
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural determination of niobium-doped silicon clusters by far-infrared spectroscopy and theory†
Xiaojun
Li
*a,
Pieterjan
Claes
b,
Marko
Haertelt
c,
Peter
Lievens
b,
Ewald
Janssens
*b and
André
Fielicke
*cd
aThe Key Laboratory for Surface Engineering and Remanufacturing in Shaanxi Province, School of Chemical Engineering, Xi'an University, Xi'an 710065, Shaanxi, P. R. China. E-mail: xjli@xawl.edu.cn
bLaboratory of Solid State Physics and Magnetism, KU Leuven, B-3001 Leuven, Belgium. E-mail: ewald.janssens@fys.kuleuven.be
cFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, D-14195 Berlin, Germany
dInstitut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstr. 36, D-10623 Berlin, Germany. E-mail: fielicke@physik.tu-berlin.de
First published on 27th January 2016
Abstract
In this work, the structures of cationic SinNb+ (n = 4–12) clusters are determined using the combination of infrared multiple photon dissociation (IR-MPD) and density functional theory (DFT) calculations. The experimental IR-MPD spectra of the argon complexes of SinNb+ are assigned by comparison to the calculated IR spectra of low-energy structures of SinNb+ that are identified using the stochastic ‘random kick’ algorithm in conjunction with the BP86 GGA functional. It is found that the Nb dopant tends to bind in an apex position of the Sin framework for n = 4–9 and in surface positions with high coordination numbers for n = 10–12. For the larger doped clusters, it is suggested that multiple isomers coexist and contribute to the experimental spectra. The structural evolution of SinNb+ clusters is similar to V-doped silicon clusters (J. Am. Chem. Soc., 2010, 132, 15589–15602), except for the largest size investigated (n = 12), since V takes an endohedral position in Si12V+. The interaction with a Nb atom, with its partially unfilled 4d orbitals leads to a significant stability enhancement of the Sin framework as reflected, e.g. by high binding energies and large HOMO–LUMO gaps.
1. Introduction
Over the past few decades, scientific interest in clusters has rapidly increased, as they constitute a class of materials with fascinating physical and chemical properties, which are distinct from those of individual atoms, molecules or bulk matter.1–5 In particular, clusters of the semiconductor material silicon have attracted a lot of theoretical and experimental attention due to their potential applications in the modern microelectronics industry.6–12 Different from carbon clusters that usually show sp2 hybridization, bonding in pure silicon clusters occurs through sp3 hybridization and thus they are more chemically reactive due to the existence of unsaturated dangling bonds on the clusters' surface, making them less suitable as nanoscale building blocks.13 However, it has been found that silicon clusters can be stabilized by doping with transition metal (TM) atoms,14–16 and by now a great number of studies, both experimentally17–29 and theoretically,30–36 have explored the structures and electronic properties of TM-doped silicon clusters for their potential use in silicon-based nanomaterials. In fact, doping silicon clusters with appropriate transition metal atoms37–41 can reduce the number of dangling bonds on the cluster surface or even fully saturate them via pd hybridization, and thereby change the geometrical structures and chemical reactivities compared to pure silicon clusters. Accordingly, novel physical and chemical properties of transition-metal doped silicon clusters, e.g., size-specific stability,30,42 magnetic,17,28,37,39,43 and optical properties,44 open up many new and exciting applications, for instance, for silicon-based nano-devices in optoelectronics, tunable lasers, and sensors.For example, Hiura et al.11 reported the formation of a series of silicon cage clusters with endohedral TM atoms, TM@Sin+ (TM = Hf, Ta, W, Re, Ir, etc.), and showed by density functional theory (DFT) calculations that W@Si12 is a very stable cluster due to both electronic and geometrical shell closures. Endohedral TM@Sin (TM = W, Zr, Os, Pt, Co) clusters have been extensively investigated theoretically, in order to test their potential as self-assembly building blocks.15,24,45,46 The properties of the endohedral clusters were shown to depend largely on the cluster size and the type of TM atoms.37,47 Silicon clusters doped with a lanthanide atom retaining a partially filled 4f shell, such as SinLn− (Ln = Ho, Gd, Pr, Sm, Eu, Yb), have been suggested as magnetic silicon-based nanomaterials.17 Interestingly, the fully TM-encapsulated silicon clusters, TM@Si16 (TM = Ti and Zr), are also predicted to exhibit luminescence in the visible range, making them attractive for optoelectronic applications,44 and the neutral TiSi16 cluster has a closed-shell electron configuration with a large gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).18 The isoelectronic TaSi16+ has been recently reported to form stable heterodimers with C60 upon deposition from the gas phase.48
Experimentally, information on relative stabilities as well as geometric and electronic structures has been obtained from mass spectrometric observations,18,49,50 reactivity experiments that probe the location of the dopant atoms,43,50–52 photodissociation experiments,53 photoelectron spectroscopy,17–20,50 and infrared spectroscopy.23–29 However, from most experimental studies detailed geometrical information cannot be directly deduced and comparison with theoretically predicted properties is required for structural assignments. For example, from experimental infrared spectra obtained using either IR multiple photon dissociation or IR-UV two-color ionization together with theoretical studies the structures for neutral SinTM (TM = V, Mn, Co)23,24 and cationic SinTM+ (TM = V, Mn, Cu, Ag, Au)25–29 have been determined. The comparison with the experimental findings also allows to test the suitability of different theoretical approaches for predicting the ground state structures.
In general, there is still only limited systematic understanding of how doping with different TMs influences structural properties and, in particular, the chemical bonding in the doped clusters, although this may be seen as a prerequisite for designing silicon-based nanomaterials with tailor-made properties. Previous studies25,27,29 on Cu, Ag, and Au doped Si clusters allow for a direct comparison of the effects of the different coinage metal dopants. Overall, for these three metals rather similar structures are assigned for the doped clusters, having the dopants at low-coordinated sites due to the similar electronic configuration with completely filled d orbitals. Gradual differences between the binding behavior of Cu Ag, and Au are due to the larger atomic radii of the latter two atoms. In contrast to the coinage metals, V with an [Ar]3d34s2 atomic configuration is able to accept more electrons to participate in the chemical bonding, which is reflected in a higher coordination of the V dopant in silicon clusters.27 In this paper, we focus on Nb, having a [Kr]4d45s1 atomic ground state, as a dopant. The question is raised in how far the different atomic configuration of Nb or the larger atomic radius (198 pm vs. 171 pm) may affect the structures of the doped clusters compared to its lighter homologue V.
In the following sections, we report on a combined experimental and theoretical study investigating the structures, stabilities, and electronic properties of cationic Nb-doped silicon clusters, SinNb+ (n = 4–12), applying infrared multiple photon dissociation (IR-MPD) spectroscopy to the cationic cluster–Ar complexes together with density functional theory (DFT) calculations.
