is collapsed. The inset in the plot reports the values of Ttr between 0 and 0.5 mol L−1, while the straight lines describe the calculated trends according to eqn (18) for the calorimetric enthalpy ΔHcal(1) = 5.5 kJ mol−1 and ΔHcal(2) = 7 kJ mol−1 reported for the NIPAM dehydration in water. | ||
Remarkably, the influence of urea on the transition temperature of PNIPAM may have different origins:36 on the one hand, the presence of urea changes the properties of the aqueous medium;29 on the other hand, the osmolyte could directly interact with the polymer, as recently demonstrated for high urea concentrations.38,82
While at high cu the direct urea binding to the polymer is very likely, it is important to verify which mechanism dominates the change of Ttr for a PNIPAM brush in presence of a low molar urea concentration. As a first hypothesis, it is assumed that urea does not directly interact with PNIPAM, i.e. it is preferentially excluded from a PNIPAM surface. Under this condition, the shift of Ttr might be reasoned by a change of the PNIPAM hydration properties.
From the decrease of the chemical potential of bulk water upon addition of urea, and from the equilibrium condition at Ttr with identical chemical potentials for H2O (bulk) and H2O (brush) phase, the following relation between the temperature shift ΔTtr and the dehydration enthalpy ΔHdehy can be derived:
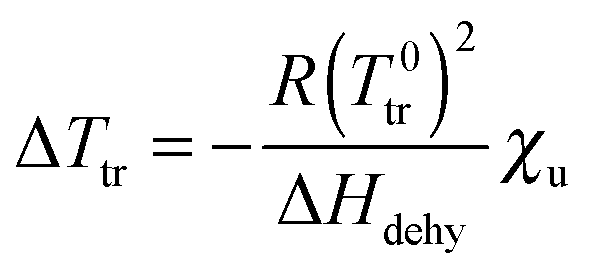 | (18) |
It is worth to mention that this approach represents a simplified picture of the system, where the transition enthalpy of the macromolecule from swollen to collapsed state is omitted. The validity of eqn (18) to describe the experimental data of Fig. 2 at low cu would confirm the dilution effect of bulk water by urea, i.e. indirect effects. Previous studies on the coil-to-globule transition of PNIPAM reported calorimetric enthalpy values ΔHcal between 5.5 and 7 kJ mol−1 for a single (NIPAM) repeating unit.83,84 This can be related to the release of one bound water molecule from each monomer unit, in agreement with previous experimental results.83,85 Other literature works report the release of a much higher number of water molecules per NIPAM unit in bulk or in gels.86,87 Nevertheless, in our evaluation only the water molecules directly bound to the monomer unit via H-bonding are considered.
Assuming that the same process occurs for the dehydration of PNIPAM brushes in presence of low molar urea concentrations, where direct urea–PNIPAM interactions are negligible, it follows that ΔHcal ∼ ΔHdehy and therefore it is possible to calculate the theoretical trend of ΔTtr as a function of cu for the reported dehydration enthalpies of ΔHcal(1) = 5.5 kJ mol−1 and ΔHcal(2) = 7 kJ mol−1. The corresponding functions are shown as solid lines in the inset of Fig. 2. The agreement between experimental data and theoretical trends corroborates the assumption that the dehydration mechanism of PNIPAM in low concentrated urea solutions is comparable to the dehydration in pure water in accordance to ref. 83 and 84. Thus, the shift of the transition temperature can be explained by the dilution of water by urea as induced by the mole fraction χu.
A different mechanism is likely to occur in the high concentration regime, where urea is known to have a significant influence on the stability of proteins and a direct osmolyte–polymer binding has been reported in previous studies.38,82 Indeed, the profile of Ttr shown in Fig. 2 is very similar to the results presented by Cremer and coworkers38 obtained by Fourier transformed infrared (FTIR) spectroscopy measurements. The reported FTIR data showed a strong amide band for PNIPAM in 6 M urea solution, with a major contribution from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(PNIPAM)–H2O hydrogen bonds and a second minor one from CO(PNIPAM)–NH(urea) bonds. An analogous effect is likely to dominate the collapse of PNIPAM brushes at cu ≥ 2 M, as indicated by our results, where the direct binding of urea with (NIPAM) monomers might introduce the aggregation of adjacent chains, such that urea acts as a cross-linking agent.38 Evidence of direct polymer–osmolyte binding was also found by Wang et al.82 for poly(N,N-dimethylacrylamide) (PDMA) and poly(N,N-diethylacrylamide) (PDEA) brushes by atomic force microscopy-based single molecule force spectroscopy (SMFS). The enhanced stiffness of the polymer chains in presence of 2 M and 8 M urea solution was explained by the formation of hydrogen bonds between the polymer side groups and urea molecules. Cross-linking of urea via hydrogen bonding was also reported by Lu et al.88 to explain urea-induced aggregation of PNIPAM chains observed by static and dynamic light scattering. According to Cremer and co-workers,32,38 a schematic representation of the bridging mechanism between PNIPAM chains in presence of urea is shown in Fig. 3. This aspect will be discussed in more detail in the next section.
O(PNIPAM)–H2O hydrogen bonds and a second minor one from CO(PNIPAM)–NH(urea) bonds. An analogous effect is likely to dominate the collapse of PNIPAM brushes at cu ≥ 2 M, as indicated by our results, where the direct binding of urea with (NIPAM) monomers might introduce the aggregation of adjacent chains, such that urea acts as a cross-linking agent.38 Evidence of direct polymer–osmolyte binding was also found by Wang et al.82 for poly(N,N-dimethylacrylamide) (PDMA) and poly(N,N-diethylacrylamide) (PDEA) brushes by atomic force microscopy-based single molecule force spectroscopy (SMFS). The enhanced stiffness of the polymer chains in presence of 2 M and 8 M urea solution was explained by the formation of hydrogen bonds between the polymer side groups and urea molecules. Cross-linking of urea via hydrogen bonding was also reported by Lu et al.88 to explain urea-induced aggregation of PNIPAM chains observed by static and dynamic light scattering. According to Cremer and co-workers,32,38 a schematic representation of the bridging mechanism between PNIPAM chains in presence of urea is shown in Fig. 3. This aspect will be discussed in more detail in the next section.
