
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Azoniaspiro salts: towards bridging the gap between room-temperature ionic liquids and molten salts†
Matthew T.
Clough
 ab,
Karolin
Geyer
c,
Patricia A.
Hunt
*a,
Alastair J. S.
McIntosh
a,
Rebecca
Rowe
a,
Tom
Welton
*a and
Andrew J. P.
White
a
ab,
Karolin
Geyer
c,
Patricia A.
Hunt
*a,
Alastair J. S.
McIntosh
a,
Rebecca
Rowe
a,
Tom
Welton
*a and
Andrew J. P.
White
a
aDepartment of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: p.hunt@imperial.ac.uk; t.welton@imperial.ac.uk
bMax-Planck-Institut für Kohlenforschung, 45470 Mülheim-an-der-Ruhr, Germany
cBASF SE, Ludwigshafen, Germany
First published on 22nd December 2015
Abstract
In a continued effort to improve the suitability of ionic liquids in applications operating at raised temperatures, novel spirocyclic ‘azoniaspiro’ salts (with cations derived from five-, six-, seven- and eight-membered rings) are prepared and characterised. The structural and thermal properties of these salts are compared against those of established analogues. The stable geometries and ion pairing behaviour of these species are investigated via a combined experimental/computational approach, employing X-ray crystallography and Density Functional Theory (DFT) methods. Subsequently, the thermal stabilities of these organic salts are characterised and compared using a broad range of techniques. Hyphenated Thermogravimetry-Mass Spectrometry investigations enable complex mechanisms underlying thermal decomposition to be elucidated. Lastly, transition state structures are optimised, corresponding to plausible decomposition mechanisms of the azoniaspiro salt, 6-azoniaspiro[6.5]dodecanium chloride, and one prototypical monocyclic species 1-butyl-1-methylpiperidinium chloride, using DFT. The observed improved thermal stabilities of the azoniaspiro salts, and their potential higher-temperature stable-liquid ranges, render them promising candidates for future ionic liquid applications.
Introduction
Ionic liquids are species constituted entirely of ions, that exist in the molten state at or marginally above room temperature.1 Primitive ionic liquids belonged to a restricted class of air-/moisture-sensitive N-alkylpyridinium halometallate ‘melts’.2–7 By contrast, contemporary room-temperature ionic liquids are stable in the presence of both moisture and oxygen; they are commonly derived from alternative unsaturated heterocycles (e.g. imidazole,8 1,8-diazabicyclo[5.4.0]undec-7-ene9) or saturated heterocycles (e.g. pyrrolidine,10 piperidine), and incorporate a molecular anion with well-defined structure. Dialkylpiperidinium ionic liquids have been identified as strong candidate electrolytes for battery technology, attributable to their wide electrochemical windows.11 The seven-membered heterocycle azepane is a by-product of polyamide synthesis; it has been suggested, therefore, that dialkylazepanium ionic liquids may represent more sustainable analogues of pyrrolidine- and piperidine-derived ionic liquids.12One common feature of the above-described cation classes is that, in each case, the positive charge is associated with a quaternary nitrogen centre. At present under-investigated, ‘azoniaspiro’-type salts represent an interesting additional potential class of ionic liquid cation, whereby two rings are fused together via a quaternary nitrogen atom.13 The presence of the two cyclic moieties offers a greater variety of possible structures, arising from combinations of normal to medium sized rings, and the substitution of heteroatoms into either or both of the rings. In the late 1970s, azoniaspiro salts were investigated as ‘soluble hydrophobic species’.14 Tetraalkyl-ammonium salts incorporating monocyclic and azoniaspiro cations have played the role of ‘structure-directing agents’ in zeolite synthesis.15–17 More recently, mixtures of azoniaspiro salts and ethylene carbonate (EC) have been investigated as electrolytes for use in Li batteries.18 To the authors knowledge, azoniaspiro salts have not yet been widely explored in the context of ionic liquid chemistry.
For any application involving a tetraalkylammonium salt at elevated temperatures, an understanding of the thermal stability limitations of this specific salt is crucial. This must include knowledge of the safe operating temperatures, the underlying chemical mechanisms of degradation, and the properties of all thermal decomposition products formed. The measurement of these thermal properties requires a combination of experimental and computational techniques.
Thermogravimetric analysis (TGA) has been widely implemented in the evaluation of ionic liquid thermal stabilities.19–30 Temperature-ramped TGA experiments measure the changes in weight% as a function of temperature, whilst maintaining a constant heating rate. Heating rates of 10 °C min−1 or 20 °C min−1 are typical. However, caution must be exercised to keep experimental conditions as uniform as possible when comparing any individual thermal stability parameter; the ‘Tonset’ parameter allows rapid comparison of compounds in a series, where care has been taken to keep experimental conditions the same.20 However, while it has been commonly used to assess ionic liquid stability, Tonset has been shown to greatly overestimate stability.21,22 Alternatively, ‘Tstart’ quantifies the first point of appreciable weight loss (dw/dt ≠ 0). Isothermal TGA experiments provide insights into the long-term thermal stability, and Arrhenius parameters of thermal degradation.31
Herein we report syntheses and thermal characterisations of a broad series of tetraalkylammonium chloride salts, 1–10, incorporating either one or two saturated rings of five, six, seven or eight atoms (Fig. 1). Thermal decomposition of ionic liquids frequently involves nucleophilic attack of the anion on pendant alkyl chains of the cation.28,32–35 It was therefore pro-posed that the inclusion of a second cyclic structure would enhance steric interactions around the nitrogen centre and α-carbons, conferring improved thermal stability on the salt.