2. Experimental and theoretical methods
2.1 Experimental methods
The experiments are performed in a molecular beam setup, which contains a dual laser vaporization source54 and a time-of-flight mass spectrometer equipped for infrared (IR) excitation experiments.55 The setup has been connected to a beamline of the Free Electron Laser for Infrared eXperiments (FELIX) in Nieuwegein, which recently has been moved to the RU Nijmegen, the Netherlands.56 The source parameters are optimized for the formation of cold singly niobium doped silicon clusters. Argon atoms, which act as messenger atoms, are attached to the clusters to record the IR spectra of the clusters in the gas phase. The formation of cluster–argon complexes is induced at ∼100 K by the addition of a fraction of ∼2% of Ar to the He carrier gas, respectively. Resonant absorption of IR photons and subsequent vibrational energy redistribution heat the clusters and may result in evaporation of the weakly bound rare gas atom. The IR-MPD spectra are constructed by recording the ion intensities of the cluster–rare gas complexes as a function of the FELIX frequency in the 260–590 cm−1 range. From the depletion spectra, IR absorption spectra are calculated as described previously.552.2 Theoretical methods
To aid structural assignments, the low-lying structures of the cationic SinNb+ (n = 4–12) clusters were searched using the stochastic ‘random kick’ procedure,57,58 interfaced with the Gaussian 09 program.59 The procedure starts with random structures for each cluster size. Then the ‘kick’ procedure is performed to generate a large number of possible starting geometries that are used for further local optimizations, within a sphere-shell with the radius between Rmin and Rmax (Rmin = 0, Rmax = 4–7 Å). This is repeated until no new minima appeared. The choice of Rmax is chosen to be 2–3 times the Si–Si or Si–Nb bond length; the minimum distance (Dmin) between two atoms is set to be 2.0 Å, which is smaller than the Si–Si or Si–Nb bond length.60 All kicked structures were optimized by DFT with the pure GGA functional BP8661,62 in conjunction with def-SVP basis sets for energy evaluation. This computational level has proven to reproduce well the experimentally observed spectra for different TM-doped silicon clusters, such as SinAg+ (n = 6–15),29 SinV+ (n = 4–11) and SinCu+ (n = 6–11).27 In the latter study, however, it was found that relative energies are better predicted by the hybrid functional B3LYP.61,63 In order to obtain a reasonable probability for predicting the global minimum (GM), each cluster size was searched two or three times by using the stochastic ‘kick’ procedure with different Rmax. All initial optimizations were carried out without symmetry constraint, and singlet and triplet spin states were examined for each starting structure. If any local minimum is found to have a specific symmetry, the structure was reoptimized again within that symmetry. Vibrational frequency analyses were employed to confirm that these structures are true minima. To aid the visual comparison with the experimental spectra the calculated infrared stick spectra were convoluted with a Gaussian function with a full width at half-maximum (FWHM) of 10 cm−1. In order to fit well with the experimental data a frequency scaling factor of 1.04 was applied, similar to previous studies.9,27,29,64The reported relative energies of the Nb-doped silicon clusters do not include zero-point vibrational energies (ZPVE) as they are small and almost provide the same energetic ordering (see the energy evaluation for BP86 and B3LYP methods