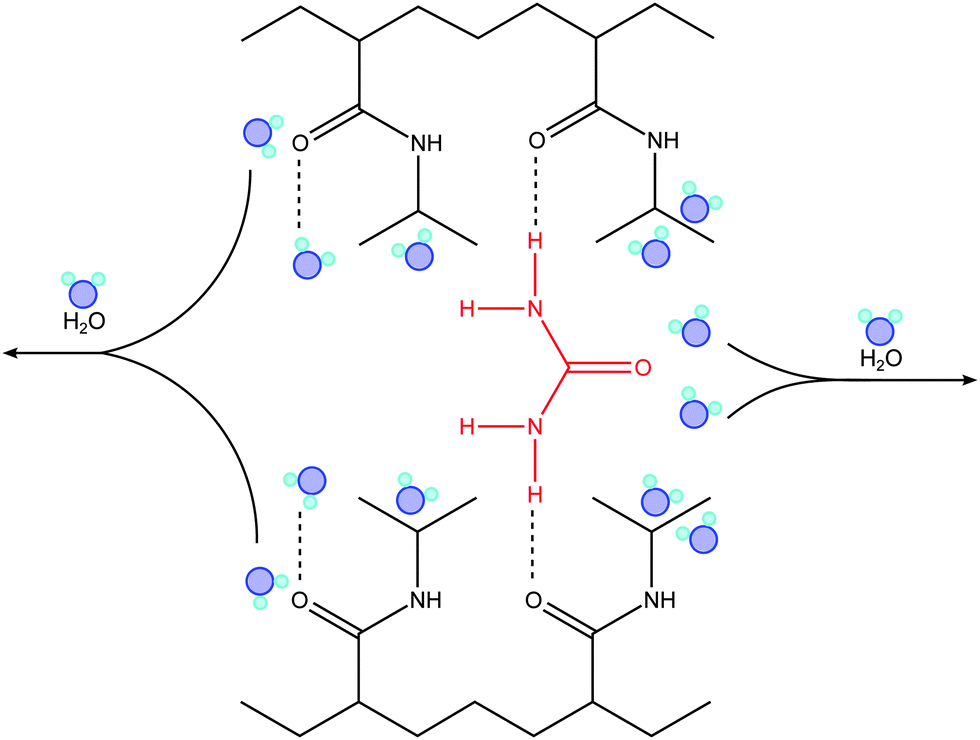 | ||
| Fig. 3 Schematic representation of the urea bridging effect between adjacent PNIPAM chains occurring in the high concentration regime (cu ≥ 2 M). Urea as a cross-linking agent significantly enhances the stability of the collapsed state. The image was inspired by ref. 32 and 38. | ||
To study the effect of urea on the conformation of grafted chains, the swelling degree ϕsw of PNIPAM brushes below (288 K) and above (328 K) the phase transition temperature was calculated according to eqn (7) for the different urea concentrations. As it is shown in Fig. 4, a stronger swelling percentage compared to pure water (cu = 0 mol L−1) is achieved for low urea concentrations both at 288 K and 328 K. In contrast, in the high concentration regime (cu ≥ 2 mol L−1) ϕsw is lower (288 K) or similar (328 K) to the swelling degree in pure water. Analogous to the previous assumption, we rule out a strong accumulation of urea inside the brush. Therefore indirect effects might be responsible for the observed enhancement of brush swelling at cu = 0.5 mol L−1. More specifically, steric effects on the chain conformation due to the presence of urea in the swelling medium might lead to higher chain stretching. This aspect is analyzed and clarified by the numerical studies presented in the next section. It is worth to mention that this effects might not be visible at concentrations below 0.5 M due to the very low amount of the osmolyte in the swelling medium. In the case of high cu, there are two important implications: at low temperature, increasing urea concentrations decrease the brush swelling, likely due to a decrease of water bonding (lower hydration) to the polymer chain; at high temperature, the presence of urea molecules around the macromolecule might be responsible for the stabilization of the collapsed state. It has to be noticed that the considered temperatures of 288 K and 328 K are significantly below and above the transition temperature, such that temperature dependent dehydration effects, which could in principle influence Ttr, can be omitted.
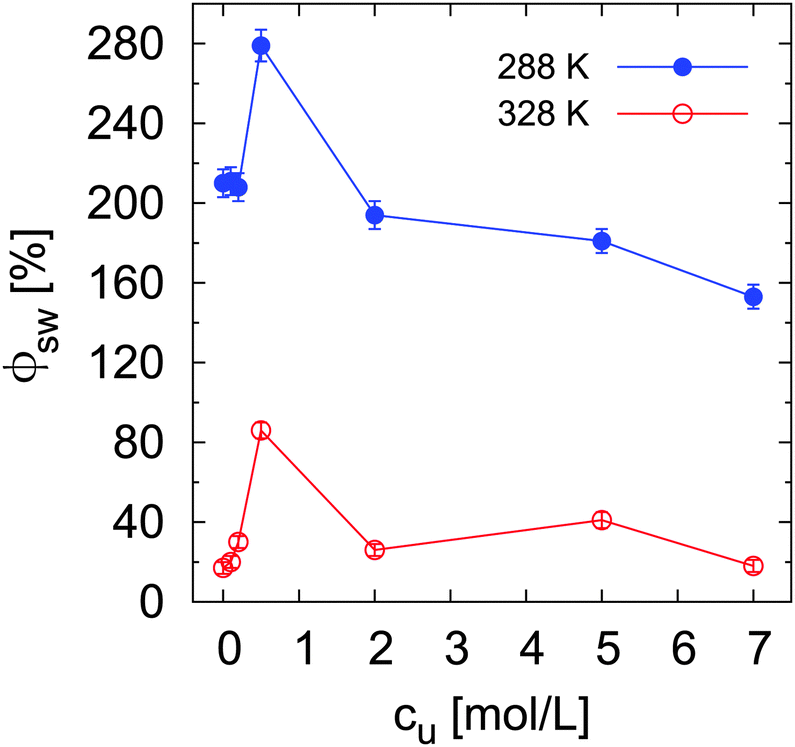 | ||
| Fig. 4 Swelling degree ϕsw of PNIPAM brushes at 288 K and 328 K for different urea concentrations. The parameter ϕsw was calculated according to eqn (7). | ||
An analogous representation of the structural transition of PNIPAM brushes across Ttr is given by the percentage of the total brush collapse (i.e. reached at 328 K), which is reported in the ESI.† The obtained trend as a function of cu demonstrates a reduced brush collapse for any urea concentration compared to pure water. While in the high concentration regime this can be reasonably explained by the reduced water content in the brush, which would confirm the direct polymer–urea interaction reported by other authors,38 in the low concentration regime it is likely the result of a more subtle interplay between hydration properties and bulk effects of urea.
