Evidence for electrohydrodynamic convection as a source of spontaneous self-ordering in porous anodic alumina films†
Received
2nd December 2015
, Accepted 24th January 2016
First published on 2nd February 2016
Abstract
A comparative study of self-ordering behaviour of anodic alumina films fabricated in a series of diluted (down to 0.05 M) oxalic acid electrolytes allowed developing a relationship between the supporting electrolyte concentration and self-ordering voltages for the formation of porous oxide materials. Besides its practical importance, this work elucidates some fundamental principles of porous alumina formation, e.g. it suggests that the cell patterning arises from the electrohydrodynamic (EHD) convection process rather than the interfacial tension gradients near the anode surface (Marangoni-type instability).
Introduction
A considerable part of contemporary research in nanotechnology is linked to self-organized materials, such as porous anodic aluminium oxide (PAOX) layers.1 For instance, porous alumina membranes can be used in nanofluidic systems,2–4 for the template synthesis of rod-like nanostructures,5 or even as nanoreactors, which can regulate the degree of polymerization by the spatial confinement effect.6 For all these applications, the facile adjustment of both the structural parameters and the non-stoichiometric chemical composition of PAOX would be highly desirable. This could be achieved by increasing the number of suitable electrolytes for the optimized aluminium anodization conditions. However, a large share of well-organized mesoporous alumina films is still prepared for further laboratory tests via methods experimentally invented by Masuda et al. in the middle of 1990's (potentiostatic anodic oxidation of aluminium at a moderate voltage, and in most cases in a fairly concentrated single-solute aqueous electrolyte).7,8 It is commonly accepted that only voltages and concentrations from a narrow ‘process window’ lead to spontaneous self-organisation of a stable highly ordered nanoporous structure.9,10 These process windows are believed to be individual for every known electrolyte type, and are called the ‘self-ordering regimes’.11 Exploration of the spontaneous self-organisation in PAOX was initially motivated by numerous advantages of this approach over nanolithographic or nanoindentation texturing methods, especially in terms of simplicity and manufacturing cost (this was also emphasized in the early work of Masuda). However, in an attempt to alter the pore size and shape, many alternative approaches based on artificial pre-patterning of pore nucleation sites on aluminium substrates have been developed.12–14 At the same time, different factors influencing the growth behaviour of self-organized nanoporous arrays, such as electrolyte type,15 concentration,16 pH,17 voltage,18 and temperature16,19 have been intensively studied. Recently, we have proposed an empirical relationship between these parameters (some of them are included indirectly):20| |  | (1) |
where P is the criterion for the appearance of PAOX cell structure (the critical value was experimentally estimated for the most widely used electrolytes to be close to 0.057), qav is the average charge of supporting electrolyte anions (modulo, qav = 10−pH/C, C is the known acid concentration), ΔU is the applied voltage, η is the dynamic viscosity, and σ is the specific electric conductivity. Many recent theoretical and experimental studies show that the hexagonal patterning in PAOX is of convective origin.20–23 As the possible driving forces for nano-convection, either the interfacial tension gradient near the anode or the interaction of the electric field with charge carriers is usually considered. Other models attempting to explain the hexagonal pore arrangement are also in partial agreement with the convective model; however, they do not completely validate the process.21,24 For instance, the study featuring the tensile stress distribution as a possible factor assisting the pore ordering24 demonstrated the diffusion of tracer elements in alumina from the pore base to the pore walls. This experimental observation fully agrees with the nano-convective mechanism and does not give rise to a severe contradiction between two approaches (it is worth mentioning though that the convection is usually defined23 as a collective flow of particles in fluids and gases, but not in solids). However, the theoretical simulation of the viscous oxide flow in this work24 rests on the overly simplistic model of PAOX: a homogeneous Al2O3 continuum without electrolyte anion inclusion. This model neglects experimental observations such as the colloidal nature of the amorphous films, structural defects and flaws, bound water (up to 5.7 wt%), and the electrolyte species content up to 9.84 wt%.25–27 It is well known