shown in Table S1 of the ESI†). To test the influence of the Ar ligand, calculations were performed on cluster–Ar complexes for selected cluster sizes. The results show that Ar does not affect the energetic ordering for most low-energy isomers, with the exception of Si11Nb+ and has only a minor effect on the vibrational frequencies and infrared intensities (see Fig. S1(a–d) of the ESI†). One of the largest Ar induced shifts is observed for the Si12Nb+ cluster, which is subjected to a small blue shift of less than 9 cm−1 for the high-frequency region (see Fig. S1(d) of the ESI†).
3. Results and discussion
In this section, we first present the experimental IR-MPD spectra in the 260–590 cm−1 range and compare them with the calculated infrared spectra of different low-lying isomers to obtain structural assignments for the cationic SinNb+ (n = 4–12) clusters. The identified low-lying structures for each cluster size are labeled as iso1, iso2, iso3, etc., along with increasing relative energies obtained at the BP86/def-SVP level of theory. For the smallest cluster sizes (n = 4–6) a favorable agreement between the experimental spectra and the ones calculated for the putative ground states is obtained and the energy gaps to higher isomers are large. Therefore these are only briefly discussed. For some cases, such as Si7Nb+ and Si12Nb+, additional calculations are performed using the B3LYP functional (full optimization) to verify the relative stabilities of the low-lying isomers. Further on, the clusters' growth mechanism, electronic structure and bonding are discussed in detail. We estimate the error in the given experimental frequencies to be about ±2 cm−1, mainly stemming from uncertainties in determining the band positions and errors in the calibration of the IR frequencies.3.1 Structure determination
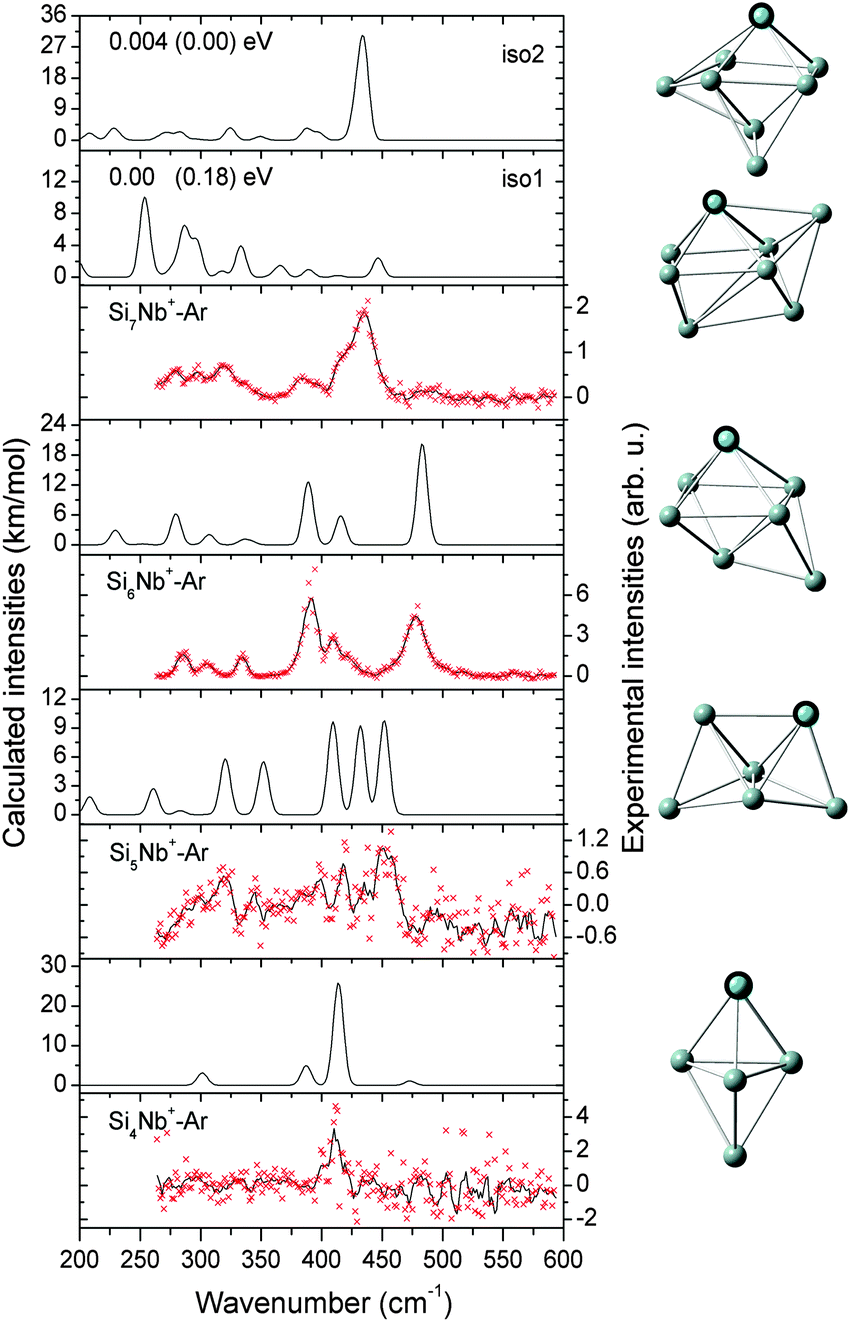 | ||
| Fig. 1 Comparison of experimental IR-MPD spectra (data points [red] together with 5-point running average [black line], right axis) of SinNb+–Ar (n = 4–7) and the corresponding calculated IR spectra (left axis). For Si7Nb+ the relative energies for two isomers are given at the BP86/def-SVP level of theory and at the B3LYP/def-SVP (in parentheses) level of theory. The assigned structures are shown on the right-side of the corresponding spectra. | ||
 | ||
| Fig. 2 Comparison of the experimental IR-MPD spectrum (data points [red] together with 5-point running average [black line], right axis) of Si8Nb+–Ar and the corresponding calculated IR spectra (left axis) of low-lying isomers. The isomers are ordered by their relative energies obtained at the BP86/def-SVP level of theory and the energies in parentheses are at the B3LYP/def-SVP level. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of iso1 and iso2 explains the experimental features well, and therefore both, iso1 and iso2, are assigned to co-exist in the experiment with similar abundances. For this size we have verified that the Ar messenger has only a minor effect on the relative energies, band positions and infrared intensities (see Fig. S1(a) of the ESI†). The assigned structures are similar to the one found for Si9V+, which is also based on a pentagonal bipyramid.27
:1 mixture of iso1 and iso2 explains the experimental features well, and therefore both, iso1 and iso2, are assigned to co-exist in the experiment with similar abundances. For this size we have verified that the Ar messenger has only a minor effect on the relative energies, band positions and infrared intensities (see Fig. S1(a) of the ESI†). The assigned structures are similar to the one found for Si9V+, which is also based on a pentagonal bipyramid.27