In summary, the experimental results indicate concentration-dependent properties of urea on transition temperatures and resulting conformations of PNIPAM brushes above and below Ttr. The understanding of the molecular origins of the observed effects on phase transition and swelling behavior motivated the numerical studies presented in the next subsection. Herewith, we mostly focus on the interactions between PNIPAM, water and urea in the low/high concentration regimes and below/above the phase transition temperature.
3.2 Numerical results
The interpretation of the experimental results presented in the previous section suffer of some speculative hypotheses to deduce a suitable molecular mechanism to explain the observed effects. This is an intrinsic limitation of most experimental methods, but as we demonstrated in a previous publication,89 it can be overcome when experiments and simulations are interpreted in a synergistic way. Following this approach, simulation results are presented to complement the experimental findings and to validate their interpretation.Typical snapshots of a collapsed and a swollen PNIPAM configuration in a low molar urea solution are presented in Fig. 5. By introducing specific position restraints to fix the PNIPAM configurations, it was possible to avoid transitions into metastable states and to obtain reliable and well-sampled statistically averaged values. A detailed discussion of this approach is given in the section Simulation details.
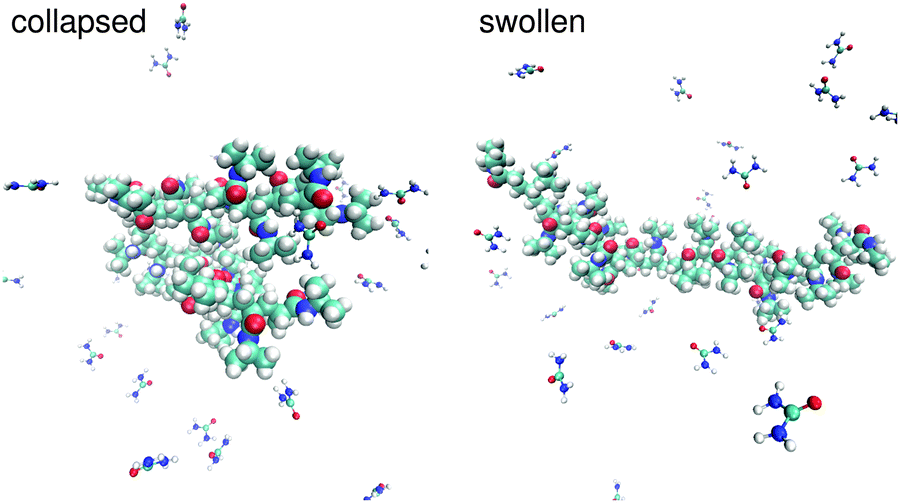 | ||
| Fig. 5 Typical simulation snapshots of PNIPAM in presence of a low molar urea solution at 288 K. The snapshot on the left side represents a collapsed PNIPAM configuration with a radius of gyration of 0.8 nm whereas the right side shows a swollen configuration with a radius of gyration of 1.4 nm. Both conformations have been used for the study of the hydration and the urea binding properties. | ||
In order to study the hydration behavior, the local Kirkwood–Buff integrals G21(r) for PNIPAM and water molecules at 288 K for cu = 0.5 mol L−1 were calculated. The values for G21(r) in pure water were also estimated for comparison. The results are presented at the top of Fig. 6. Only small differences between the G21(r) in urea solution and the value in pure water can be observed. Hence, it becomes clear that low molar urea concentrations lead to a weak change in the PNIPAM hydration behavior at low temperatures, as it was also concluded from the experimental results.