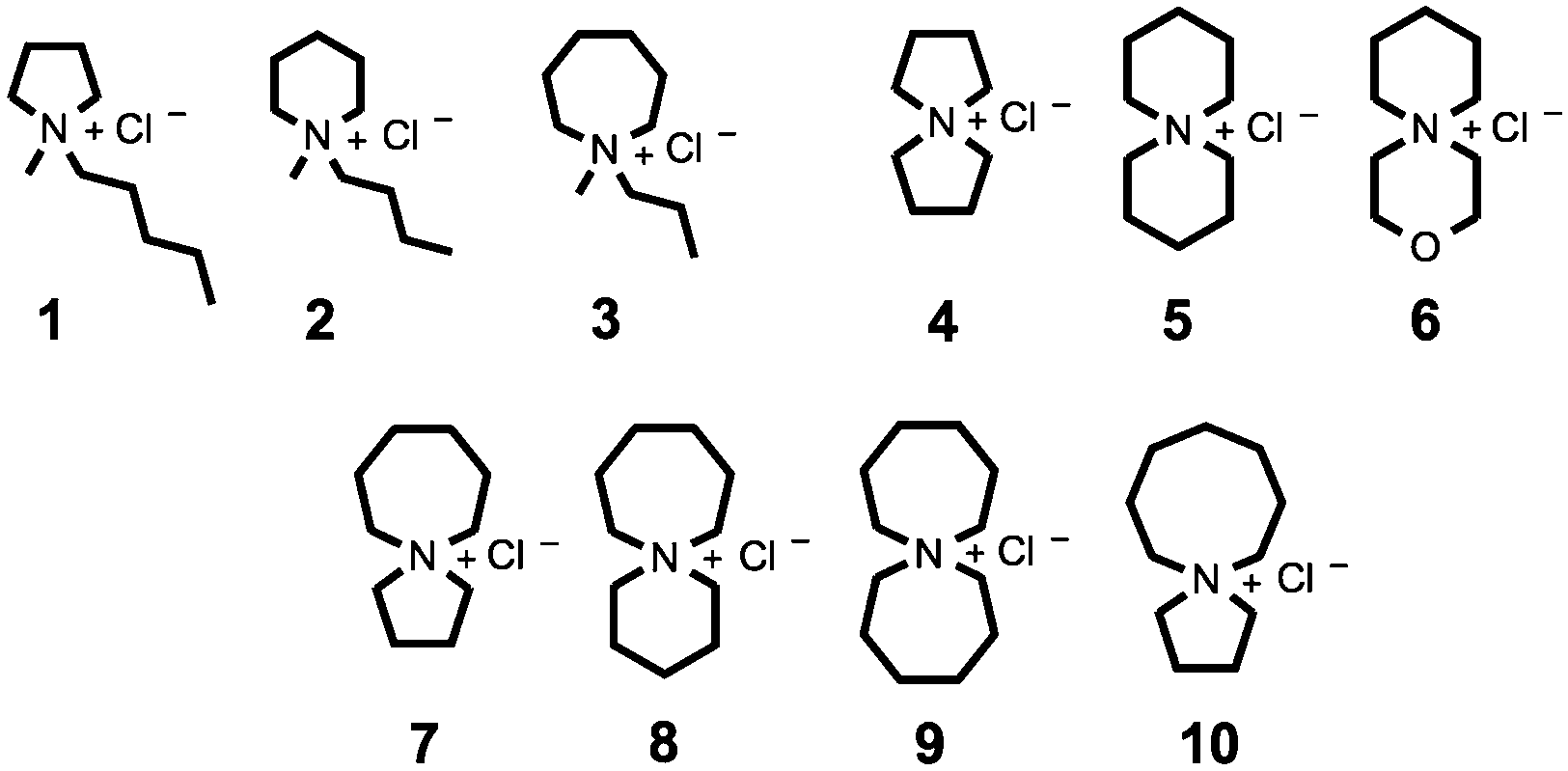 | ||
| Fig. 1 Investigated single-ring (1–3) and azoniaspiro (4–10) tetraalkylammonium chloride salts incorporating five, six, seven and eight-membered saturated rings. | ||
For clarity, azoniaspiro chloride salts included in this study are assigned abbreviated names according to the total number of ring atoms, including the quaternary nitrogen, e.g. 5-azoni-aspiro[4.4]nonanium chloride (4) is ‘[5+5]Cl’. Compound 6, incorporating the oxygen-substituted ring, is denoted ‘[3-oxa-6+6]Cl’. Monocyclic salts, 1–3, are instead named according to established literature convention, for example ‘[CnCmpyrr]Cl’, where integers ‘n’/‘m’ denote the number of carbon atoms in the alkyl chains.1
The structural properties of selected salts were investigated using X-ray crystallography and Density Functional Theory (DFT). Relative thermal stabilities and decomposition thermodynamics were compared by TGA. Furthermore, the thermal decomposition mechanisms of [C4C1pip]Cl, 2, and [7+6]Cl, 8 were studied using hyphenated thermogravimetric techniques (Thermogravimetric Analysis-Mass Spectrometry ‘TGA-MS’, and Thermogravimetric Analysis-Gas Chromatography/Mass Spectrometry ‘TGA-GCMS’); degradation mechanisms were subsequently rationalised using DFT.
‘Room-Temperature Ionic Liquids’ (RTILs) have been of particular interest, being in the molten state at, or below room temperature. However, RTILs often suffer from poor thermal stability, limiting utility at higher temperatures.36,37 Fully-inorganic molten salts are stable at elevated temperatures, but have high melting points that limit their utility at lower temperatures. It was proposed that azoniaspiro salts may partially bridge this gap with respect to the stable-liquid range.
One common degradation pathway for ionic liquids is the nucleophilic attack of the anion on the cation.28,32–35 Since we were interested in the contributions of an array of azoniaspiro cations to the relative stabilities of ionic liquids, we selected the chloride anion due to its ability to act as a nucleophile, the relative ease of crystallisation (enabling crystal structure data to be obtained) and for simplicity in conducting a computational survey of low-energy conformers. Nevertheless, for one azoniaspiro salt, metathetic reactions were performed so as to exchange chloride for bulky, charge-diffuse, non-nucleophilic anions (triflate and bis(trifluoromethanesulfonyl)imide), with the intention of drastically lowering the melting points and therefore demonstrating the potential of using simple azonia-spiro salts, or mixtures, at (close to) ambient temperatures.
Experimental
The salts included in this study are shown in Fig. 1. All compounds were prepared from commercial starting materials. Single-ring chloride salts, 1–3, were prepared by alkylation of an N-methylheterocycle with the relevant 1-chloroalkane according to established literature protocols.1N-methylazepane was prepared from commercial hexamethyleneimine (azepane) via Eschweiler–Clarke methylation with formic acid and formaldehyde.38 Azoniaspiro salts, 4–10, were prepared by heating under reflux of a biphasic toluene–water solution, containing a nitrogen heterocycle (i.e. piperidine, morpholine, azepane or heptamethyleneimine), a 1,n-dichloroalkane, and one molar equivalent of sodium hydroxide. The full synthesis of [7+6]Cl, 8, is described below. Synthesis and characterisation of 1–10 and intermediate compounds is described fully in the ESI.†Thermogravimetric Analysis (TGA) experiments were performed with PerkinElmer ‘Pyris 1’ TGA apparatus. Temperature-ramped TGA experiments were carried out using 4–8 mg of the salt, with a heating rate of 10 °C min−1, and employing platinum pans. In all circumstances, nitrogen was used as the inert carrier gas at a flow rate of 20 ml min−1. Isothermal TGA was performed using 7–8 mg of the compound. Thermogravimetric Analysis-Mass Spectrometry (TGA-MS) experiments were performed on 20–60 mg of the salt, employing ceramic pans. A heating rate of 10 °C min−1 and a CP Grade helium flow of 20 ml min−1 were employed. ‘Pyris 1’ TGA apparatus was connected to a ‘Hiden Analytical HPR 20’ mass spectrometer via a ceramic heated capillary. Electron Ionisation (EI) mass spectrometry was used, with 70 eV ionisation energy. TGA, TGA-MS and TGA-GCMS experimental conditions are fully described in the ESI.† All calculations were performed at the B3LYP-D2/6-311++G(d,p) level of theory. ΔG, ΔH, TΔS and E (zero-point-corrected) energies are provided in the ESI.†
Synthesis of 6-azoniaspiro[6.5]dodecanium chloride (8)