that the presence of grain boundaries and structural defects can significantly change the mechanisms and velocity of ion diffusion (the so-called grain-boundary diffusion). Thus, the concept of colloidal self-organisation20,26 is more consistent with experimental facts. It is very likely that the hexagonal cell arrangement in PAOX arises similarly to the patterns observed upon establishing the electrohydrodynamic (EHD) convection in colloidal systems (in the case of PAOX, this can be an in situ formed suspension layer of mobile charged alumina particles).23 Another convincing experimental fact that counts in favour of the structure-forming role of convection is the periodic damped I–t (electric current versus time) oscillation, which is typical for PAOX growth and anodic dissolution of some other metals where surfaces are etched in the form of hexagons due to a convective process.23 For instance, the shapes of I–t curves are almost identical for the aluminium anodization in a mixed electrolyte containing cerium salt28 and the potentiostatically controlled Cu|CuSO4|Cu electrolytic cell where the electrochemical analogue of Rayleigh–Bénard convection takes place.29
In this study, we explore the self-ordering behaviour of PAOX layers formed at new low concentrations of oxalic acid electrolyte (decreased down to 0.05 mol L−1, which is six times lower than that conventionally used for the well-established mild anodization regimes7). We systematically change the electrolyte concentration and explore new process windows for these nontrivial electrolyte solutions. This will provide the experimental test of our earlier hypothesis that the convective currents responsible for the hexagonal pattern can occur even at very low electrolyte concentrations, approaching deionised water.22 However, a sufficient amount of electrolyte is needed to ensure acidic pH < 4, since only electrolytes in which alumina is slightly soluble are known to produce porous solid films.30 This specific condition is concerned with the chemistry of PAOX formation (stabilisation of the in situ formed colloidal alumina particles) rather than establishing nano-convection, which is responsible for self-organisation.20 In addition, we test the interval of applicability of eqn (1), as well as the validity of the earlier found critical value of the criterion P, for different solutions. The clarified relation of concentration and self-ordering voltages will facilitate the design of further electrochemical experiments with free choice of electrolyte composition. Our experiments can improve our knowledge of the PAOX formation mechanism, particularly, of the possible driving force for nano-convection.
Results and discussion
Theoretical prediction of self-ordering voltages for new electrolyte concentrations using the earlier found value of P
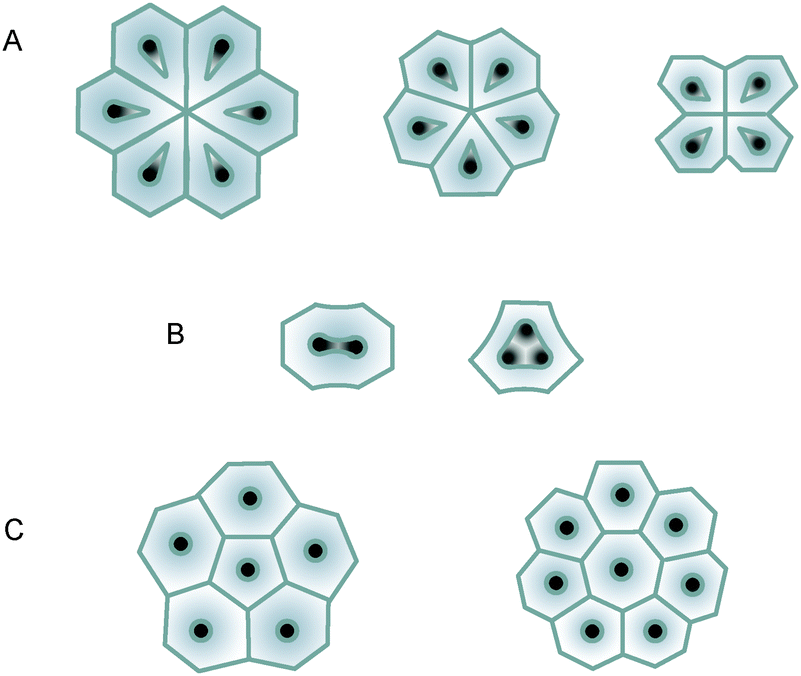
In order to determine the new self-ordering regimes, it is helpful to start with the measurement of macroscopic parameters of diluted electrolyte solutions and estimation of the new self-ordering voltages using eqn (1), and then refining these voltages experimentally. Before we begin the analysis of the electrolyte properties, one important detail must be mentioned regarding eqn (1) and the criterion P from ref. 20. The critical number P for honeycomb structure formation was found using the measured parameters of three well-known classical electrolytes for PAOX synthesis, i.e. 0.3 M oxalic acid, 0.3 M sulphuric acid, and 10 wt% phosphoric acid solutions. These compositions displayed pH close to 1.00, and thus were measured in the so-called ‘acidic error range’, where a glass electrode shows noticeably overestimated pH values. Although these readings cannot very largely influence the calculated values of qav and P, we must bear in mind this general shortcoming of glass electrodes. Table 1 summarizes the measured properties of the diluted oxalic acid solutions, including the dynamic viscosities, specific electric conductivities, and pH. As one can see, the average absolute anionic charge |qav|, which is derived from pH measurements, decreases with higher concentrations. This effect is entirely clear because the degree of dissociation is larger for diluted solutions. However, 0.3 M composition slightly deviates from the general dependence. This observation is very likely caused by the non-linear response of the glass pH electrode when approaching low pH values of about 1.00 (the above-mentioned instrumental ‘acidic error’). Eqn (1) suggests voltages appropriate for the transition from non-porous to honeycomb alumina for all the oxalic acid concentrations under study; the specific electric conductivities are above the minimal required value of the self-assembly (σ = 4–5 mS cm−1) in all cases as well.22 Thus, our calculations indicate that the