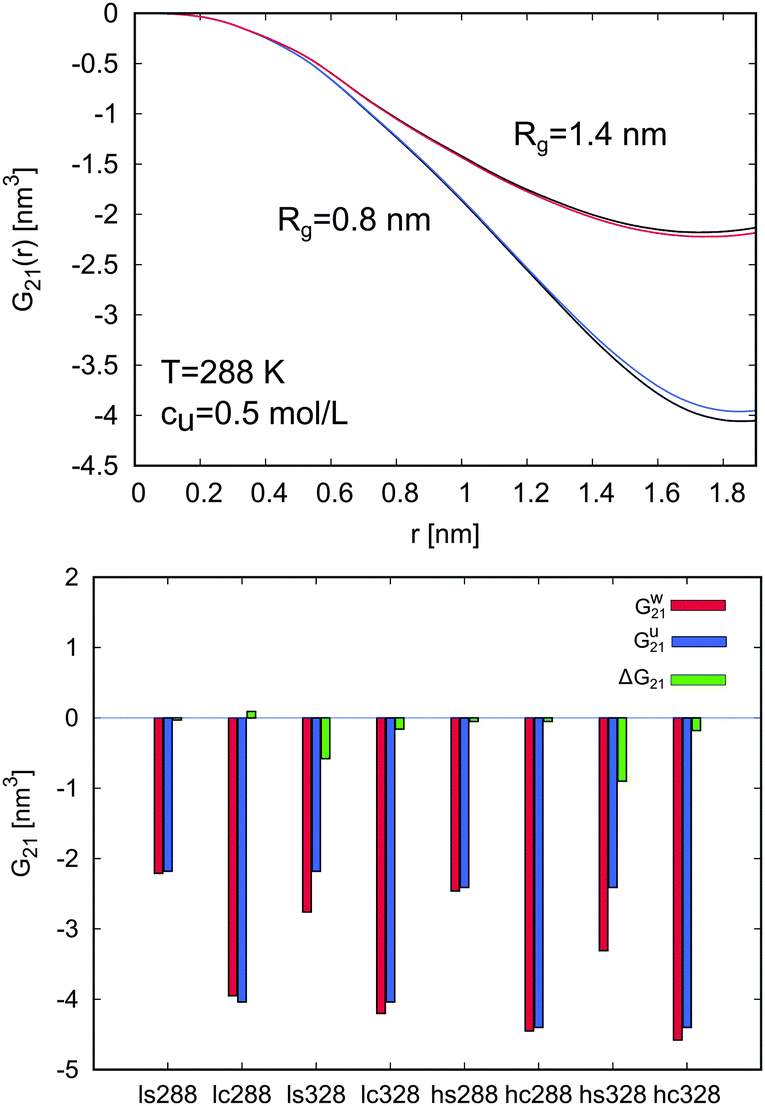 | ||
| Fig. 6 Top: Kirkwood–Buff integrals G21(r) for water molecules around the swollen (Rg = 1.4 nm: red and upper black line) and the collapsed PNIPAM configuration (Rg = 0.8 nm: blue and lower black line) in a 0.5 M urea solution and pure water at 288 K. The black lines represent the Kirkwood–Buff integral values around the swollen and the collapsed configuration in pure water, while the blue and the red line correspond to the values for water molecules in a 0.5 M urea solution. Bottom: Values for the Kirkwood–Buff integrals G21 at rc = 1.8 nm for water molecules around all considered PNIPAM configurations and system parameters (288 K and 328 K, (h)igh and (l)ow molar urea concentrations, (s)wollen and (c)ollapsed PNIPAM configuration). The red bars for Gw21 denote the values in pure water, the blue bars for Gu21 represent the values in presence of urea, and the green bars for ΔG21 correspond to the difference in the KB integrals according to eqn (19). | ||
Changes in the PNIPAM–water interactions in presence of urea can be easily evaluated considering the differences of the Kirkwood–Buff integrals according to
| ΔG21 = Gu21 − Gw21 | (19) |
To study the direct and local interactions between PNIPAM and urea or water molecules, the number of hydrogen bonds was calculated. The corresponding results and further findings are shown in Fig. 7 and in Table S3 of the ESI.† At low molar urea concentrations, the number of water–PNIPAM hydrogen bonds is comparable to pure water (ΔH-bonds (water)) for both temperatures (ls288, lc288, ls328 and lc328). This means that the first solvent shell around PNIPAM at low cu has a similar composition as in pure water, and only a small number of urea–PNIPAM hydrogen bonds (H-bonds (urea)) are formed. These results validate the previous assumptions of a minor PNIPAM–urea interaction in presence of low cu, and support the hypothesis of indirect effects from the water–urea network on the chain conformation at low cu.
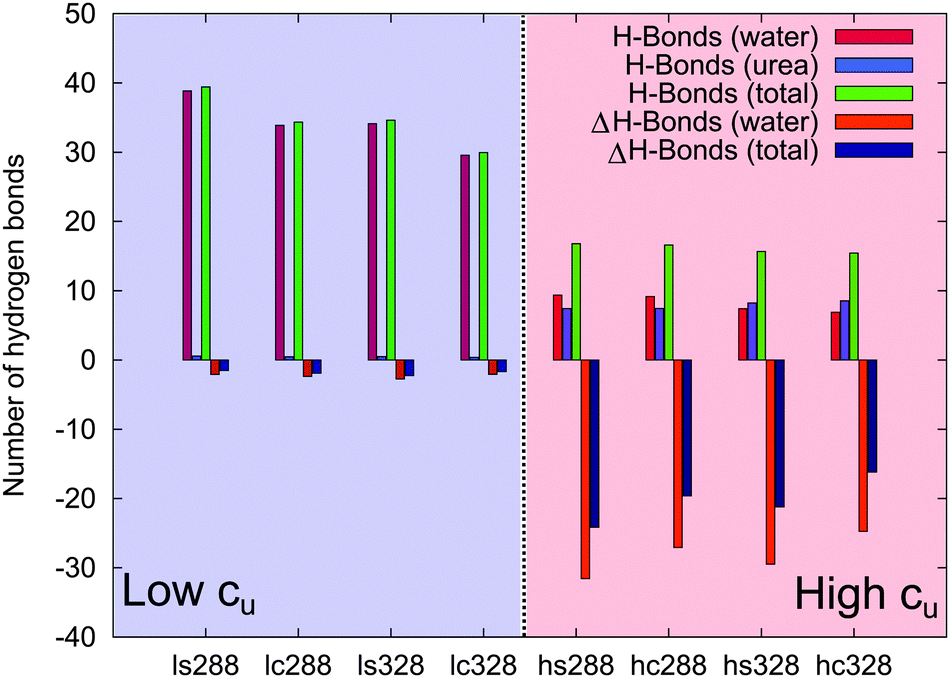 | ||
| Fig. 7 Number of PNIPAM hydrogen bonds in urea solutions with urea (H-bonds (urea)), water (H-bonds (water)), total number of hydrogen bonds (H-bonds (total)) and the difference of H-bonding for the same configuration and temperature in pure water (ΔH-bonds (water) and ΔH-bonds (total)). | ||
In contrast, a significant decrease of water hydrogen bonds compared to pure water (ΔH-bonds (water)) is found at high cu (hs288, hc288, hs328 and hc328), and also the total number of hydrogen bonds is smaller compared with pure water (i.e. large negative values of ΔH-bonds (total)). Interestingly, at high temperature (hs328 and hc328) the number of urea hydrogen bonds even exceeds the number of water hydrogen bonds (H-bonds (urea) > H-bonds (water)) meaning that in this condition urea replaces water at molecular interfaces.88 Moreover, from the data reported in Table S3 of the ESI† it can be seen that urea hydrogen bonds are energetically stronger than water hydrogen bonds. From these results it can be concluded that at high cu urea binding dominates the behavior of PNIPAM chains across the phase transition, while the effects arising from the change of PNIPAM–water interactions are negligible.