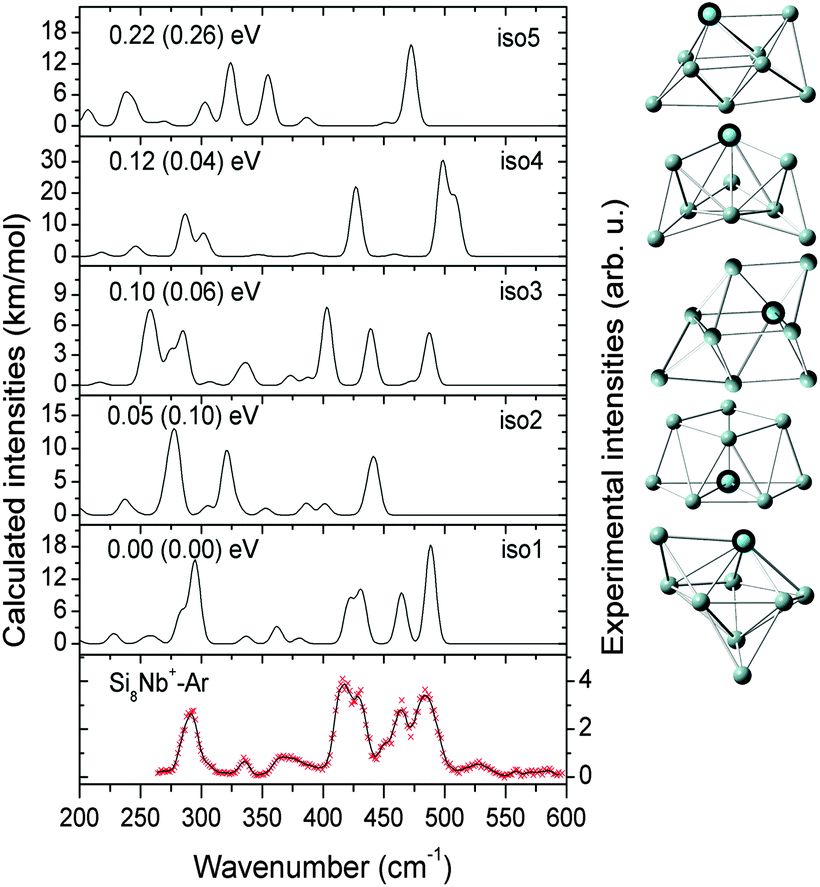
 | ||
| Fig. 3 Comparison of the experimental IR-MPD spectrum (data points [red] together with 5-point running average [black line], right axis) of Si9Nb+–Ar and the corresponding calculated IR spectra (left axis) of low-lying isomers. The mixing ratio of the mix (iso1:2) spectrum is 1:1. The relative energies are calculated at the BP86/def-SVP level of theory and at the B3LYP/def-SVP level of theory (in parentheses). The transition structure (TS) between iso1 and iso2 is also shown. | ||
:1 mixture of iso1 and iso2 (BP86: +0.10 eV, B3LYP: +0.06 eV) may explain the IR-MPD spectrum. While the higher energy band observed at ∼475 cm−1 can only stem from iso2, the most intense experimental feature at ∼400 cm−1 may be a signature of the presence of iso1. However, the intense band of iso1 at 312 cm−1 leads to some discrepancy, as such a band is not observed in the experiment. Furthermore, the presence of iso3 (BP86: +0.12 eV, B3LYP: +0.17 eV) cannot be excluded. In particular below 375 cm−1 its calculated IR spectrum shows some features that may have their counterparts in the experimental spectrum. This gives some indications that multiple isomers may coexist for the Si10Nb+ cluster in the experiment. Structurally, the lower-energy isomers iso1 and iso3 have a special form of a basket-like cage with the Nb atom in a central position of high coordination. The structure of iso1 is similar to the previously reported low-lying iso6 structure of the Si10V+ cluster (for this V-doped cluster no definite structural assignment has been made).27 Isomer iso2 of Si10Nb+ can be formed by adding two Si atoms on two faces of the Si8Nb+ bicapped pentagonal bipyramid.
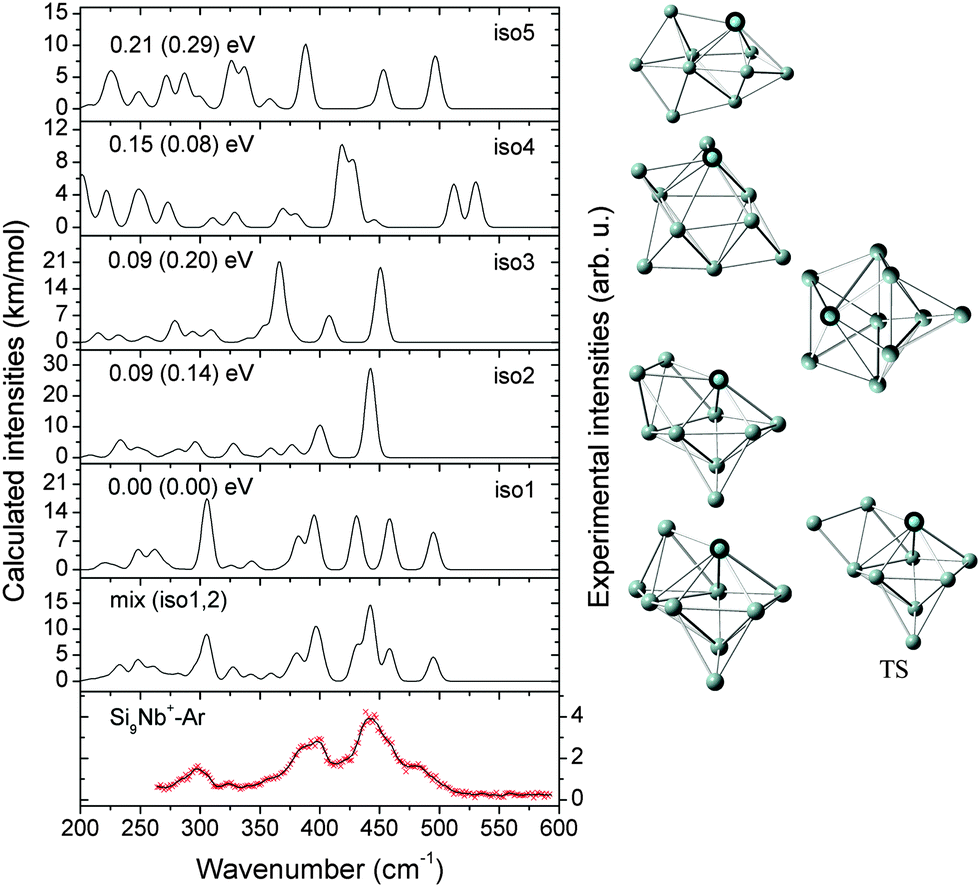
 | ||
| Fig. 4 Comparison of the experimental IR-MPD spectrum (data points [red] together with 5-point running average [black line], right axis) of Si10Nb+–Ar and the corresponding calculated IR spectra (left axis) of low-lying isomers. The mixing ratio of the mix (iso1:2) spectrum is 2:1. The relative energies are calculated at the BP86/def-SVP level of theory and at the B3LYP/def-SVP level of theory (in parentheses). | ||
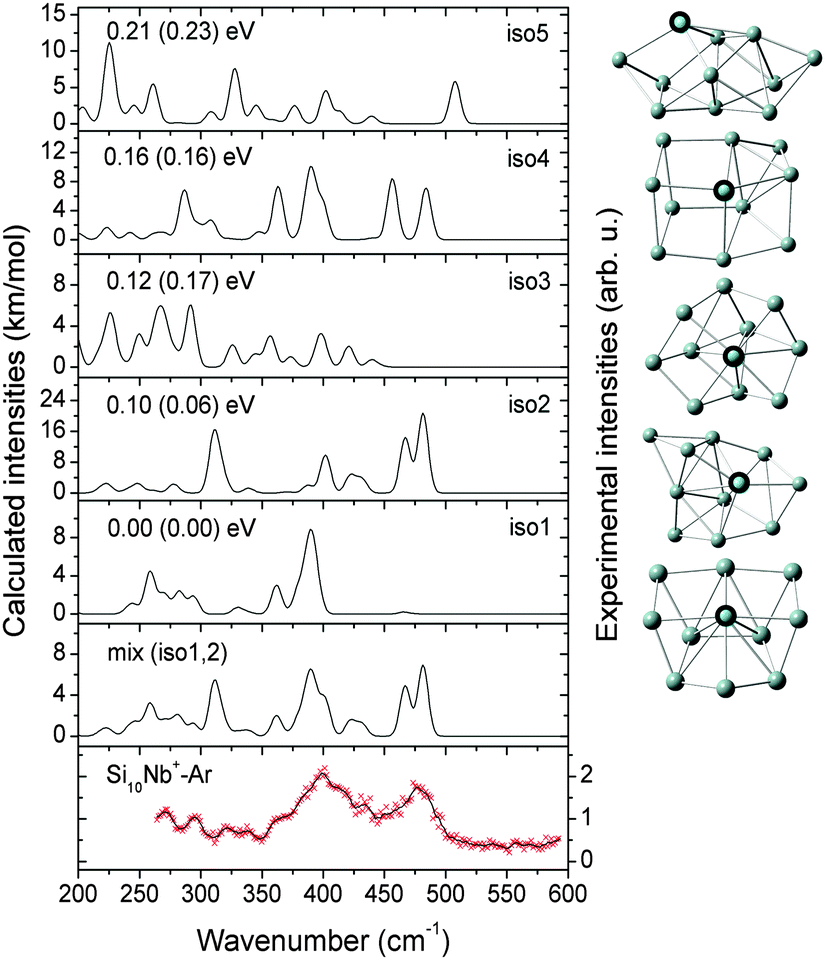
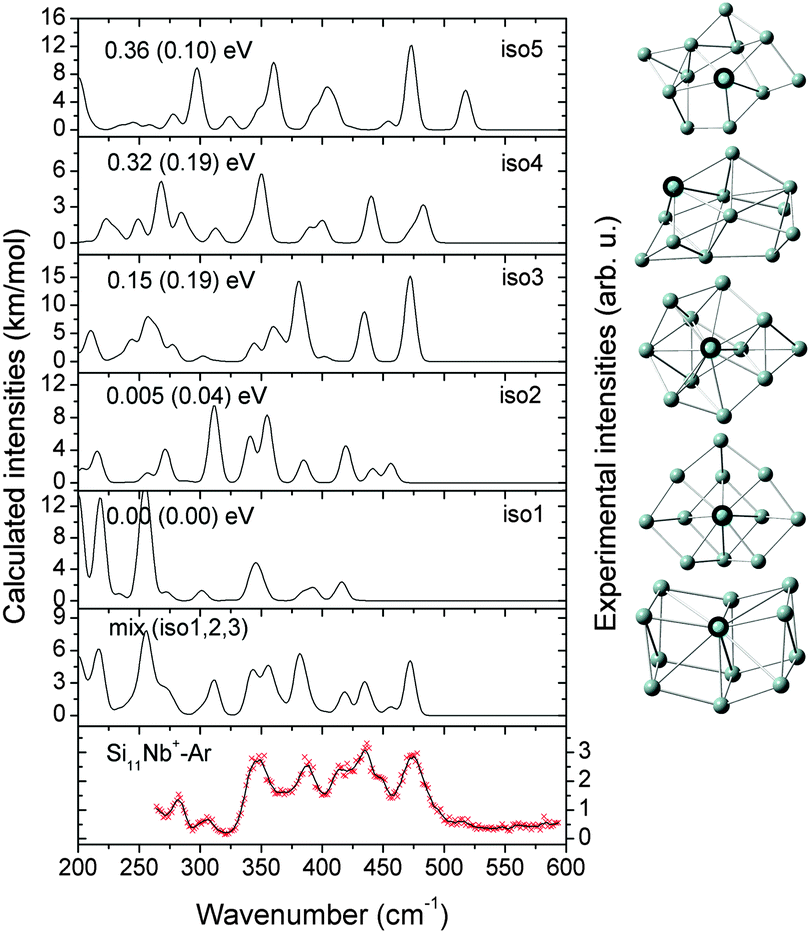 | ||
| Fig. 5 Comparison of the experimental IR-MPD spectrum (data points [red] together with 5-point running average [black line], right axis) of Si11Nb+–Ar and the corresponding calculated IR spectra (left axis) of low-lying isomers. The mixing ratio of the mix (iso1:2:3) spectrum is 1:1:1. The relative energies are calculated at the BP86/def-SVP level of theory and at the B3LYP/def-SVP level of theory (in parentheses). | ||
Notably, the Nb atom is exohedral in all low energy isomers of Si11Nb+, while for the vanadium doped clusters this size marks the transition from exohedral to endohedral structures. The predicted ground state for Si11V+ is a metal encapsulating Si cage, though experimentally it is found that at least a significant fraction (∼50%) of it contains an exohedral V atom.27,52
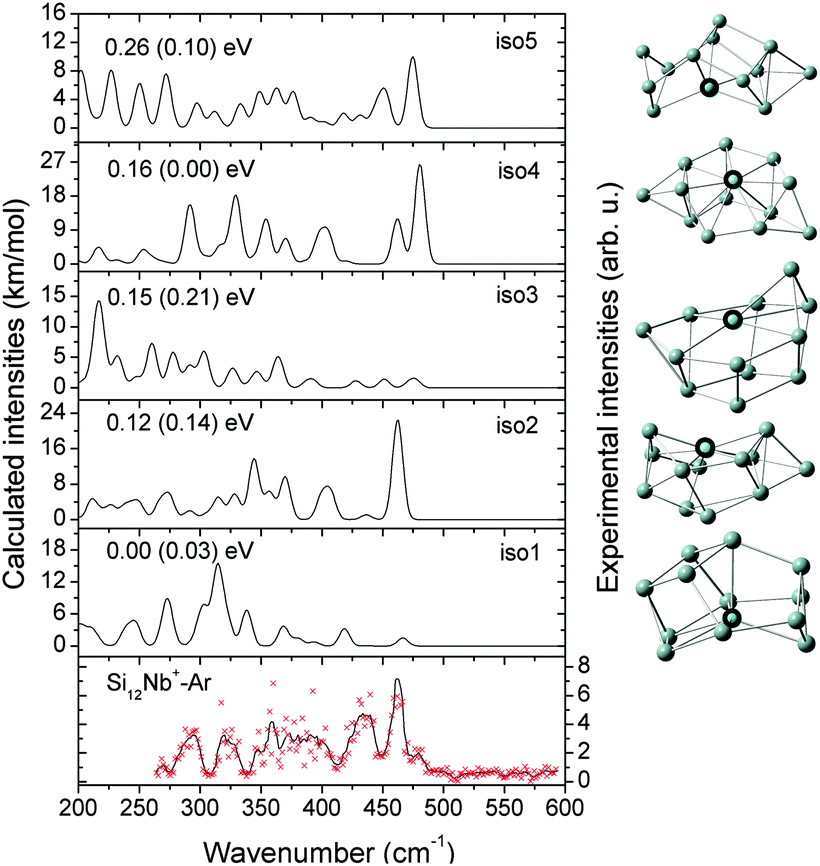 | ||
| Fig. 6 Comparison of the experimental IR-MPD spectrum (data points [red] together with 5-point running average [black line], right axis) of Si12Nb+–Ar and the corresponding calculated IR spectra (left axis) of low-lying isomers. The relative energies are calculated at the BP86/def-SVP level of theory and at the B3LYP/def-SVP level of theory (in parentheses). | ||
3.2 Structural evolution with size