Azepane (15.0 g, 151 mmol) was diluted with toluene (70 ml) in a 250 ml, three-necked round-bottomed flask, fitted with a reflux condenser, a dropping funnel and a thermometer. Separately, an aqueous solution of NaOH was prepared by dissolving solid pellets of NaOH (6.05 g, 151 mmol) in deionized water (20 ml). The NaOH solution was carefully added to the toluene solution, and the dropping funnel was charged with 1,5-dichloropentane (21.33 g, 151 mmol). The biphasic mixture was heated to 80 °C under a nitrogen atmosphere and the 1,5-dichloropentane was added dropwise to the toluene/water mixture over a period of 15 minutes, with vigorous stirring. The solution was maintained at 80 °C for four days and was subsequently allowed to cool to room temperature. Toluene (100 ml) and deionized water (100 ml) were added, and the two layers were separated. The (upper) toluene layer was washed with deionized water (2 × 25 ml), and the three aqueous fractions were combined. Water was removed by rotary evaporation. To the resultant crude residue was added cold ethanol (200 ml), in which the product was found to be more soluble than NaCl. The solution was filtered, and the filtrate was treated with anhydrous MgSO4. The solution was filtered again, and the ethanol was removed by rotary evaporation, to yield 6-azoniaspiro[6.5]dodecanium chloride (18.6 g, 60%) as a white, free-flowing solid. Found: m.p. 272–273 °C with decomposition (lit. 270–272 °C with decomposition).131H NMR (400 MHz, DMSO-d6): δ 3.44 (4H, t, J = 5 Hz), 3.34 (4H, t, J = 6 Hz), 1.88–1.68 (8H, m), 1.65–1.47 (6H, m). 13C {1H} NMR (100 MHz, DMSO-d6): δ 61.4, 60.3, 27.1, 20.8, 20.3, 19.4. ν(neat)/cm−1 2933, 2862 (aliphatic C–H stretch, w), 1449 (aliphatic CH2 bend, w), 870 (aliphatic C–N stretch, m). m/z (LSIMS+): 168 (100%) [(CH2)6N(CH2)5]+. m/z (LSIMS−): 35 (100%) 35Cl−, 37 (23%) 37Cl−. Calc. for C11H22ClN: C, 64.84; H, 10.88; N, 6.87%. Found: C, 64.81; H, 10.77; N, 6.89%.Results and discussion
Initially, for selected compounds within the series (Fig. 1), a combination of X-ray crystallography and DFT techniques was employed in order to evaluate the preferential sites of the chloride anion around the tetraalkylammonium cation. Knowledge of the low-energy chloride bonding sites, and the stable geometries of single-ring and azoniaspiro cations, is valuable when proposing thermal decomposition mechanisms (and, in addition, when using these salts as structure-directing agents in zeolite synthesis). Moreover, structural information of this type allows for the prediction of possible solvating properties of the azoniaspiro salts.The gas phase ion pairing behaviour of dialkylimidazolium chloride ionic liquids has been extensively investigated.39–42 Hunt and co-workers demonstrated that for the prototypical ionic liquid 1-butyl-3-methylimidazolium chloride, [C4C1im]Cl, Cl− preferentially interacts with both the acidic C2 proton on the dialkylimidazolium cation and with an α-proton on either the methyl or butyl side chains.39 More recently ‘dimer’ structures (stable clusters incorporating two cations and two anions) of [C1C1im]Cl have been investigated in detail, yielding a complex array of geometries.43–45
The fully-saturated tetraalkylammonium chloride salts included in this investigation lack the acidic aromatic C2 proton of the dialkylimidazolium analogues; the low-energy ion pair geometries are therefore anticipated to differ substantially. Single, colourless crystals of [5+5]Cl, 4, [6+6]Cl, 5, [7+5]Cl, 7, and [7+6]Cl, 8, were grown via the vapour diffusion of n-pentane (in which the salts were largely insoluble) into a saturated isopropanol solution of the azoniaspiro salt. For salt 5, containing two six-membered rings, an X-ray structure was obtained (shown in Fig. 2, Fig. E6 and E7, ESI†).
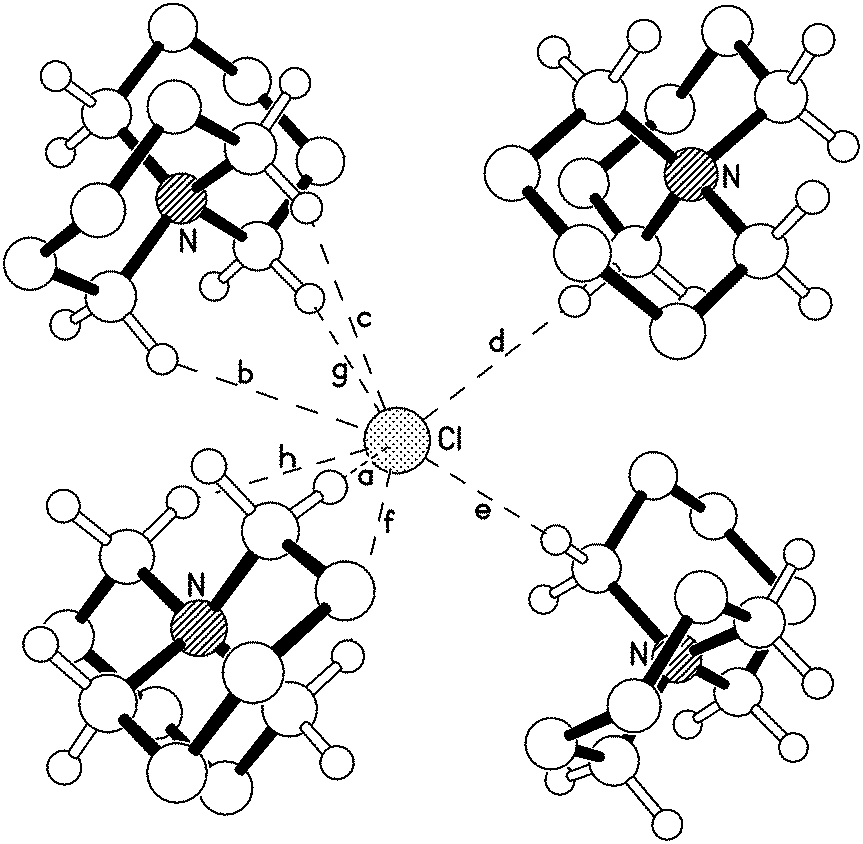 | ||
| Fig. 2 X-ray crystal structure of [6+6]Cl, 5, demonstrating apparent C–H⋯Cl hydrogen-bonding interactions between chloride and neigh-bouring azoniaspiro cations (only α-H atoms are shown, for clarity). | ||
The X-ray crystal structure of 5 (Fig. 2) indicates that both six membered rings adopt a ‘chair’-type conformation around the central nitrogen atom of the azoniaspiro cation. Chloride anions are then preferentially located in multiple ‘tripod’-like arrangements interacting with three α (or β) protons on nearby azoniaspiro cations. The unit cell structure of 5 highlights an alternating array of cations and anions, each chloride anion interacting with tripods of α and β hydrogen atoms on each of four neighbouring cations (C–H⋯Cl distances 2.75–2.95 Å).