In order to classify the hydrogen bonds with respect to their related energetic contributions according to the transition state theory with the Luzar–Chandler approach,90,91 it is possible to calculate the forward rate constants k of hydrogen bonds via k ∼ exp(−ΔF*/kBT), where ΔF* denotes the activation free binding energy. The corresponding values for water–PNIPAM and urea–PNIPAM hydrogen bonds are shown in Table S3 of the ESI.† The most relevant result is the decrease of the activation free energies of water for increasing urea concentrations. The replacement of water–PNIPAM hydrogen bonds with urea–PNIPAM hydrogen bonds is therefore energetically favorable. Furthermore, for increasing urea concentration, the strength of water hydrogen bonds is slightly lowered, which facilitates the release of water molecules from the first solvent shell, in agreement with the experimental results (Fig. 2). Moreover, the increasing amount of urea molecules favors PNIPAM dehydration also due to the replacement of water by urea in the first solvent shell. These conclusions are additionally supported by the stagnating values of ϕsw at high urea concentrations (Fig. 4).
In order to evaluate the influence of urea binding on the shift of the chemical equilibrium towards the swollen or the collapsed state, the local/bulk partition coefficients were estimated according to eqn (16). The results reported in Fig. 8 prove that the local/bulk partition coefficients Kp(r) for low cu at both temperatures are smaller than 1 at all distances. This confirms that urea is preferentially excluded from the PNIPAM backbone at low concentrations, in perfect agreement with the small number of urea–PNIPAM hydrogen bonds reported in Fig. 7. The highly stretched PNIPAM conformations observed at low urea concentrations (cu = 0.5 mol L−1) in Fig. 4 can be therefore attributed to the weaker urea exclusion around the swollen conformation in accordance to eqn (13). Although urea is very poorly bound to PNIPAM, the interaction with the water molecules around the macromolecule leads to volume exclusion effects, which contribute to enhance the chain stretching. The swollen conformation is therefore more favored compared to the collapsed state. A slightly stronger urea accumulation at short distances (r ≤ 1 nm) is found for high cu, which is more pronounced at high temperature, and demonstrates the preferential binding of urea to PNIPAM chains. However, the preferential exclusion of urea for low cu results in a slight dehydration, as demonstrated by the water accumulation behavior according to the KB integrals presented in Fig. 6 and the negative values for ΔH-bonds (water). This finding is in good agreement with the general explanation for the occurrence of a coil to globule transition for PNIPAM33–35 and the shift to lower transition temperatures. It can be assumed that urea molecules are preferentially excluded from PNIPAM at low cu where it is energetically unfavorable to replace water with urea molecules. The situation changes for high cu at high temperature, where a significant dehydration upon phase transition occurs and the strong binding of urea molecules is energetically favorable to compensate the loss of the water molecules. These two mechanisms are able to explain the two different slopes for the decrease of the transition temperature observed for different urea concentrations in Fig. 3.
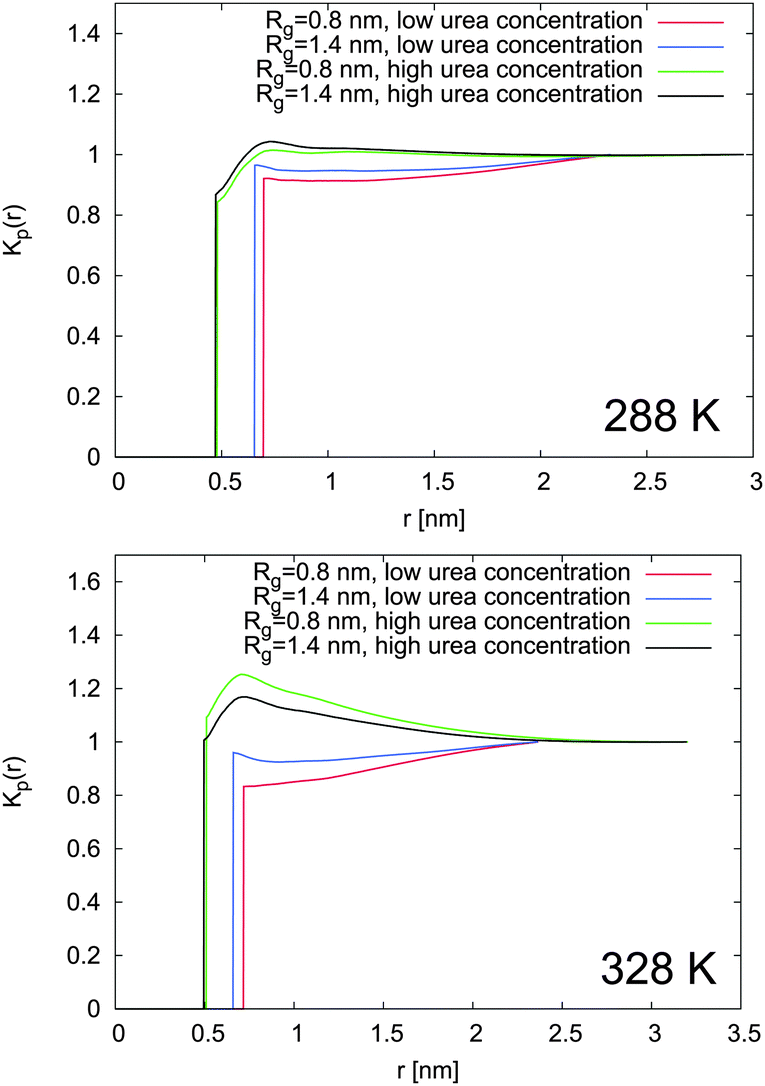 | ||
| Fig. 8 Local partition coefficient for swollen (Rg = 1.4 nm) and collapsed (Rg = 0.8 nm) PNIPAM conformations in presence of high and low molar urea concentrations at 288 K (top) and at 328 K (bottom). | ||
The shift of the chemical equilibrium towards swollen or collapsed PNIPAM conformations as shown in Fig. 4 can be rationalized with regard to the local/bulk partition coefficient free energy difference (eqn (17)). It becomes obvious that only the high molar urea concentration at 328 K indicates a positive value of ΔΔFKp ≈ 0.2 kJ mol−1 at a PNIPAM distance of r = 0.5 nm, which validates the energetic stability of the collapsed PNIPAM conformation. For all the other temperatures and concentrations, swollen conformations are preferred. Based on these findings, it can be concluded that the chemical equilibrium is slightly shifted towards the more swollen conformations for low cu at both temperatures, in agreement with the experimental results (Fig. 4), whereas only high temperatures and high cu induce a shift of the chemical equilibrium towards the collapsed state. The strong attraction of urea to PNIPAM in high concentrated solutions and at high temperatures may rationalize the previously discussed effects for urea in terms of a cross-linking agent between adjacent PNIPAM chains27,32,38,40,88 above a critical urea concentration. The presence of a direct urea binding can be also assumed with regard to the stagnating values for ϕsw at cu ≥ 2 mol L−1 which can be rationalized by a fully saturated urea shell around PNIPAM.