formation of nanoporous alumina films is possible in all the selected electrolyte compositions. The theoretically predicted self-organization voltages from Table 1 are calculated for P = 0.057, i.e. for the centre of the confidence interval defined in ref. 20. Since this confidence interval (0.057 ± 0.024) was broadened due to only three values used in statistical treatment, variations of the required voltages in a certain range are possible. Therefore, the initial experiments were followed by the empirical refinement of the applied voltages to optimize the pore size uniformity and the degree of ordering. The refined self-ordering voltages ΔU were determined by the optimum combination of morphological features such as the size of long-range hexagonally ordered domains (single six-fold coordinated pores are not counted, although highlighted in the SEM-micrographs), regular hexagonal shape of cells, and a single pore per cell. Main types of point defects, which distort the hexagonal pattern and constitute the disordered areas, are illustrated in Scheme 1.
Table 1 Measured parameters of H2C2O4 solutions at 0 °C, calculated and experimentally refined voltages, observed pore diameters dp, and revised values of criterion P
| Concentration (M) |
Measured macroscopic solution parameters |
Calculated ΔUb (V) |
Initial dp (nm) |
Critical ΔU (V) refined by experimentc |
d
p (nm) after optimization |
Specific numerical values of P |
|
η (cP) |
σ (mS cm−1) |
pH |
|qav| |
|
Solution properties are taken from ref. 20, optimized ΔU and the corresponding dp from ref. 7.
Approximated to whole numbers.
Determined by the optimal combination of morphological features such as the size of long-range ordered domains and the regular hexagonal shape of defect-free cells containing single pores.
|
| 0.3a |
1.89 |
64.0 |
1.30 |
0.167 |
— |
— |
40a |
40a |
0.055 |
| 0.2 |
1.88 |
40.5 |
1.54 |
0.144 |
30 |
18 ± 2 |
43 |
27 ± 2 |
0.081 |
| 0.1 |
1.86 |
26.1 |
1.69 |
0.204 |
14 |
9 ± 1 |
44 |
24 ± 2 |
0.185 |
| 0.05 |
1.81 |
16.1 |
1.80 |
0.317 |
5 |
6 ± 1 |
45 |
32 ± 2 |
0.490 |
 |
| | Scheme 1 Basic types of point defects in PAOX: (A) missing pores, surrounded by a different number of satellites; (B) multiple pores within one cell; (C) pores surrounded by 5 or 7 neighboring pores (rarely 8) instead of 6. The five-fold and seven-fold coordinated pores are usually adjacent to each other to annihilate the strain. Besides the depicted defects, cells with six satellites and single pores within, but with strongly distorted geometry can also be mentioned as local imperfections. | |
Experimental refinement of anodizing voltage in 0.2 M oxalic acid
Calculation of the required voltage using the earlier found confidence interval for P (0.057 ± 0.024)20 sets a very broad process window (30 ± 13 V) for self-ordering in 0.2 M oxalic acid. Anodization at 30 V resulted in porous film formation with mainly short-range hexagonal pore arrangement (see Fig. 1, above left). As the applied voltage increases, individual cells acquire a well-defined hexagonal shape. However, even at the 40 V potential difference (which yields reproducibly good results for routinely used 0.3 M concentration7), the disordered areas still prevail over the hexagonally ordered ensembles. The size of ordered domains undoubtedly reaches a micrometer-scale at 42 V, and the best results could be achieved at 43–45 V. As the voltage reaches 46 V (Fig. S1 of the ESI†), the ordered domains are still up to 2.5–3 μm large, but double pores within a cell start to occur frequently. Such point defects often denote that the oxidation voltages exceed the optimal values. For instance, if anodization is conducted in 0.3 M solution, but at an increased voltage of 60 V, the majority of cells in the resulting PAOX layers will have double or triple pore openings (see Fig. S2, ESI†). Thus, we can consider 42–43 V as the suitable critical voltages for 0.2 M oxalic acid, above which the convection preferentially appears in the form of long-range ordered hexagonal cells, and 44–45 V as the upper limit of the optimal process window. Due to irregular shapes of hexagonally ordered domains, an accurate comparison of their sizes at different oxidation voltages is difficult. Thus, the found process window of 42–45 V has slightly uncertain limits because of the mainly qualitative assessment of the degree of ordering. It should also be noted that the PAOX layers formed in a 0.2 M electrolyte solution demonstrated a higher stability of the hexagonal pattern than layers from more diluted compositions, which are discussed later on in this work, as well as higher tolerance to moderate voltage variations. So, even at 32 V (which is 10–11 V lower than the estimated critical ΔU), the ordered domain size was still 0.2–0.8 μm. Of course, a smaller specific electric conductivity of 0.2 M