In the large crystals of [5+5]Cl, 4, the azoniaspiro salt was found to have co-crystallised with H2O. A crystal structure of 4·H2O is displayed in the ESI† (Fig. E5). Moreover, the crystalline material was found to be highly hygroscopic, and deliquesced rapidly when exposed to air. Attempts to remove the water by drying of an ethanol solution of 4 with MgSO4 and subsequent recrystallisation of the sample proved unsuccessful. Therefore compound 4 is omitted from further discussion.
For salts 7 and 8 (containing a seven-membered ring), X-ray crystal structures were difficult to obtain. The scattering patterns suggested fundamental disorder in the crystals, which may indicate the presence of multiple cation structures arising from a high degree of conformational flexibility of the seven-membered rings.
Subsequently, B3LYP-D2/6-311++G(d,p) calculations were employed to locate low-energy ion pair conformers, initially for the single-ring salt [C4C1pip]Cl, 2, and then for azoniaspiro salt [7+6]Cl, 8. Cation structures were initially optimised, in the absence of the chloride anion, with differing combinations of ring conformation (chair and boat type). The chloride anion was then positioned in chemically-sensible locations around the lowest-energy cation structures, and ion pair optimisation calculations were performed. Ion pair structures and ΔE/ΔG values are summarised below for 2 (Fig. 3, Table 1) and 8 (Fig. 4, Table 2).
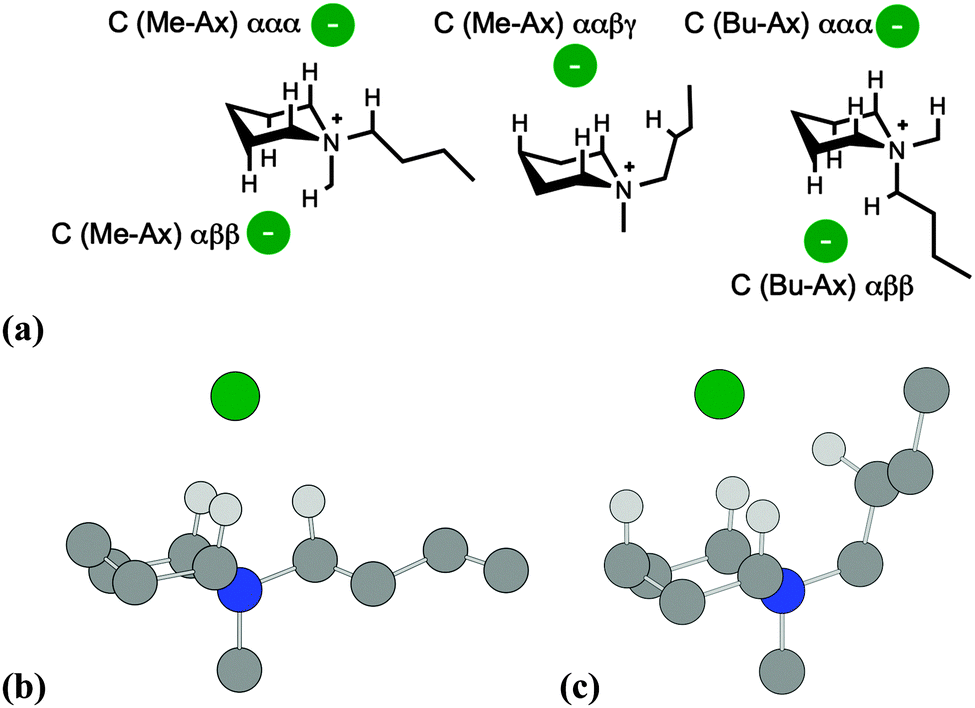 | ||
| Fig. 3 (a) Primary cation–anion interactions for chair conformers of [C4C1pip]Cl, 2. Structures of the: (b) ‘C (Me-Ax) ααα 1’, and; (c) ‘C (Me-Ax) ααβγ’ ion pair conformers of 2, showing primary H-bond interactions. All other hydrogen atoms are omitted, for clarity. | ||
| Ion pair [C4C1pip]Cl | ΔE (kJ mol−1) | ΔG (kJ mol−1) |
|---|---|---|
| C (Me-Ax) ααα 1 | 0.0 | 0.0 |
| C (Bu-Ax) ααα 1 | 1.8 | 2.0 |
| C (Bu-Ax) ααα 2 | 4.7 | 4.4 |
| C (Me-Ax) ααα 2 | 5.1 | 5.2 |
| C (Me-Ax) ααβγ | 13.2 | 16.5 |
| C (Bu-Ax) αββ | 17.4 | 19.2 |
| C (Bu-Ax) ααβ | 21.6 | 21.1 |
| B ααβ 1 | 23.9 | 24.3 |
| B ααα 1 | 25.1 | 25.9 |
| C (Me-Ax) αββ | 29.7 | 29.6 |
| B ααα 2 | 30.3 | 31.8 |
| B ααα 3 | 31.1 | 30.8 |
| B ααα 4 | 31.3 | 31.2 |
| B ααβ 2 | 46.3 | 45.5 |
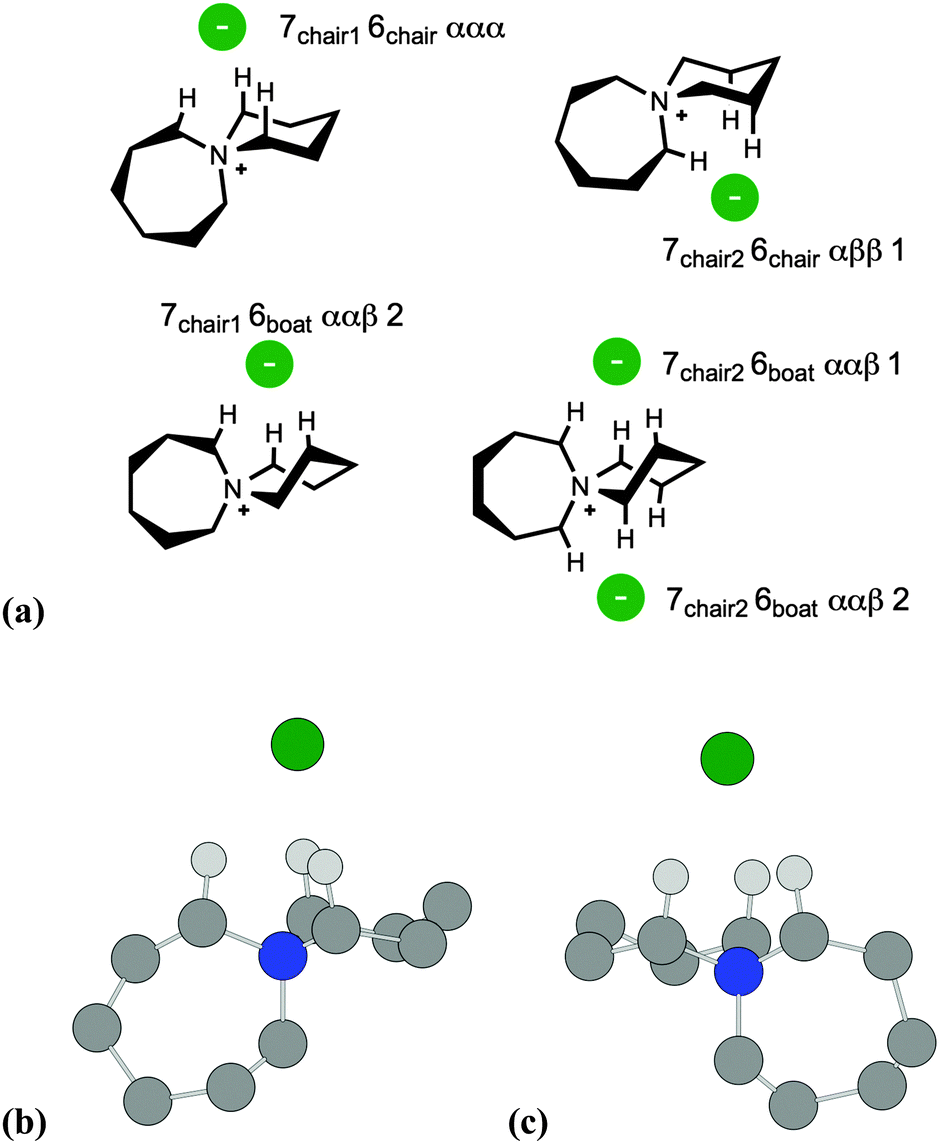 | ||
| Fig. 4 (a) Diagram representing primary cation–anion interactions for [7+6]Cl, 8. Structures of the low-energy (b) ‘7chair1 6chair ααα’ and (c) ‘7chair2 6chair ααα 1’ ion pair conformers of 8, showing primary H-bond interactions. All other hydrogen atoms are omitted, for clarity. | ||
| Ion pair 6-azoniaspiro[6.5]dodecanium chloride | ΔE (kJ mol−1) | ΔG (kJ mol−1) |
|---|---|---|
| 7chair1 6chair ααα | 0.0 | 0.0 |
| 7chair2 6chair ααα 1 | 1.6 | 2.0 |
| 7chair1 6chair ααβ 1 | 4.3 | 4.0 |
| 7chair2 6chair ααα 2 | 4.7 | 4.1 |
| 7chair1 6chair ααβ 2 | 10.2 | 9.9 |
| 7chair2 6boat ααα | 24.7 | 23.8 |
| 7chair2 6chair αββ 1 | 25.9 | 23.9 |
| 7chair2 6chair αββ 2 | 26.0 | 24.2 |
| 7chair2 6boat ααβ 1 | 30.7 | 28.8 |
| 7chair1 6boat ααβ 1 | 35.9 | 33.9 |
| 7chair1 6boat ααβ 2 | 36.6 | 33.9 |
| 7chair2 6boat ααβ 2 | 38.2 | 35.7 |
| 7chair2 6boat αββ | 45.5 | 43.4 |
A naming convention was chosen to describe the different ion pair conformers of 2. Chair conformers are denoted ‘C’, and boat conformers as ‘B’. Ion pairs of 2 were distinguished also according to whether the methyl/butyl chains were in the axial position (‘Me-Ax’ and ‘Bu-Ax’, respectively).
Furthermore, conformers were described in terms of the chloride anion location and the protons of the cation with which it interacts. Hence, the lowest-energy ‘C (Me-Ax) ααα 1’ ion pair conformer of 2 (Table 1) is a structure whereby the methyl group is in the axial position; the chloride anion interacts with three α-protons on the [C4C1pip]+ cation. Eight ion pair geometries of 2 with a cation chair (‘C’) conformation were located, and six boat (‘B’) conformers were also optimised (Table 1). All boat conformers were higher in energy than chair equivalents, with the exception of the ‘C (Me-Ax) αββ’ structure.