4 Summary and conclusions
In this work, the effect of urea on the conformational behavior of PNIPAM brushes was studied by combining optical experiments and atomistic molecular dynamics simulations. The reported results indicated a concentration-dependent binding behavior between urea and PNIPAM. In the low concentration regime (cu ≤ 0.5 mol L−1), urea is preferentially excluded from the PNIPAM backbone. Hence, the linear decrease of Ttr was rationalized by indirect effects of urea, which influence the dehydration process at high temperature by dilution of bulk water. However, despite the low number of urea molecules around PNIPAM in the first solvent shell, it was found that a more stretched conformation is stabilized due to a more favorable urea accumulation around the swollen conformation and excluded volume effects, as a consequence of the water–urea network around the macromolecule.92 In contrast, in the high concentration regime a preferential binding of urea to PNIPAM was found at both temperatures. Due to the small number of water molecules bound to PNIPAM, it can be assumed that the shift of the transition temperatures at high cu is mostly dominated by direct urea interactions with PNIPAM. Moreover, the high positive local/bulk partition coefficient of urea at 328 K supports the hypothesis of urea acting as a crosslinking agent between adjacent PNIPAM chains.32,38,88Thus, two basic mechanisms for different urea concentrations are proposed: (1) a preferential exclusion of urea from PNIPAM surface at low molar concentrations. This was rationalized by the experimental results, which supported a similar dehydration mechanism at low cu as in pure water, proving the absence of a direct binding; (2) a preferential binding of urea to PNIPAM above a critical concentration (cu ≥ 2 mol L−1). This produces a further decrease of the transition temperature in agreement to previous findings,38,82 and a cross-linking effect due to a favorable binding between adjacent chains with a similar mechanism as proposed earlier.38,88 Although several differences between numerical and experimental studies exist, for instance the consideration of a single chain in contrast to polymer brushes, a reasonable agreement between the results was found. This qualitative coincidence is of crucial importance, as it demonstrates that the experimental outcomes are strongly influenced by single chain properties and not by collective effects of the brush. Specific effects like cross-linking between the polymer chains which have been reported for high urea concentrations38,88 are not detectable by the simulations. However, indirect hints were found, like the strong accumulation of urea molecules around PNIPAM chains by a preferential binding mechanism, providing a reasonable explanation of the experimental observations. With regard to the two distinct accumulation regimes of urea, it has to be mentioned that explicit reasons for a similar behavior have been also recently discussed,22 and with regard to charged co-solutes, the presence of specific binding mechanisms for chaotropic ions was demonstrated.93,94
In conclusion, the conformational behavior and the transition temperatures of PNIPAM brushes are strongly affected by the presence of urea, either by indirect effects or by direct polymer–osmolyte interactions. This article demonstrates that the combined results of experiments and simulations point towards the most reliable methodology to study molecular mechanisms and their consequences for macromolecular conformations.
Acknowledgements
The authors greatly acknowledge helpful discussions with Anand Narayanan Krishnamoorthy, Ewa Anna Oprzeska-Zingrebe, Nico van der Vegt, Francisco Rodriguez-Ropero, Jan Heyda, Dominik Horinek and Pavel Jungwirth. The authors J. S., J. M. and C. H. thank the Deutsche Forschungsgemeinschaft through the cluster of excellence initiative ‘Simulation Technology’ (EXC 310) and the SFB 716 for financial support. M. A. S. thanks the excellence cluster “The Hamburg Centre for Ultrafast Imaging – Structure, Dynamics, and Control of Matter at the Atomic Scale” of the DFG. The authors S. M. and R. v. K. acknowledge the International graduate School IRTG 1524 (DFG) for the financial support.References
- P. H. Yancey, J. Exp. Biol., 2005, 208, 2819–2830 CrossRef CAS PubMed.
- D. R. Canchi and A. E. Garca, Annu. Rev. Phys. Chem., 2013, 64, 273–293 CrossRef CAS PubMed.
- D. Harries and J. Rösgen, Methods Cell Biol., 2008, 84, 679–735 CAS.
- R. Politi and D. Harries, Chem. Commun., 2010, 46, 6449–6451 RSC.
- B. J. Bennion and V. Daggett, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5142–5147 CrossRef CAS PubMed.
- R. Zangi, R. Zhou and B. Berne, J. Am. Chem. Soc., 2009, 131, 1535–1541 CrossRef CAS PubMed.
- D. A. Beck, B. J. Bennion, D. O. Alonso and V. Daggett, Methods Enzymol., 2007, 428, 373–396 CAS.