oxalic acid and weaker ionic currents during the electrolysis (in comparison with the commonly used 0.3 M concentration) cannot provide the same degree of aluminium substrate pre-texturing (in the form of hexagonally ordered uniform concaves, underlying the alumina cells), and the remaining natural defects, such as pits, ridges and scratches, will bring distortions to the hexagonal layout. Although the experimentally refined critical voltage belongs to the initially specified confidence interval 30 ± 13 V, P evidently needs recalculation for the new electrolyte concentration. If we accept 43 V as the new critical voltage, the value of P is equal to 0.081, which considerably exceeds P = 0.055 for the classical 0.3 M oxalic acid.20 As was established in our preceding paper,22 the earlier found value of criterion P can be actually underestimated for weaker diluted organic acids, and very likely needs a correction for a better hexagonal pore arrangement. Taking into account the large mismatch between the previous and the current values of P (47.3%), this result cannot be solely ascribed to the moderate instrumental error during pH-measurements, and has another underlying reason. As we show later on in this work, the criterion P appears to be indeed non-constant (systematically changed) for different supporting electrolyte concentrations.
 |
| | Fig. 1 SEM micrographs of the porous alumina films formed in 0.2 M oxalic acid electrolyte solution after the variation of voltages in the 30–45 V range (all the SEM images from this work are taken at consistent 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× and 500000× magnifications). Hexagonally ordered domains are colored in cyan, while disordered areas are highlighted in red. An extended version of this figure, including several additional tested voltages in the interval between 30 and 41 V, as well as at ΔU exceeding 45 V, is available in the ESI† in Fig. S1. 000× and 500000× magnifications). Hexagonally ordered domains are colored in cyan, while disordered areas are highlighted in red. An extended version of this figure, including several additional tested voltages in the interval between 30 and 41 V, as well as at ΔU exceeding 45 V, is available in the ESI† in Fig. S1. | |
Experimental refinement of anodizing voltage in 0.1 M oxalic acid
According to prior calculations, the predicted self-ordering voltage for 0.1 M anodizing electrolyte lies presumably within the interval 14 ± 6 V. Oxidation at 14 V resulted in an entirely disordered pore layout (see Fig. 2). No hexagonal structure was observed at the upper confidence limit as well (see the SEM images of the samples obtained at 19 V and 21 V in Fig. 2). The first sporadically ordered fragments occurred only at 25 V and had dimensions up to 0.4 μm. As the voltage increases, the regularity in the pore layout slightly improves. However, PAOX layers formed at practically all tested voltages showed high randomness in the pore arrangement, and the tendency to ordering was noticeably lower than during the experiments in 0.2 M oxalic acid. The most stable and reproducible hexagonal patterns were obtained at the voltages of 43–44 V. This incipient hexagonal arrangement was fully lost already at the voltage of 45 V. Moreover, the 45 V PAOX layers frequently demonstrated the same type of point imperfections as earlier discussed for the films formed in 0.2 M solution at excess voltages, i.e. double pores within one cell (see the corresponding close-up SEM image in Fig. S3, ESI†). Thus, the 44 V potential difference can also be considered as the upper limit of the narrow optimized process window. As in the case of 0.2 M anodizing electrolyte, the experimentally refined self-ordering voltage for the 0.1 M composition is beyond the theoretically predicted range, and the criterion P certainly needs a correction. If we take 44 V as the critical voltage for the 0.1 M electrolyte concentration, the new value of P is equal to 0.185, and thus exceeds both previous results for the more concentrated 0.2 M and 0.3 M compositions. The discrepancy between the predicted self-ordering voltages and the experimental critical ΔU for 0.1 M oxalic acid is about 23–24 V, and the deviation from the previously found value of the critical number P is now 236%. Now, it is beyond any doubt that the gap between the values calculated using eqn (1) and the empirical values results from reasons other than the measuring error during the pH determination. On the other hand, the mismatch between the predicted and experimentally optimized critical voltages and the criterion P systematically increases, when approaching low electrolyte concentrations. This suggests that the critical parameters for the self-ordering can depend on the anodizing electrolyte concentration, and even have a functional correlation with it.