A similar naming convention was employed to distinguish different ion pair structures of [7+6]Cl, 8. Two distinct chair-type conformers were observed for the seven-membered ring. The six-membered ring could adopt either a chair or boat conformation. In addition, the conformers were also described in terms of the aliphatic ring protons that interact closely with the chloride anion, as for compound 2. Hence, for the lowest-energy ‘7chair1 6chair ααα’ ion pair conformer of 8, the seven-membered ring adopts the first of two chair conformers, and the six-membered ring also adopts a chair structure. Chloride then interacts with three α protons on the azoniaspiro cation.
For the majority of ion pair conformers of 2, the chloride anion interacts with a tripod array of α and β protons radiating out of the cation. Higher-energy conformers (ΔE = 13–30 kJ mol−1) involve bonding interactions of Cl− with β/γ protons on the piperidinium cation (C–H⋯Cl distances 2.36–2.49 Å).
The four lowest-energy ion pairs of salt 2 are very close in energy (ΔE = <6 kJ mol−1), and involve the interaction of the chloride anion with three α protons on the piperidinium cation. There appears to be no substantial energy increase associated with the 1,3-diaxial interactions of an axial butyl substituent.
One particular ion pair structure of 2 (‘C (Me-Ax) ααβγ’) is approximately 13 kJ mol−1 higher in energy than the lowest-energy conformer and involves interaction of a butyl β proton (facilitated by –Bu rotation) and also a ring γ proton (Fig. 3c).
Ion pair conformers of salt 8 with ‘boat’-conformation six-membered rings (ΔE = 24–46 kJ mol−1) typically lie higher in energy than those with ‘chair’-conformation rings (ΔE = 0–26 kJ mol−1). For all optimised ion pairs of 8, the chloride anion interacts preferentially with a tripodal arrangement of α and β protons in close proximity to the central quaternary nitrogen atom of the azoniaspiro cation (C–H⋯Cl distances 2.35–2.50 Å, cf. 2.75–2.95 Å for the X-ray structure of related [6+6]Cl, 5, Fig. 2). The two lowest-energy ion pair conformers of salt 8, ‘7chair1 6chair ααα’ and ‘7chair2 6chair ααα’, (Fig. 4(b) and (c)) are isoenergetic within the computational error, despite having different core cation geometries. Indeed, relative energies of the four lowest-energy conformers (Table 1) may become re-ordered upon a change in the computational method. The X-ray investigation of 8 demonstrated fundamental disorder for cation structure, possibly in accordance with the observed co-existence of analogous degenerate structures in the crystalline material.
Therefore DFT calculations of [C4C1pip]Cl, 2 and [7+6]Cl, 8, indicate that Cl− preferentially adopts a position close to the quaternary nitrogen atom, interacting with three protons in a tripodal array. Stable computed conformers of 8 closely match those observed in the X-ray structure of the analogous salt 5. Furthermore the experimental (X-ray) C–H⋯Cl bond distances are not dissimilar from the calculated (DFT) distances. These observations therefore offer valuable insights into low-energy geometries of tetraalkylammonium salts, representing ‘ground state’ conformers when calculating thermal decomposition re-action profiles.
Subsequently, thermal stabilities of salts 1–10 were compared by employing temperature-ramped Thermogravimetric Analysis (TGA). Thermographs are displayed in Fig. 5 and Tonset, Tstart and melting point values (Tm) are listed in Table 3.