- A. M. Bhattacharyya and P. Horowitz, J. Biol. Chem., 2000, 275, 14860–14864 CrossRef CAS PubMed.
- A. K. Bhuyan, Biochemistry, 2002, 41, 13386–13394 CrossRef CAS PubMed.
- M. A. Schroer, M. Paulus, C. Jeworrek, C. Krywka, S. Schmacke, Y. Zhai, D. C. F. Wieland, C. J. Sahle, M. Chimenti, C. A. Royer, B. Garcia-Moreno, M. Tolan and R. Winter, Biophys. J., 2010, 99, 3430–3437 CrossRef CAS PubMed.
- M. A. Schroer, Y. Zhai, D. C. F. Wieland, C. J. Sahle, J. Nase, M. Paulus, M. Tolan and R. Winter, Angew. Chem., Int. Ed., 2011, 50, 11413–11416 CrossRef CAS PubMed.
- J. Smiatek, R. K. Harishchandra, O. Rubner, H.-J. Galla and A. Heuer, Biophys. Chem., 2012, 160, 62–68 CrossRef CAS PubMed.
- J. Smiatek, R. K. Harishchandra, H.-J. Galla and A. Heuer, Biophys. Chem., 2013, 180, 102–109 CrossRef PubMed.
- R. K. Harishchandra, S. Wulff, G. Lentzen, T. Neuhaus and H.-J. Galla, Biophys. Chem., 2010, 150, 37–46 CrossRef CAS PubMed.
- K. D. Collins, G. W. Neilson and J. E. Enderby, Biophys. Chem., 2007, 128, 95–104 CrossRef CAS PubMed.
- D. Horinek and R. R. Netz, J. Phys. Chem. A, 2011, 115, 6125–6136 CrossRef CAS PubMed.
- E. Schneck, D. Horinek and R. R. Netz, J. Phys. Chem. B, 2013, 117, 8310–8321 CrossRef CAS PubMed.
- A. Narayanan Krishnamoorthy, C. Holm and J. Smiatek, J. Phys. Chem. B, 2014, 118, 11613–11621 CrossRef CAS PubMed.
- K. D. Collins, Biophys. J., 1997, 72, 65–76 CrossRef CAS PubMed.
- K. D. Collins, Methods, 2004, 34, 300–311 CrossRef CAS PubMed.
- J. Smiatek, J. Phys. Chem. B, 2014, 118, 771–782 CrossRef CAS PubMed.
- J. Heyda and J. Dzubiella, J. Phys. Chem. B, 2014, 118, 10979–10988 CrossRef CAS PubMed.
- W. Lim, J. Roesgen and S. Englander, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2595–2600 CrossRef CAS PubMed.
- J. Dunbar, H. P. Yennawar, S. Banerjee, J. Luo and G. K. Farber, Protein Sci., 1997, 6, 1727–1733 CrossRef CAS PubMed.
- E. P. O'Brien, R. I. Dima, B. Brooks and D. Thirumalai, J. Am. Chem. Soc., 2007, 129, 7346–7353 CrossRef PubMed.
- T. Street, D. Bolen and G. Rose, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17064–17065 CrossRef CAS PubMed.
- F. Rodriguez-Ropero and N. F. A. van der Vegt, Phys. Chem. Chem. Phys., 2015, 17, 8491–8498 RSC.
- R. Breslow and T. Guo, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 167–169 CrossRef CAS.
- S. N. Timasheff, Biochemistry, 1992, 31, 9857–9864 CrossRef CAS PubMed.
- E. I. Tiktopulo, V. N. Uversky, V. B. Lushchik, S. I. Klenin, V. E. Bychkova and O. B. Ptitsyn, Macromolecules, 1995, 28, 7519–7524 CrossRef CAS.
- G. Graziano, Int. J. Biol. Macromol., 2000, 27, 89–97 CrossRef CAS PubMed.
- Y. Zhang and P. S. Cremer, Annu. Rev. Phys. Chem., 2010, 61, 63–83 CrossRef CAS PubMed.
- S. A. Deshmukh, S. K. Sankaranarayanan, K. Suthar and D. C. Mancini, J. Phys. Chem. B, 2012, 116, 2651–2663 CrossRef CAS PubMed.
- H. Du, S. R. Wickramasinghe and X. Qian, J. Phys. Chem. B, 2013, 117, 5090–5101 CrossRef CAS PubMed.
- H. Du, R. Wickramasinghe and X. Qian, J. Phys. Chem. B, 2010, 114, 16594–16604 CrossRef CAS PubMed.
- C. Hofmann and M. Schönhoff, Colloid Polym. Sci., 2009, 287, 1369–1376 CAS.
- C. H. Hofmann and M. Schönhoff, Colloid Polym. Sci., 2012, 290, 689–698 CAS.
- L. B. Sagle, Y. Zhang, V. A. Litosh, X. Chen, Y. Cho and P. S. Cremer, J. Am. Chem. Soc., 2009, 131, 9304–9310 CrossRef CAS PubMed.
- E. A. Algaer and N. F. van der Vegt, J. Phys. Chem. B, 2011, 115, 13781–13787 CrossRef CAS PubMed.
- F. Rodriguez-Ropero and N. F. van der Vegt, J. Phys. Chem. B, 2014, 118, 7327–7334 CrossRef CAS PubMed.
- Y. Gao, J. Yang, Y. Ding and X. Ye, J. Phys. Chem. B, 2014, 118, 9460–9466 CrossRef CAS PubMed.
- Y. Fang, J.-C. Qiang, D.-D. Hu, M.-Z. Wang and Y.-L. Cui, Colloid Polym. Sci., 2001, 279, 14–21 CAS.
- L. Liu, Y. Shi, C. Liu, T. Wang, G. Liu and G. Zhang, Soft Matter, 2014, 10, 2856–2862 RSC.
- K. Fuchise, R. Kakuchi, S.-T. Lin, R. Sakai, S.-I. Sato, T. Satoh, W.-C. Chen and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6259–6268 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- J. Gensel, T. Borke, N. P. Pérez, A. Fery, D. V. Andreeva, E. Betthausen, A. H. E. Müller, H. Möhwald and E. V. Skorb, Adv. Mater., 2012, 24, 985–989 CrossRef CAS PubMed.