 |
| | Fig. 2 SEM examination of the PAOX films obtained from 0.1 M oxalic acid anodizing electrolyte at voltages within the range of 14–44 V (close-up images are taken at 500000× magnification, and the overall views at 100000×). The extended version of this microscopic analysis with additional tested voltages is demonstrated in Fig. S3 of the ESI.† The color scheme showing ordered and disordered areas on the PAOX layers is identical to the one used in Fig. 1: ordered domains are highlighted with cyan, and disordered fields with red. | |
Experimental refinement of anodizing voltage in 0.05 M oxalic acid
Calculations using P from ref. 20 suggest that the self-ordering in the most diluted 0.05 M electrolyte should occur at approximately 6 ± 1 V. The films obtained at the lowest test voltage of 5 V have a well-defined porous structure. However, instead of a cell pattern, they revealed a spongy morphology (Fig. 3). No hexagonal ordering could be observed even at voltages about 10–20 V higher than initially predicted by eqn (1). For the 0.05 M anodizing electrolyte, an optimal combination of the regular hexagonal shape of individual cells and the large size of ordered domains (up to 1 μm) occurs at the voltage of 45 V, which very likely should be considered as critical for this concentration. At 46 V, the cells acquire even a clearer and sharper hexagonal form, but the size of ordered domains becomes noticeably smaller. At higher voltages, the hexagonal pattern disappears completely, and individual cells begin to suffer from defects such as double and triple pores (see the corresponding SEM images of the sample obtained at 48 V in Fig. S4 of the ESI†). If we consider 45 V as the new critical voltage for 0.05 M oxalic acid, the corrected new value of P is 0.490. Thus, it went up by a factor of 8.9 in comparison with the critical number for 0.3 M oxalic acid electrolyte. It should be mentioned that the comparison of the samples obtained from 0.05 M oxalic acid is fairly difficult because of the poor self-ordering in practically all cases. Very low supporting electrolyte concentration cannot provide sufficient current densities for the efficient structuring of the Al substrate in the form of hexagons, and for maintaining a reasonable reaction velocity for spontaneous rearrangement into a well-organized pattern. In the whole range of tested voltages, self-ordered domains were observed as sporadic structures in mainly disordered PAOX, and only somewhat intuitive selection based on qualitative assessment was possible. When determining the critical voltage for the most diluted solution, we should also pay attention to the general trend in the whole survey of all other tested concentrations.