In Fig. 7 we present the structural evolution of SinNb+ (n = 4–12) clusters based on the structures for the individual sizes as discussed above, in comparison with the bare Sin+1+ and SinV+ clusters.9,27,67,68 V and Nb are both in group 5 of the periodic table and possess five valence electrons but having different atomic ground state configurations, i.e. [Ar]3d34s2 and [Kr]4d45s1, with closed and open s-shell, respectively, therefore it a priori is not clear if they would induce a similar geometrical structure in the doped silicon clusters. From the assignments given before, it is obvious that for the smaller sizes (SinM+ with n = 4, 5 for M = V and Nb) the trigonal bipyramidal framework is maintained.27 Different from the pentagonal bipyramidal Si6V+,27 the Si6Nb+ cluster is a capped octahedron. One can see that the larger sizes (SinM+ with n = 7–10 for M = V, and n = 7–9 for M = Nb) appear as multi-capped pentagonal bipyramidal structures with a metal substitution on an axial position. When directly adding one Si atom to a side face of Si6Nb+ or substituting one Si atom by Nb at an axial position of Si8+,9 the capped pentagonal bipyramidal Si7Nb+ cluster is obtained after structural relaxation. Actually, for n = 7–9, one Si atom always caps a face of the pentagonal bipyramid building block, while the Nb atom occupies the opposite apex of the bipyramid. Starting from n = 10, one finds new structural forms with the Nb atom embedded in the surface in a 4-fold (sometimes square) coordination, such structures are different from the V-doped silicon clusters, e.g., for which the Si12V+ cluster is a distorted V-doped hexagonal prism.26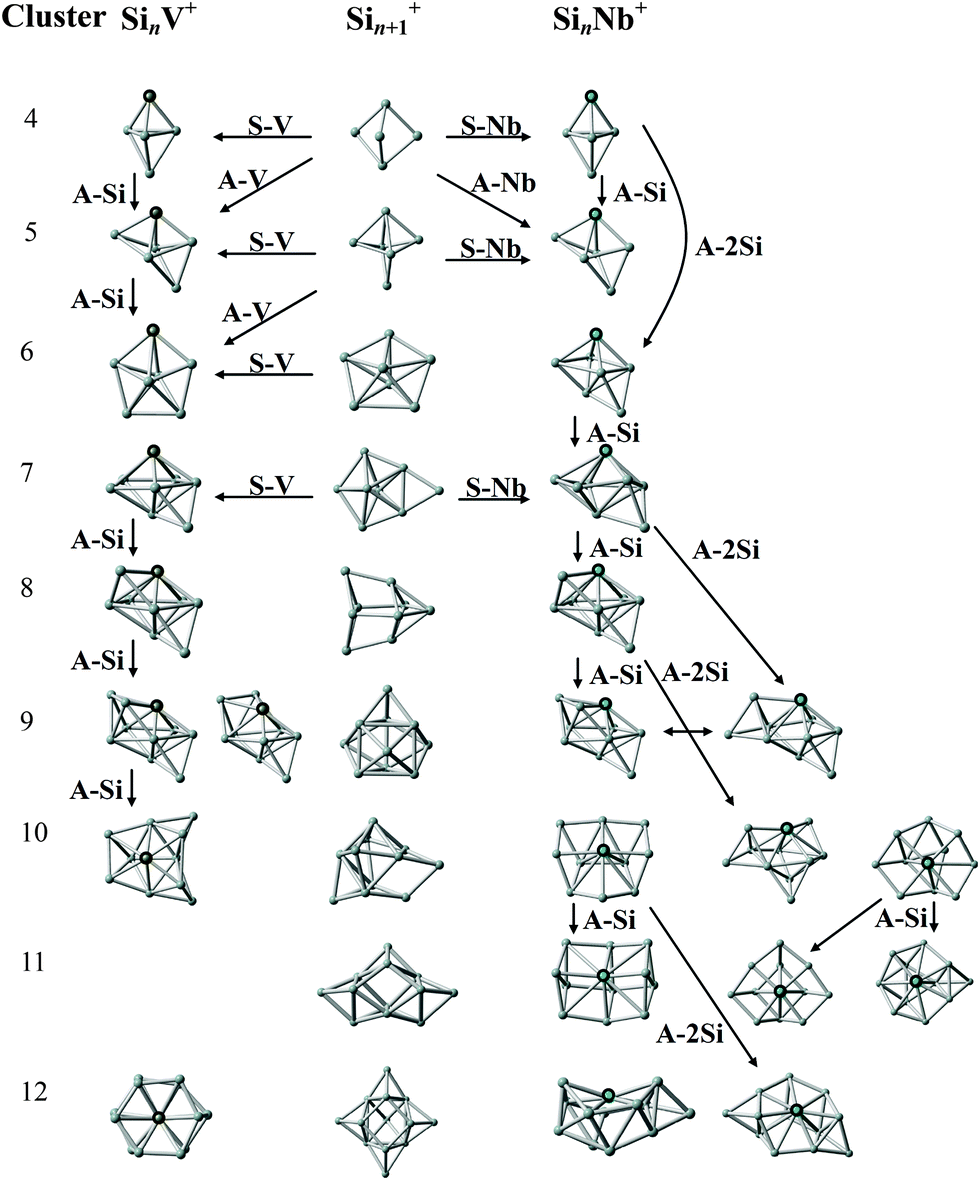 | ||
| Fig. 7 Structural evolution of SinNb+ (n = 4–12) clusters (right) in comparison with that of bare Sin+1+ (middle) and SinV+ (left) clusters. Arrows indicate structurally related clusters. A-Nb, A-Si, and A-V stand for the formal addition of a niobium, silicon, and vanadium atom. S-Nb and S-V stand for the substitution of a Si atom by niobium or vanadium. The structures of the bare Si5+, Sin+1+ (n = 5–10, 12), and Si12+ clusters are taken from ref. 9, 67 and 68, respectively. The structures of SinV+ (n = 4–10) and Si12V+ clusters are taken from ref. 27 and 26, respectively. | ||
In general, the Nb and V doped silicon clusters show similar structures, except for n = 10 and 12. Probably, the larger atomic radius of Nb becomes a relevant factor for the structures of the larger clusters. Structurally, the Nb dopant prefers to adsorb to an apex position of the Si framework for n = 4–9 and takes a surface embedded position for n = 10–12, whereas, for instance, Ag-doped clusters show similar Si frameworks but with the Ag dopants in lower coordinated apex or edge positions.29
3.3 Relative stability of the doped clusters
To further understand the size dependent structure and stability of the SinNb+ (n = 4–12) clusters in comparison to SinV+ (n = 4–10, 12), we evaluated the average binding energies (Eb) per atom, coordination numbers (CNs) and fragmentation energies (E1f, E2f, and E3f) for different fragmentation pathways, defined by:| Eb(n) = [ET(Nb+) + nET(Si) − ET(SinNb+)]/(n + 1) | (1) |
| E1f(n) = ET(Sin) + ET(Nb+) − ET(SinNb+) | (2) |
| E2f(n) = ET(Sin+) + ET(Nb) − ET(SinNb+) | (3) |
| E3f(n) = ET(Sin−1Nb+) + ET(Si) − ET(SinNb+) | (4) |