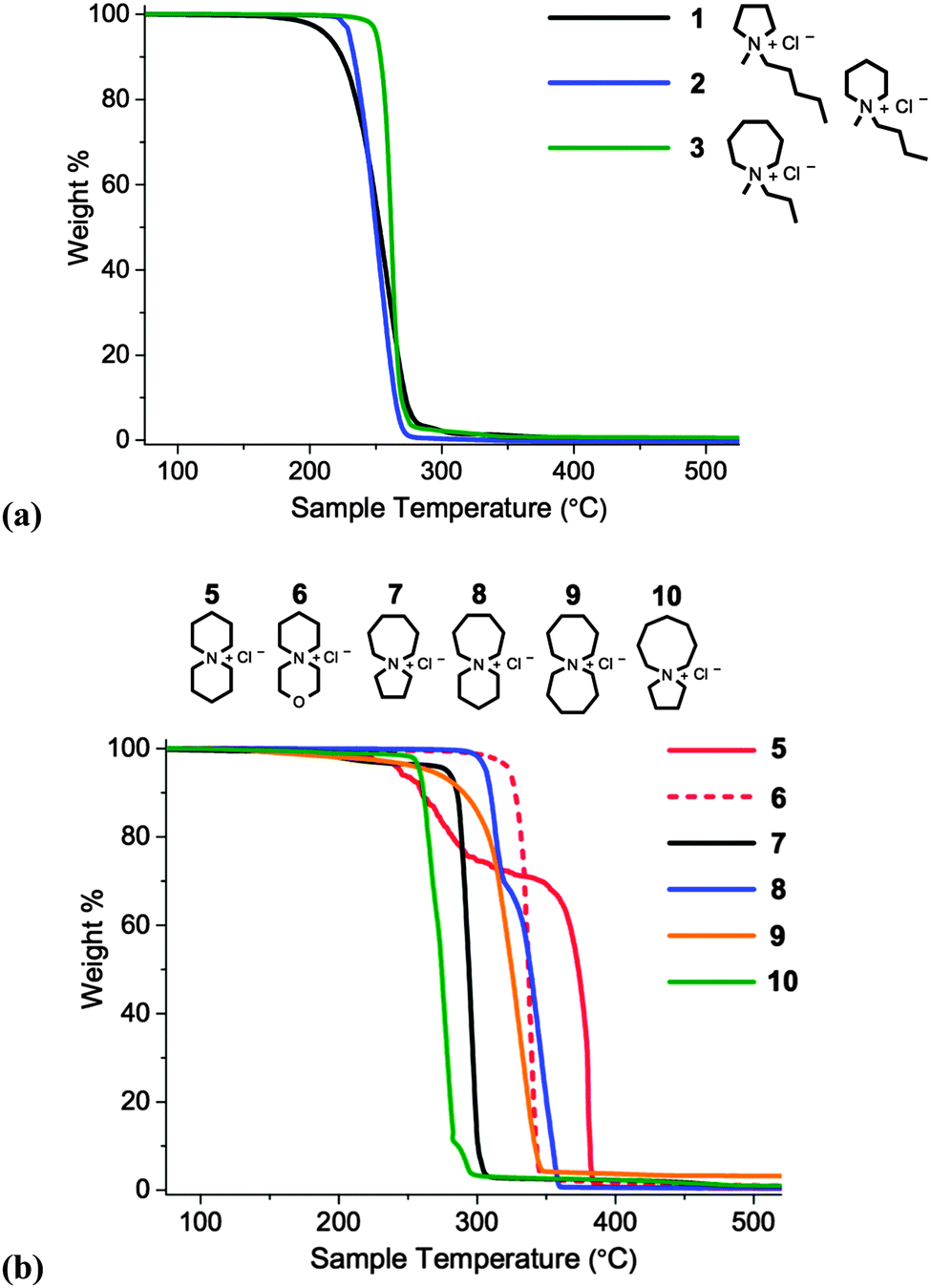 | ||
| Fig. 5 Temperature-ramped TGA thermographs for: (a) single-ring (1–3), and; (b) azoniaspiro (5–10) chloride salts, measured with heating rate 10 °C min−1, and shown in the range 75–525 °C for clarity. | ||
| Salt | T onset (°C) | T start (°C) | T m (°C) |
|---|---|---|---|
| * = melting with decomposition. | |||
| 1 [C5C1pyrr]Cl | 228 | 198 | 127–129 |
| 2 [C4C1pip]Cl | 232 | 221 | >210 |
| 3 [C3C1azp]Cl | 254 | 241 | 244–245* |
| 5 [6+6]Cl | 245 | 189 | 310–312* |
| 6 [3-oxa-6+6]Cl | 330 | 310 | >280* |
| 7 [7+5]Cl | 286 | 274 | 262–263* |
| 8 [7+6]Cl | 323 | 297 | 272–273* |
| 9 [7+7]Cl | 304 | 263 | >305* |
| 10 [8+5]Cl | 263 | 253 | 256–257* |
Spirocyclic tetraalkylammonium chloride salts (5–10) are, in general, more thermally stable than single-ring analogues (1–3), exhibiting higher Tonset and Tstart values; the increased rigidity of the cation, and the increased steric crowding in the proximity of the quaternary nitrogen atom, may partly explain this observation. For single-ring salts 1–3, a slight enhancement in thermal stability with increasing ring size was observed, from the five-membered pyrrolidinium salt 1 (Tonset = 228 °C) to the seven-membered azepanium salt 3 (Tonset = 254 °C). Within the investigated series of azoniaspiro species, 5–10, a significant variation in thermal decomposition behaviour was observed. Salts incorporating five-membered rings (7 and 10) exhibited poorer thermal stabilities than the salts composed of six- and seven-membered rings.
Two of the investigated salts exhibit clear, multi-step weight loss curves: [6+6]Cl, 5, and [7+6]Cl, 8. For these two species, approximately 30 wt% is lost in the first step, followed by the remaining 70% in the second thermal event. This decomposition profile was also observed with a slower TGA scan rate of 2 °C min−1; this indicates that the phenomenon is unlikely to be the result of a heat transfer effect. It is instead possible that two or more crystalline polymorphs exist in the solid material, accounting for the two distinct thermal degradation events.
For certain compounds in the series, the first appreciable loss of mass (Tstart, dw/dt ≠ 0) occurs at a similar temperature to the measured melting point (Tm). This is most apparent for [C3C1azp]Cl 3, and for the two azoniaspiro salts incorporating a five-membered ring, 7 and 10. The thermographs for these salts are also noticeably steeper than for other compounds in the series. This may indicate that decomposition of these salts is restricted by the reduced mobility of the ions until melting occurs; once the salt melts, decomposition occurs rapidly.