 |
| | Fig. 3 SEM micrographs of the PAOX films formed in 0.05 M oxalic acid electrolyte at different anodizing voltages. While the regular hexagonal shape of individual cells can be observed at a number of voltages above 40 V, the size of ordered domains is relatively small at all tested voltages, and reaches the largest reproducible values at 45 V. The tendency to self-ordering at the voltages of 40–42 V seems surprising because of the conflict with the general trend of critical voltages for other tested electrolyte concentrations. It is still not entirely clear, whether the self-ordering can improve repeatedly in the same solution, as the voltage increases. | |
Theoretical analysis of the experimental data
At first glance, there is a colossal mismatch between the theoretical calculations and the experimental results at low supporting electrolyte concentrations (see Fig. 4 for comparison), which may suggest that the empirical eqn (1) is invalid. However, we can easily explain this seeming difference if we assume that the honeycomb structure in PAOX is a product of the electrohydrodynamic (EHD) convection, which is driven by the interaction of the electric field E with excess space charge density qex in the solution (volume electric forces). Here, it would be helpful to start with the comparative analysis of eqn (1) and another rigorously theoretically derived and experimentally confirmed expression for the electrical analogue of Rayleigh number Rae, which determines the critical conditions, above which the EHD convection sets in:23,31| |  | (2) |
where εr is the relative permittivity of the fluid, ε0 is the permittivity of vacuum, us is the ionic mobility of species s (i.e. of the charge carriers, contributing to the Faraday current), and the rest of the parameters in the second part of eqn (2) entirely corresponds to the parameters of eqn (1). Now, if we look at the physical meaning of the ionic mobility us, it can be defined as the electric conductivity at infinite dilution. Ionic mobility was initially considered for the description of the solution electric properties in eqn (1), but specific electric conductivity σ was introduced instead because it is easily measurable and suitable for the routine laboratory prediction of new self-ordering regimes.20 The part of the numerator εrε0 is linked to the excess space charge density carried by the liquid, as expressed by the Poisson equation:
| qex = εrε0divE |
which has a close relation to the parameter qav from eqn (1) (qav is determined as the ratio of the total negative charge of all anions in the solution to the supporting electrolyte concentration C). Hence, although eqn (1) was found and confirmed only empirically, it appears to be written strikingly similar to the theoretically derived expression for the critical dimensionless number for the onset of the EHD convection.
 |
| | Fig. 4 Comparison of the calculated process windows using P = 0.057 ± 0.024 for all tested concentrations, and experimentally optimized self-ordering voltages for aluminium anodization in 0.05–0.3 M oxalic acid electrolytes. | |
For the appearance of the EHD convection in a non-conducting non-electroactive medium (i.e. in a purely physical system), a high voltage of the order of 104 V is required.23 However, in the presence of even trace amounts of conducting species, the required voltages for the development of sufficient spatial charge and the onset of the EHD convection drop to a few volts. Moreover, the required critical voltage decreases further, if the unipolar charge injection (i.e. the injection of ions of the same sign from the side of only one electrode) is substituted by the bipolar charge injection (when charge carriers of both signs are injected into the solution from the opposite electrodes). For instance, the initially experimentally confirmed critical value of Rae from eqn (2) appeared to be invalid for the systems with symmetrical bipolar charge injection. In such electrochemical systems, lower critical voltages were needed for the onset of the EHD convection, and the new critical value of Rae was approximately two times smaller than the previously determined. This situation is totally identical to what we observe in our current investigation of the electrochemical system for the PAOX growth: as summarized in Table 1, the critical value of the criterion P drops from 0.490 to 0.055, as the supporting electrolyte concentration increases from 0.05 M to 0.3 M (also see Fig. 5 for graphic comparison). Here, however, the complexity of the electrochemical cell for PAOX formation, as well as the complexity of water as solvent, must be mentioned. While physical systems seem to be the perfect models for isolation and a separate study of different factors that can affect the onset of instability (in other words, one can start with the simplest plain non-conducting non-electroactive fluid, then add charge carriers in different proportions, then test unipolar, bipolar injections etc.), the electrochemical cell for PAOX synthesis always includes a tight indivisible combination of all parameters, with a very complex interplay between them. Firstly, although pure ‘deionised’ water has a high dielectric constant, strictly speaking, it is never completely ion-free because of the dissociation of water molecules. Secondly, water plays the roles of the fluid medium and the electro-active species at the same time, and the electrode potentials of the redox reactions with participation of water depend on pH (i.e. the concentration of supporting electrolyte). Finally, the opposite electrodes during aluminium anodization make only one kind of charge carrier injection possible, which results from the generation of protons at the anode and their consumption at the cathode and conversion into gaseous H2. In addition, if not only the characteristics of the anode/solution interface are considered as the source of dynamic instabilities, but the whole electrochemical circuit, many additional external factors may come into play. For instance, if the thermoelectric (Seebeck, Peltier, and Thomson) effects cannot be neglected, they will surely depend on the ambient temperature, the material of electrode clamps, or the height of Al electrodes above the cooled solution, in which they are immersed. Furthermore, if we consider the PAOX pattern as a product of EHD convection in a colloidal system (combining the nano-convective model with the idea of colloidal gel proposed by Michelson26), more parameters, such as the mobility of colloidal particles or the thickness of the suspension layer over the anode surface, must be regarded.
 |
| | Fig. 5 Dependence of the critical values of criterion P on the oxalic acid anodizing electrolyte concentration, corresponding to moderate (mild) self-ordering voltages. Note that the value of P is equal to 0.055 for 0.3 M oxalic acid electrolyte (on the plot), whereas the average P for three mostly used electrolytes in ref. 20 is equal to 0.057. | |
The interpolated P–C curve from Fig. 5 can be expressed as:
where 0.055 is equal to the critical value of
P for 0.3 M anodizing electrolyte. Although the physical meaning of the concentration-dependent summand still needs to be clarified, all experimentally observed points obtained for the critical values of the criterion
P coincide with the exponential fitting curve with practically no scattering. Thus, there is obviously a strong functional correlation between the critical parameter
P for cell pattern appearance and the electrolyte concentration.