Fig. 8(a) shows that the average binding energies Eb of the Nb-doped silicon clusters increase up to the size of n = 8 and then are nearly constant at ∼3.8 eV for larger sizes. This goes along with the larger doped clusters having the Nb-atoms interacting with more Si atoms leading to a smaller number of dangling Si-bonds thereby stabilizing the Sin framework. This is confirmed by the large coordination numbers of the Nb atom, as shown in Fig. 8(b) (also see Table S2 of the ESI†), in which fractional coordination numbers as proposed by Grimme et al. are evaluated.71 The average binding energies of the SinNb+ clusters are higher than those of the bare Sin+ clusters by at least 0.13 eV (n = 10), indicating that the Nb dopant enhances significantly the stability of the Sin framework. By comparison, the isoelectronic V dopant enhances the stability (in terms of Eb) only slightly and makes relatively weaker chemical bonds with Si than the Nb. Further, the low-coordinated Cu/Ag dopants with filled 3d/4d orbitals are weaker bound than the Nb atom leading to a lower average binding energy of the doped clusters.27,29
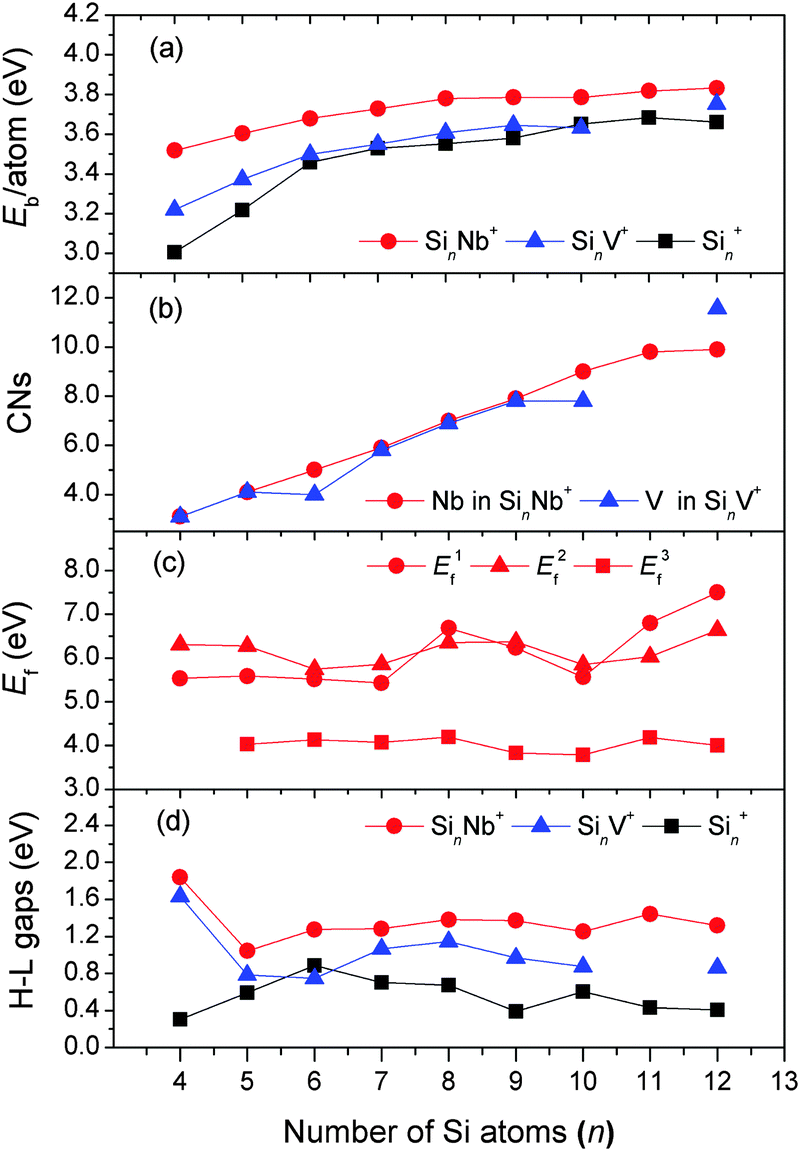 | ||
| Fig. 8 Evolution of different energy parameters and of the coordination numbers as a function of cluster size. (a) The average binding energy (Eb) per atom for SinNb+, SinV+, and Sin+; (b) the coordination number (CN) of Nb in SinNb+ and V in SinV+; (c) the fragmentation energies (Ef) for three fragmentation pathways of SinNb+: removal of (i) a Nb+ cation (E1f), (ii) a Nb atom (E2f), and (iii) a Si atom (E3f); (d) HOMO–LUMO energy gaps (H–L gaps) of SinNb+, SinV+, and Sin+. The energies of SinV+ (n = 4–10, 12) are obtained from the reoptimization at the BP86/def-SVP level of theory, based on the structures in ref. 26 and 27. Similarly, the structures of the bare Sin+ and Sin (n = 4–12) clusters are taken from ref. 9, 12, and 67–70, and reoptimized at the same level of theory. | ||
The calculated fragmentation energies of the SinNb+ clusters, plotted in Fig. 8(c), show that the loss of the Nb+ cation or the Nb atom is in all cases unfavorable compared to the loss of a single Si atom. This is different compared to SinV+ clusters where for most sizes the loss of the V cation is found to be the most facile fragmentation channel.27 As the atomic ionization energies of V and Nb are nearly identical (6.746 vs. 6.759 eV) this once more shows the significant stronger stabilization of the Si clusters by Nb doping.
The stabilization by Nb doping also becomes evident in the HOMO–LUMO gaps that are distinctly larger by up to 1.5 eV for the SinNb+ clusters than those of the bare Sin+ clusters (Fig. 8(d)). For the SinV+ clusters the gap is slightly smaller and for Si6V+ even lower compared to the bare Si clusters. Local maxima for the HOMO–LUMO gaps are found for Si4Nb+ and Si11Nb+. To further understand the effect of Nb doping, we have performed a detailed analysis of the frontier molecular orbitals by probing the partial density of states (PDOS, plotted in Fig. S6 of the ESI†), in which the contributions of different atomic shells (s, p, d) to the molecular orbitals are analyzed. From the NBO analysis performed for the Si4Nb+ cluster one recognizes that its HOMO originates mainly from the Si 3pz-states (53%) mixed with Si 3s (23%) and Nb 4dz2 (7%) states, whereas the LUMO state contains mainly Si 3py (33%) and Nb 4dx2–y2 (43%) states. The Si 3s-states contribute only little. Interestingly, for the Si11Nb+ cluster, the HOMO state is mainly composed of Si 3px (27%) and Si 3py (25%) states mixed with a relatively large contribution of Nb 4dz2 (15%) states, whereas the LUMO states are largely delocalized around the Si surface of the structure. The large HOMO–LUMO gaps may be related to this pronounced hybridization of Si 3p and Nb 4d states. A similar behavior is also observed for other cluster sizes.