Two of the organic chloride salts, [7+6]Cl, 8, and [3-oxa-6+6]Cl, 6, display particularly high thermal stabilities. Tonset values for these salts (323 and 330 °C, respectively) compare favourably with chloride ionic liquids in the chemical literature measured under similar experimental conditions; for example [C4C1im]Cl (Tonset = 257 °C),30 [C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2C1im]Cl (Tonset = 254 °C),30 [(C2)4N]Cl (Tonset = 264 °C).20 Despite the prohibitively high melting points of azoniaspiro species 6 and 8, and their lack of a stable-liquid range, these results nevertheless indicate that selected azoniaspiro cations confer a promising improvement in thermal stability. Exchanging chloride for alternative asymmetric, charge-diffuse anions (e.g. [OAc]−, [CH3SO4]−, [NTf2]−) is likely to yield compounds with lower melting temperatures, yet that retain the beneficial property of high thermal stability imparted by the azoniaspiro cation.
C2C1im]Cl (Tonset = 254 °C),30 [(C2)4N]Cl (Tonset = 264 °C).20 Despite the prohibitively high melting points of azoniaspiro species 6 and 8, and their lack of a stable-liquid range, these results nevertheless indicate that selected azoniaspiro cations confer a promising improvement in thermal stability. Exchanging chloride for alternative asymmetric, charge-diffuse anions (e.g. [OAc]−, [CH3SO4]−, [NTf2]−) is likely to yield compounds with lower melting temperatures, yet that retain the beneficial property of high thermal stability imparted by the azoniaspiro cation.
One potential degradation product of 6 is the highly toxic oxygen mustard, bis(2-chloroethyl)ether, generated by attack of two chloride anions at the α-carbons of the morpholinium ring. The potential of generating this oxygen mustard presents a significant risk when handling this compound, and renders compound 6 an unattractive candidate for raised-temperature applications. Therefore on this basis, salt 6 was not investigated further. For this reason, azoniaspiro chloride salts based on thiomorpholine were also not studied. Instead, 6-azoniaspiro-[6.5]dodecanium chloride, [7+6]Cl, 8, was identified to be the most thermally stable (and safe) azoniaspiro salt in the series, and was selected for further investigation.
It is well-established that the Tonset parameter, as obtained from temperature-ramped TGA evaluation, represents a pro-found overestimate of the long-term thermal stability of ionic liquids and related compounds; the ‘T0.01/10’ value, proposed by MacFarlane and colleagues, represents the temperature at which exactly 1% decomposition occurs in 10 hours, and is a rudimentary estimate of the long-term upper operating temperature.21,22T0.01/10 values have been measured and compared for a broad spectrum of ionic liquids.21,22,28,30
Isothermal TGA experiments were performed for [7+6]Cl, 8. Graphs representing the long-term thermal stability of 8 are displayed in Fig. 6. Details of the isothermal TGA procedure for determining T0.01/10 are described in the ESI.†
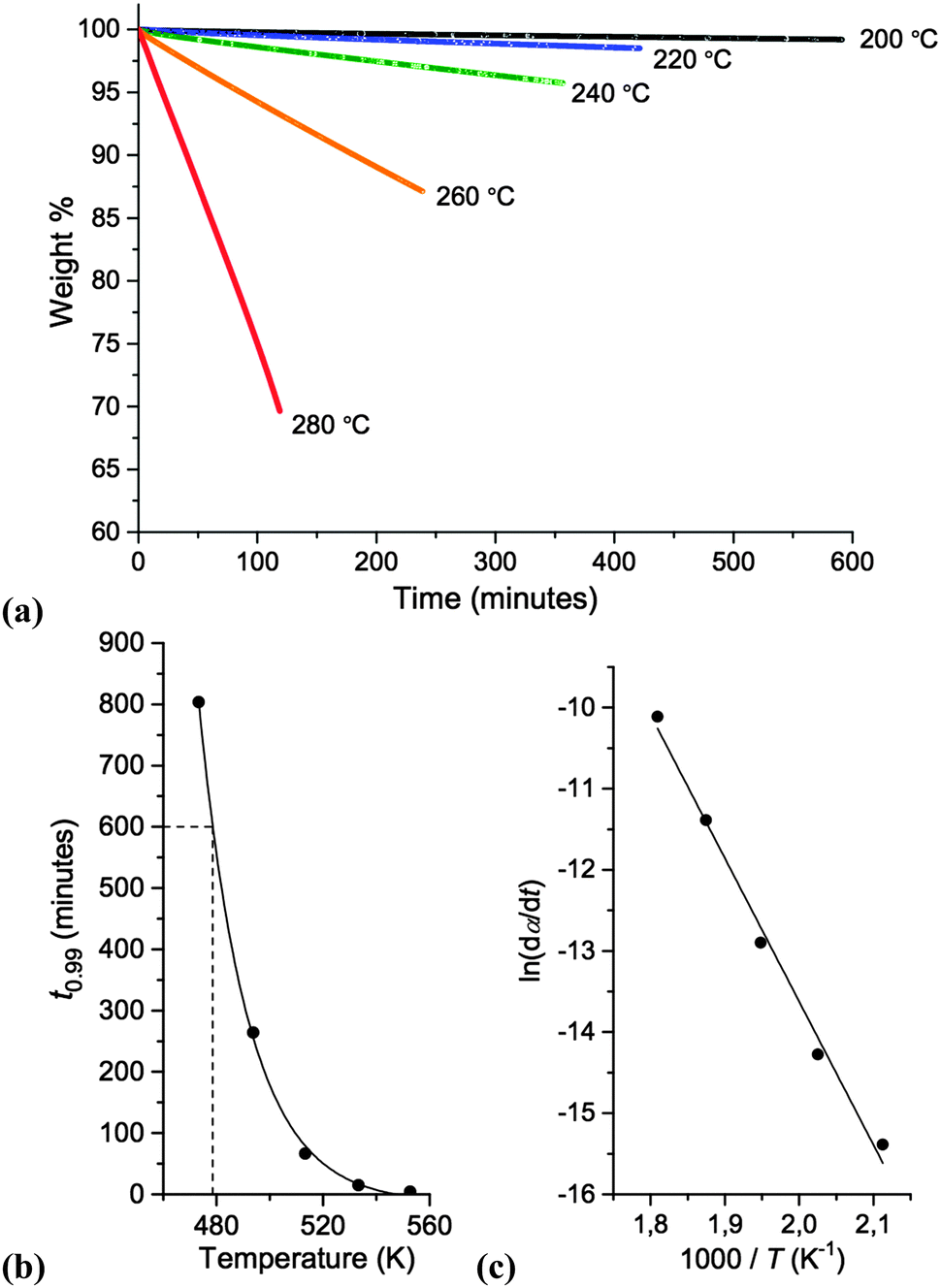 | ||
| Fig. 6 Determination of the long-term thermal stability of [7+6]Cl, 8: (a) isothermal TGA thermographs; (b) exponential fit of T (K) vs. t0.99 (mins) to determine T0.01/10; (c) determination of Ea and A. | ||
The T0.01/10 value of [7+6]Cl, 8, was measured as 205 °C, sub-stantially lower than both the Tonset (323 °C) and Tstart (297 °C) measurements from temperature-ramped TGA in accordance with the chemical literature.21 As for Tonset the obtained T0.01/10 value for 8 compares favourably with chloride ionic liquids in a previous comprehensive study by Cao and Mu; for example, [C4C1im]Cl (T0.01/10 = 140 °C), [C8C1im]Cl (T0.01/10 = 132 °C), [(HO)C2C1im]Cl (T0.01/10 = 155 °C).30 Hence, [7+6]Cl, 8, can be employed for extended periods at temperatures over 50 °C higher than more traditional chloride-containing ionic liquids.