- P. Akkahat and V. P. Hoven, Colloids Surf., B, 2011, 86, 198–205 CrossRef CAS PubMed.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- P. Laurent, G. Souharce, J. Duchet-Rumeau, D. Portinha and A. Charlot, Soft Matter, 2012, 8, 715–725 RSC.
- L. C. H. Moh, M. D. Losego and P. V. Braun, Langmuir, 2011, 27, 3698–3702 CrossRef CAS PubMed.
- S. Sanjuan, P. Perrin, N. Pantoustier and Y. Tran, Langmuir, 2007, 23, 5769–5778 CrossRef CAS PubMed.
- S. Christau, T. Möller, Z. Yenice, J. Genzer and R. von Klitzing, Langmuir, 2014, 30, 13033–13041 CrossRef CAS PubMed.
- X. Lu, L. Zhang, L. Meng and Y. Liu, Polym. Bull., 2007, 59, 195–206 CrossRef CAS.
- J. Xia and K. Matyjaszewski, Macromolecules, 1997, 30, 7697–7700 CrossRef CAS.
- J. B. Theeten and D. E. Aspnes, Annu. Rev. Mater. Sci., 1981, 11, 97–122 CrossRef CAS.
- Handbook of Ellipsometry, ed. H. G. Tompkins and E. A. Irene, William Andrew, Inc., Norwich, 2005, vol. 30, pp. 1–902 Search PubMed.
- J. R. Warren and J. A. Gordon, J. Biol. Chem., 1970, 245, 4097–4104 CAS.
- D. V. D. Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef CAS PubMed.
- A. Laio and F. L. Gervasio, Rep. Prog. Phys., 2008, 71, 126601 CrossRef.
- J. Smiatek and A. Heuer, J. Comput. Chem., 2011, 32, 2084–2096 CrossRef CAS PubMed.
- M. Bonomi, D. Branduardi, G. Bussi, C. Camilloni, D. Provasi, P. Raiteri, D. Donadio, F. Marinelli, F. Pietrucci and R. A. Broglia, Comput. Phys. Commun., 2009, 180, 1961–1972 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- H. J. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- J. Smiatek, A. Heuer, H. Wagner, A. Studer, C. Hentschel and L. Chi, J. Chem. Phys., 2013, 138, 044904 CrossRef PubMed.
- J. Smiatek, D. Janssen-Müller, R. Friedrich and A. Heuer, Physica A, 2014, 394, 136–144 CrossRef CAS.
- J. G. Kirkwood and F. P. Buff, J. Chem. Phys., 1951, 19, 774–777 CrossRef CAS.
- A. Y. Ben-Naim, Statistical thermodynamics for chemists and biochemists, Springer, 1992 Search PubMed.
- B. M. Baynes and B. L. Trout, J. Phys. Chem. B, 2003, 107, 14058–14067 CrossRef CAS.
- V. Pierce, M. Kang, M. Aburi, S. Weerasinghe and P. E. Smith, Cell Biochem. Biophys., 2008, 50, 1–22 CrossRef CAS PubMed.
- J. Smiatek, A. Wohlfarth and C. Holm, New J. Phys., 2014, 16, 25001 CrossRef.
- P. E. Smith, Biophys. J., 2006, 91, 849–856 CrossRef CAS PubMed.
- P. E. Smith, J. Phys. Chem. B, 1999, 103, 525–534 CrossRef CAS.
- S. Weerasinghe and P. E. Smith, J. Phys. Chem. B, 2003, 107, 3891–3898 CrossRef CAS.
- E. Courtenay, M. Capp, C. Anderson and M. Record, Biochemistry, 2000, 39, 4455–4471 CrossRef CAS PubMed.
- C. Wang, W. Shi, W. Zhang, X. Zhang, Y. Katsumoto and Y. Ozaki, Nano Lett., 2002, 2, 1169–1172 CrossRef CAS.
- H. Schild and D. Tirrell, J. Phys. Chem., 1990, 94, 4352–4356 CrossRef CAS.
- I. Bischofberger, D. C. E. Calzolari, P. De Los Rios, I. Jelezarov and V. Trappe, Sci. Rep., 2014, 4, 4377 CAS.
- H. G. Schild, M. Muthukumar and D. A. Tirrell, Macromolecules, 1991, 24, 948–952 CrossRef CAS.
- Y. Ono and T. Shikata, J. Am. Chem. Soc., 2006, 128, 10030–10031 CrossRef CAS PubMed.
- M. Shibayama, M. Shibayama, S.-Y. Mizutani, S.-Y. Mizutani, S. Nomura and S. Nomura, Macromolecules, 1996, 29, 2019–2024 CrossRef CAS.
- Y. Lu, X. Ye, K. Zhou and W. Shi, J. Phys. Chem. B, 2013, 117, 7481–7488 CrossRef CAS PubMed.
- S. Micciulla, P. A. Sánchez, J. Smiatek, B. Qiao, M. Sega, A. Laschewsky, C. Holm and R. von Klitzing, Soft Mater., 2014, 12, S14–S21 CrossRef.
- A. Luzar and D. Chandler, Nature, 1996, 379, 55–57 CrossRef CAS.
- D. van der Spoel, P. J. van Maaren, P. Larsson and N. Timneanu, J. Phys. Chem. B, 2006, 110, 4393–4398 CrossRef CAS PubMed.
- A. Soper, E. Castner and A. Luzar, Biophys. Chem., 2003, 105, 649–666 CrossRef CAS PubMed.
- K. B. Rembert, J. Paterová, J. Heyda, C. Hilty, P. Jungwirth and P. S. Cremer, J. Am. Chem. Soc., 2012, 134, 10039–10046 CrossRef CAS PubMed.
- P. Jungwirth and P. S. Cremer, Nat. Chem., 2014, 6, 261–263 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp07544k |
| This journal is © the Owner Societies 2016 |