This increasing electron transfer towards the Nb dopant becomes apparent also in the occupation of the 4d orbitals of Nb. The occupation of these orbitals increases with cluster size from 4.41 e for n = 5 (4.41) to 5.58 e for n = 11, which is higher than in the 4d orbitals (4.0) of the isolated Nb atom (for details see Table S3 of the ESI†). The Nb atoms (similar for V) always locate at high coordination positions in the Si frameworks as shown in Fig. 7. Therefore, bonding to the Si framework must involve the 4d orbitals leading to a rise of their occupation along with an increase of the coordination number. Thereby the high coordination numbers appear to be a reason for the increasing stabilization of the doped clusters. In comparison, for Cu and Ag doped silicon clusters the natural populations of the metals' d orbitals are about 9.8 and 9.9, respectively, indicating that the d orbitals are not significantly involved in chemical bonding, and the Cu and Ag atoms favor low coordination in the doped silicon clusters.27,29
4. Conclusions
In conclusion, we have performed a systematic experimental and DFT study on cationic Nb-doped silicon clusters, SinNb+ (n = 4–12), to obtain insights into their structures and stabilities. Infrared spectra are obtained using multiple photon dissociation of the cluster–argon complexes and are compared to calculated harmonic IR spectra. In most cases, good agreement is found when using the BP86 functional and the spectra are assigned to the lowest-energy structures identified in the calculations. However, for certain sizes, higher energy isomers need to be considered to explain the experimental spectra. Here, the hybrid functional B3LYP is found to give a more reliable energy ordering. While up to the size of Si8Nb+ the experimental spectra are well explained by considering a single isomer, for the larger clusters multiple isomers appear to be present in the experiment. Structurally, it is found that the Nb dopant tends to adsorb to an apex position of the Si framework for n = 4–9, showing the same structural evolution like the SinV+ clusters with the only exception of Si6V+, and a high-coordination surface position for n = 10–12. Thus, at least up to the size of Si12Nb+, where the V doped species is already endohedral, only exohedral structures are identified. The analysis of binding energies and HOMO–LUMO gaps indicates that doping with Nb atoms leads to a significant stabilization compared to the pure silicon cluster, as observed for the V-dopant. Furthermore, the predicted lowest energy fragmentation channel for these clusters is the loss of Si atoms, due to the high stability of the Nb-doped clusters.Note added in proof
A recent publication77 also reported the structures of cationic niobium-doped silicon clusters, but some of their predicted structures are not supported by our analysis, based on the experimental IR-MPD spectroscopy and DFT calculations. In that paper published by Xia et al.,77 the lowest-energy structures and spin states of the cationic niobium-doped silicon clusters are in good agreement with those with n = 4, 6, and 11 reported here, but disagree with those with n = 5, 7, 8, 9, 10, and 12, as identified by IR-MPD spectroscopy.Acknowledgements
The authors gratefully acknowledge the support from the Stichting voor Fundamenteel Onderzoek der Materie (FOM) in providing beam time on FELIX and highly appreciate the skillful assistance of the FELIX staff. This work is supported by the European Community's FP7/2007-2013 (Grant No. 226716), the Research Foundation-Flanders (FWO, grant G.0B41.15), the KU Leuven Research Council (GOA/14/007) program, and the Deutsche Forschungsgemeinschaft within FOR 1282 (FI 893/4). We gratefully acknowledge the financial support from the Natural Science Foundation of Shaanxi Province (No. 2014JM2-2019), the Natural Science Foundation of Education Department of Shaanxi Province (No. 14JK2128), and the Special Natural Science Foundation of Science and Technology Bureau of Xi'an City (No. CXY1352WL20).References
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- P. Gruene, D. M. Rayner, B. Redlich, A. F. G. van der Meer, J. T. Lyon, G. Meijer and A. Fielicke, Science, 2008, 321, 674–676 CrossRef CAS PubMed.
- S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fässler, Angew. Chem., Int. Ed., 2011, 50, 3630–3670 CrossRef CAS PubMed.
- E. C. Honea, A. Ogura, C. A. Murray, K. Raghavachari, W. O. Sprenger, M. F. Jarrold and W. L. Brown, Nature, 1993, 366, 42–44 CrossRef CAS.
- A. H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- L. X. Benedict, A. Puzder, A. J. Williamson, J. C. Grossman, G. Galli, J. E. Klepeis, J.-Y. Raty and O. Pankratov, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 085310 CrossRef.
- K.-M. Ho, A. A. Shvartsburg, B. Pan, Z.-Y. Lu, C.-Z. Wang, J. G. Wacker, J. L. Fye and M. F. Jarrold, Nature, 1998, 392, 582–585 CrossRef CAS.
- M. Scheer, R. C. Bilodeau, C. A. Brodie and H. K. Haugen, Phys. Rev. A: At., Mol., Opt. Phys., 1998, 58, 2844 CrossRef CAS.
- J. T. Lyon, P. Gruene, A. Fielicke, G. Meijer, E. Janssens, P. Claes and P. Lievens, J. Am. Chem. Soc., 2009, 131, 1115–1121 CrossRef CAS PubMed.
- D. A. Götz, S. Heiles and R. Schäfer, Eur. Phys. J. D, 2012, 66, 293 CrossRef.
- H. Hiura, T. Miyazaki and T. Kanayama, Phys. Rev. Lett., 2001, 86, 1733 CrossRef CAS PubMed.
- M. Haertelt, J. T. Lyon, P. Claes, J. de Haeck, P. Lievens and A. Fielicke, J. Chem. Phys., 2012, 136, 064301 CrossRef PubMed.
- U. Röthlisberger, W. Andreoni and M. Parrinello, Phys. Rev. Lett., 1994, 72, 665 CrossRef PubMed.
- S. M. Beck, J. Chem. Phys., 1987, 87, 4233 CrossRef CAS.
- S. M. Beck, J. Chem. Phys., 1989, 90, 6306 CrossRef CAS.
- H. T. Pham, T.-T. Phan, N. M. Tam, L. V. Duong, M. P. Pham-Ho and M. T. Nguyen, Phys. Chem. Chem. Phys., 2015, 17, 17566–17570 RSC.
- A. Grubisic, Y. J. Ko, H. Wang and K. H. Bowen, J. Am. Chem. Soc., 2009, 131, 10783–10790 CrossRef CAS PubMed.
- K. Koyasu, M. Akutsu, M. Mitsui and A. Nakajima, J. Am. Chem. Soc., 2005, 127, 4998–4999 CrossRef CAS PubMed.
- H.-G. Xu, M. M. Wu, Z.-G. Zhang, J. Yuan, Q. Sun and W. Zheng, J. Chem. Phys., 2012, 136, 104308 CrossRef PubMed.
- X. Kong, H.-G. Xu and W. Zheng, J. Chem. Phys., 2012, 137, 064307 CrossRef PubMed.
- X. Huang, H.-G. Xu, S. Lu, Y. Su, R. B. King, J. Zhao and W. Zheng, Nanoscale, 2014, 6, 14617–14621 RSC.
- X. Huang, S.-J. Lu, X. Liang, Y. Su, L. Sai, Z.-G. Zhang, J. Zhao, H.-G. Xu and W. Zheng, J. Phys. Chem. C, 2015, 119, 10987–10994 CAS.
- P. Claes, V. T. Ngan, M. Haertelt, J. T. Lyon, A. Fielicke, M. T. Nguyen, P. Lievens and E. Janssens, J. Chem. Phys., 2013, 138, 194301 CrossRef PubMed.
- Y. Li, N. M. Tam, P. Claes, A. P. Woodham, J. T. Lyon, V. T. Ngan, M. T. Nguyen, P. Lievens, A. Fielicke and E. Janssens, J. Phys. Chem. A, 2014, 118, 8198–8203 CrossRef CAS PubMed.
- Y. Li, J. T. Lyon, A. P. Woodham, P. Lievens, A. Fielicke and E. Janssens, J. Phys. Chem. C, 2015, 119, 10896–10903 CAS.
- P. Claes, E. Janssens, V. T. Ngan, P. Gruene, J. T. Lyon, D. J. Harding, A. Fielicke, M. T. Nguyen and P. Lievens, Phys. Rev. Lett., 2011, 107, 173401 CrossRef CAS PubMed.
- V. T. Ngan, P. Gruene, P. Claes, E. Janssens, A. Fielicke, M. T. Nguyen and P. Lievens, J. Am. Chem. Soc., 2010, 132, 15589–15602 CrossRef CAS PubMed.