Experimental
Aqueous oxalic acid electrolytes of different concentrations (0.2, 0.1, and 0.05 mol L−1) were prepared from oxalic acid dihydrate (≥99 wt%, extra pure, purchased from Acros Organics) and distilled water (σ ≈ 2 μS cm−1) using standard analytical volumetric flasks. The kinematic viscosities of solutions were measured with Schott Viscoclock Micro-Ubbelohde Viscometer (capillary diameter 0.53 mm). Samples' temperature was controlled with Schott CT 1650 thermostat and CK 160 cooler. The densities of solutions at 0 °C were obtained on an Anton Paar DMA 5000 density meter. The specific conductivity was measured using a WTW LF 538 conductivity meter using a Lauda RM6 thermostat. pH values were determined using a WTW Digital pH Meter, pH 525. Aluminium sheets (PURALUX®, purity 99.5–99.93%) were anodized in the electrolyte solutions under potentiostatic conditions at 0–0.5 °C using a Sorensen XEL 250 DC power supply. After the first oxidation for 20 hours, alumina was removed in an etching aqueous solution (0.16 M K2Cr2O7 and 1.5 M H3PO4). Then, the main oxidation step was performed for another 20 hours. The aluminium substrates were used ‘as is’, without prior electropolishing. Scanning electron microscopy (SEM) studies of the resulting porous films were performed using a Philips XL-30 FEG, operated at 20–25 kV. Samples were mounted on conductive carbon-rich polymer films and sputtered with Pt/Pd alloy. The pore sizes (dp) and interpore distances (Dint) were evaluated using measuring tools of microscope control software or image processing programs. During the statistical treatment, confidence intervals were defined by the significance level α = 0.05 from 15 random measurements; dp were measured in high resolution SEM images between the contrasting pore margins (without special visualization techniques), and Dint between centres of randomly chosen neighbouring pores. To bring the confidence intervals in conformity with the electron-scan microscope resolution, the mean dp and Dint values were approximated to whole numbers.
Conclusions
As exemplified by our study of aluminium anodization in a series of diluted oxalic acid electrolytes, the formation of a spatial hexagonal pattern in porous anodic alumina layers conforms to the same rules as the self-ordering in other electrochemical systems, where the electrohydrodynamic (EHD) convection sets in. The dependence of the critical voltages ΔU and the critical values of the criterion P on supporting electrolyte concentration can be used for the experimental confirmation of the structure-forming role of the EHD convection, in analogy with the earlier studied similar physical and electrochemical cells. In this work, we clarify the nature of possible differences between the theoretically predicted process windows using eqn (1) and the actual experimental observations for some weak or highly diluted electrolytes, which may result from the misconception of the PAOX formation mechanism, and show that the critical value of the criterion P needs to be adjusted individually for every new supporting electrolyte concentration (the optimized values of P for any oxalic acid concentration in the range between 0.05 M and 0.3 M at the temperatures close to 0 °C can be obtained from the interpolated data in this work). We have demonstrated that the explanation for the hexagonal cell structure in PAOX by the onset of an EHD convective pattern is more viable than the currently prevailing hypothesis suggesting the convective instability of Marangoni type, which is caused by the interfacial tension gradients near the anode during the formation of porous films.
Acknowledgements
The contribution of Nalan Kalyon and Sharon Jeziorowski to the experimental part of this work is gratefully acknowledged.
References
-
H. Masuda, in Ordered Porous Nanostructures and Applications, ed. R. B. Wehrspohn, Springer, New York, 2005, vol. 1, pp. 37–55 Search PubMed.
- J. P. Fu, P. Mao and J. Han, Trends Biotechnol., 2008, 26, 311–320 CrossRef CAS PubMed.
- P. W. Bohn, Annu. Rev. Anal. Chem., 2009, 2, 279–296 CrossRef CAS PubMed.