- V. T. Ngan, E. Janssens, P. Claes, J. T. Lyon, A. Fielicke, M. T. Nguyen and P. Lievens, Chem. – Eur. J., 2012, 18, 15788–15793 CrossRef CAS PubMed.
- Y. Li, J. T. Lyon, A. P. Woodham, A. Fielicke and E. Janssens, ChemPhysChem, 2014, 15, 328–336 CrossRef CAS PubMed.
- Q. Peng and J. Shen, J. Chem. Phys., 2008, 128, 084711 CrossRef PubMed.
- L.-J. Guo, X. Liu, G.-F. Zhao and Y.-H. Luo, J. Chem. Phys., 2007, 126, 234704 CrossRef PubMed.
- J. Yang, J. Wang and Y. Hao, Theor. Chem. Acc., 2015, 134, 81 CrossRef.
- L. Lin and J. Yang, J. Mol. Model., 2015, 21, 155 CrossRef PubMed.
- J.-R. Li, G.-H. Wang, C.-H. Yao, Y.-W. Mu, J.-G. Wan and M. Han, J. Chem. Phys., 2009, 130, 164514 CrossRef PubMed.
- J.-P. Dognon, C. Clavaguéra and P. Pyykkö, Chem. Sci., 2012, 3, 2843–2848 RSC.
- J. M. Goicoechea and J. E. McGrady, Dalton Trans., 2015, 6755–6766 RSC.
- L. Ma, J. Zhao, J. Wang, B. Wang, Q. Lu and G. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 125439 CrossRef.
- J. Wang, J. Zhao, L. Ma, B. Wang and G. Wang, Phys. Lett. A, 2007, 367, 335–344 CrossRef CAS.
- J. Wang, Q.-M. Ma, Z. Xie, Y. Liu and Y.-C. Li, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 035406 CrossRef.
- F.-C. Chuang, Y.-Y. Hsieh, C.-C. Hsu and M. A. Albao, J. Chem. Phys., 2007, 127, 144313 CrossRef PubMed.
- S. Furuse, K. Koyasu, J. Atobe and A. Nakajima, J. Chem. Phys., 2008, 129, 064311 CrossRef PubMed.
- H. Kawamura, V. Kumar and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 245433 CrossRef.
- V. Zamudio-Bayer, L. Leppert, K. Hirsch, A. Langenberg, J. Rittmann, M. Kossick, M. Vogel, R. Richter, A. Terasaki, T. Möller, B. v. Issendorff, S. Kümmel and J. T. Lau, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 115425 CrossRef.
- V. Kumar, T. M. Briere and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 155412 CrossRef.
- V. Kumar and Y. Kawazoe, Phys. Rev. Lett., 2001, 87, 045503 CrossRef CAS PubMed.
- V. Kumar and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 073404 CrossRef.
- J. Lu and S. Nagase, Phys. Rev. Lett., 2003, 90, 115506 CrossRef PubMed.
- M. Nakaya, T. Iwasa, H. Tsunoyama, T. Eguchi and A. Nakajima, J. Phys. Chem. C, 2015, 119, 10962–10968 CAS.
- Z. Chen, S. Neukermans, X. Wang, E. Janssens, Z. Zhou, R. E. Silverans, R. B. King, P. von R. Schleyer and P. Lievens, J. Am. Chem. Soc., 2006, 128, 12829–12834 CrossRef CAS PubMed.
- M. Ohara, K. Koyasu, A. Nakajima and K. Kaya, Chem. Phys. Lett., 2003, 371, 490–497 CrossRef CAS.
- M. Ohara, K. Miyajima, A. Pramann, A. Nakajima and K. Kaya, J. Phys. Chem. A, 2002, 106, 3702–3705 CrossRef CAS.
- E. Janssens, P. Gruene, G. Meijer, L. Wöste, P. Lievens and A. Fielicke, Phys. Rev. Lett., 2007, 99, 063401 CrossRef PubMed.
- J. B. Jaeger, T. D. Jaeger and M. A. Duncan, J. Phys. Chem. A, 2006, 110, 9310–9314 CrossRef CAS PubMed.
- W. Bouwen, P. Thoen, F. Vanhoutte, S. Bouckaert, F. Despa, H. Weidele, R. E. Silverans and P. Lievens, Rev. Sci. Instrum., 2000, 71, 54–55 CrossRef CAS.
- A. Fielicke, G. von Helden and G. Meijer, Eur. Phys. J. D, 2005, 34, 83–88 CrossRef CAS.
- D. Oepts, A. F. G. van der Meer and P. W. van Amersfoort, Infrared Phys. Technol., 1995, 36, 297–308 CAS.
- M. Saunders, J. Comput. Chem., 2004, 25, 621–626 CrossRef CAS PubMed.
- J. Tong, Y. Li, D. Wu, Z.-R. Li and X.-R. Huang, J. Chem. Phys., 2009, 131, 164307 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D. 01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. B. Tai and M. T. Nguyen, J. Chem. Theory Comput., 2011, 7, 1119–1130 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. Gruene, A. Fielicke, G. Meijer, E. Janssens, V. T. Ngan, M. T. Nguyen and P. Lievens, ChemPhysChem, 2008, 9, 703–706 CrossRef CAS PubMed.
- C. W. Bauschlicher, Chem. Phys. Lett., 1995, 246, 40–44 CrossRef CAS.
- K. P. Jensen, B. O. Roos and U. Ryde, J. Chem. Phys., 2007, 126, 014103 CrossRef PubMed.
- S. Nigam, C. Majumder and S. K. Kulshreshtha, J. Chem. Phys., 2004, 121, 7756 CrossRef CAS PubMed.
- M. Vogel, C. Kasigkeit, K. Hirsch, A. Langenberg, J. Rittmann, V. Zamudio-Bayer, A. Kulesza, R. Mitrić, T. Möller, B. v. Issendorff and J. T. Lau, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 195454 CrossRef.
- A. Fielicke, J. T. Lyon, M. Haertelt, G. Meijer, P. Claes, J. de Haeck and P. Lievens, J. Chem. Phys., 2009, 131, 171105 CrossRef PubMed.
- A. A. Shvartsburg, B. Liu, M. F. Jarrold and K.-M. Ho, J. Chem. Phys., 2000, 112, 4517 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1.
- P. Guo, Z.-Y. Ren, F. Wang, J. Bian, J.-G. Han and G.-H. Wang, J. Chem. Phys., 2004, 121, 12265 CrossRef CAS PubMed.
- J. Wang and J.-G. Han, J. Chem. Phys., 2005, 123, 064306 CrossRef PubMed.
- P. Guo, Z.-Y. Ren, A.-P. Yang, J.-G. Han, J. Bian and G.-H. Wang, J. Phys. Chem. A, 2006, 110, 7453–7460 CrossRef CAS PubMed.
- Q. Peng and J. Shen, J. Chem. Phys., 2008, 128, 084711 CrossRef PubMed.
- X. X. Xia, A. Hermann, X. Y. Kuang, Y. Y. Jin, C. Lu and X. D. Xing, J. Phys. Chem. C, 2016, 120, 677–684 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison of calculated relative energies without and with the inclusion of zero-point vibrational energy for selected clusters; average Si–Si and Nb–Si bond distances and coordination numbers of Nb and Si atoms in the doped clusters; natural charge population and natural electron configuration on the Nb dopant; comparison of the calculated (scaled) IR spectra of the low-lying isomers and their cluster–Ar complexes for selected clusters; comparison of experimental IR-MPD spectra of SinNb+–Ar (n = 4–7) and the corresponding calculated IR spectra of the low-lying isomers; the total density of states (DOS) and the partial density of states (PDOS) for Si4Nb+ and Si11Nb+ clusters; and the optimized Cartesian coordinates of low-lying isomers of all doped clusters. See DOI: 10.1039/c5cp07298k |
| This journal is © the Owner Societies 2016 |