- S. M. Wu, F. Wildhaber, O. Vazquez-Mena, A. Bertsch, J. Brugger and P. Renaud, Nanoscale, 2012, 4, 5718–5723 RSC.
- M. Pashchanka, J. Engstler, J. J. Schneider, V. Siozios, C. Fasel, R. Hauser, I. Kinski, R. Riedel, S. Lauterbach, H. J. Kleebe, S. Flege and W. Ensinger, Eur. J. Inorg. Chem., 2009, 3496–3506 CrossRef CAS.
- C. B. Gorman, R. J. Petrie and J. Genzer, Macromolecules, 2008, 41, 4856–4865 CrossRef CAS.
- H. Masuda and K. Fukuda, Science, 1995, 268, 1466–1468 CAS.
- H. Masuda, F. Hasegwa and S. Ono, J. Electrochem. Soc., 1997, 144, L127–L130 CrossRef CAS.
- A. P. Li, F. Muller, A. Birner, K. Nielsch and U. Gosele, J. Appl. Phys., 1998, 84, 6023–6026 CrossRef CAS.
- F. Y. Li, L. Zhang and R. M. Metzger, Chem. Mater., 1998, 10, 2470–2480 CrossRef CAS.
- K. Nielsch, J. Choi, K. Schwirn, R. B. Wehrspohn and U. Gösele, Nano Lett., 2002, 2, 677–680 CrossRef CAS.
- H. Asoh, K. Nishio, M. Nakao, T. Tamamura and H. Masuda, J. Electrochem. Soc., 2001, 148, B152–B156 CrossRef CAS.
- Y. Matsui, K. Nishio and H. Masuda, Small, 2006, 2, 522–525 CrossRef CAS PubMed.
- H. Asoh, K. Nishio, M. Nakao, A. Yokoo, T. Tamamura and H. Masuda, J. Vac. Sci. Technol., B, 2001, 19, 569–572 CAS.
- S. Shingubara, K. Morimoto, H. Sakaue and T. Takahagi, Electrochem. Solid-State Lett., 2004, 7, E15–E17 CrossRef CAS.
- M. A. Kashi and A. Ramazani, J. Phys. D: Appl. Phys., 2005, 38, 2396–2399 CrossRef CAS.
- A. L. Friedman, D. Brittain and L. Menon, J. Chem. Phys., 2007, 127, 154717 CrossRef PubMed.
- G. D. Sulka and K. G. Parkola, Thin Solid Films, 2006, 515, 338–345 CrossRef CAS.
- G. D. Sulka and K. G. Parkola, Electrochim. Acta, 2007, 52, 1880–1888 CrossRef CAS.
- M. Pashchanka and J. J. Schneider, J. Mater. Chem., 2011, 21, 18761–18767 RSC.
- J. Sha, S. J. Lu, Z. S. Su and W. Z. Zhou, Chem. Commun., 2009, 5639–5641 Search PubMed.
- M. Pashchanka and J. J. Schneider, Phys. Chem. Chem. Phys., 2013, 15, 7070–7074 RSC.
-
M. Orlik, Self-organization in Electrochemical Systems II, Springer-Verlag, Berlin, Heidelberg, 2012 Search PubMed.
- J. E. Houser and K. R. Hebert, Nat. Mater., 2009, 8, 415–420 CrossRef CAS PubMed.
- G. E. Thompson, Thin Solid Films, 1997, 297, 192–201 CrossRef CAS.
- C. E. Michelson, J. Electrochem. Soc., 1968, 115, 213–219 CrossRef CAS.
- M. E. Mata-Zamora and J. M. Saniger, Rev. Mex. Fis., 2005, 51, 502–509 CAS.
- H. Wang and H. W. Wang, Mater. Chem. Phys., 2006, 97, 213–218 CrossRef CAS.
- B. Baranowski, J. Non-Equilib. Thermodyn., 1980, 5, 67–72 CAS.
- J. W. Diggle, T. C. Downie and C. W. Goulding, J. Electroanal. Chem., 1968, 18, 192–193 CrossRef CAS.
- J. M. Schneider and P. K. Watson, Phys. Fluids, 1970, 13, 1948–1954 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A more detailed set of microscopic analysis data for PAOX samples obtained at all studied electrolyte concentrations and all tested voltages in sequence. See DOI: 10.1039/c5cp07436c |
|
| This journal is © the Owner Societies 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a