The observed linear isotherms for 8 indicate pseudo zeroth-order thermal decomposition, whereby one cation reacts with one anion to yield wholly volatile products, and thus the concentrations of ions do not change. In this circumstance, Arrhenius parameters of thermal decomposition can be determined from the isothermal TGA data, according to eqn (1), below:31
dα/dt = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ea/RT) exp(−Ea/RT) | (1) |
Neither the temperature-ramped or isothermal TGA experiments yield any information pertaining to the precise chemical mechanisms that occur upon thermal decomposition of the chloride salts. However, TGA ‘hyphenated’ with Mass Spectrometry (TGA-MS) enables detection and characterization of volatile degradants; a flow of inert helium gas carries thermal decomposition products from the TGA apparatus, via a heated ceramic capillary, to be analysed by Electron Ionisation Mass Spectrometer (EIMS). At intervals, EI mass spectra are recorded, allowing the intensities of select m/z values to be plotted as a function of the sample temperature.
It is likely that EIMS will bring about the fragmentation of small-molecule thermal decomposition species. Therefore for certain products, the parent ion ([M]+) may not necessarily be observed. The National Institute of Standards and Technology (NIST) Chemistry WebBook was employed in order to identify observed thermal decomposition products, based upon their expected fragmentation patterns.46
Initially one single-ring compound, [C4C1pip]Cl 2, and one azoniaspiro species, [7+6]Cl 8, were investigated using TGA-MS. Experiments were performed on 20–60 mg of the salt and with the same heating rate of 10 °C min−1 employed for the regular temperature-ramped TGA study (Fig. 5). Helium was used as the carrier gas. Electron Ionisation (EI) mass spectra were collected at periodic intervals throughout the experiment in the range m/z 10–300 (ionisation energy 70 eV). The primary peaks of interest were of m/z 40 and above. Detailed TGA-MS procedures are described in the ESI.†
Plausible decomposition pathways were initially proposed for the single-ring salt [C4C1pip]Cl, 2 (Fig. 7). Chloride is a moderate nucleophile but a poor base; thermal decomposition mechanisms are therefore likely to involve action of chloride as a nucleophile (also in agreement with previous literature on halide ionic liquids35). Decomposition may occur through SN2 nucleophilic attack of the chloride anion at a carbon atom α to the quaternary nitrogen atom, bringing about a dealkylation of the methyl or butyl chains (Fig. 7, A1, B1 and C1) or opening of the six-membered ring (Fig. 7, D1 and E1). TGA-MS data for 2 is shown in graphical form (Fig. 8).
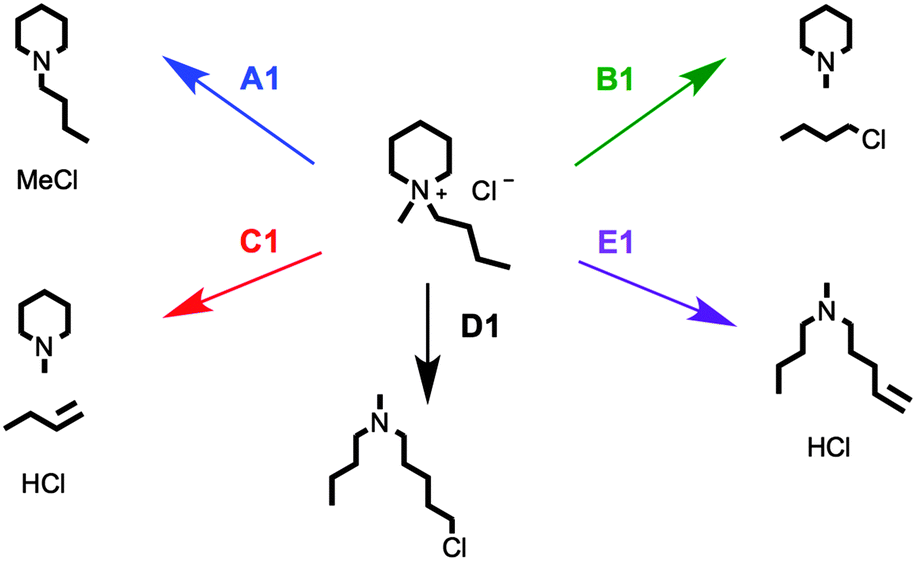 | ||
| Fig. 7 Proposed SN2 (A1, B1 and D1) and E2 Hofmann elimination (C1 and E1) thermal decomposition pathways for [C4C1pip]Cl, 2. | ||
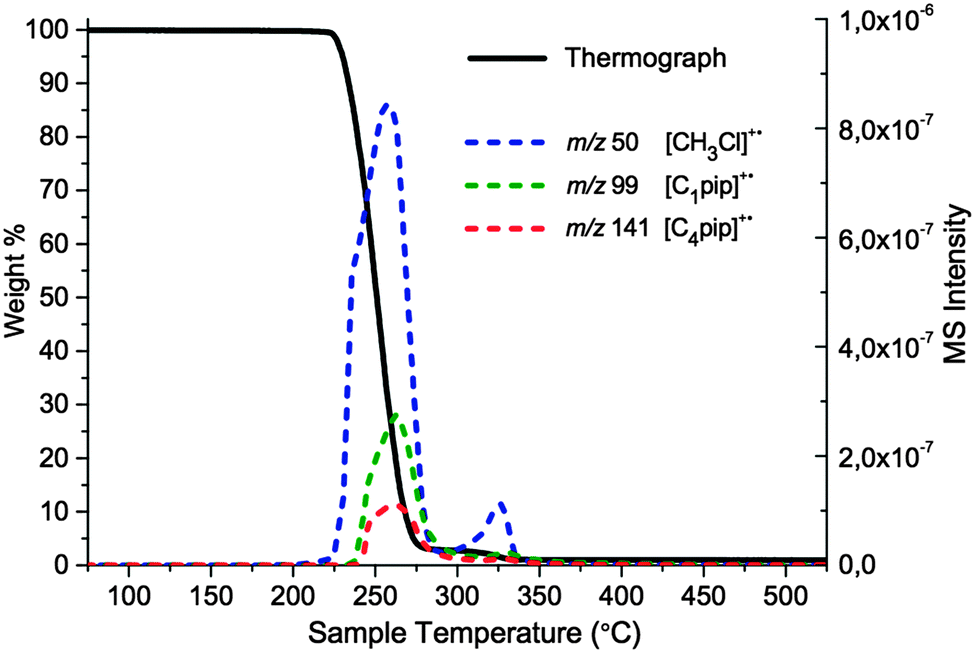 | ||
| Fig. 8 TGA-MS evaluation of [C4C1pip]Cl, 2. The continuous line denotes the TGA thermograph, the coloured dashed lines denote MS intensities for select m/z values. The assigned products are indicated. The thermograph is shown in the range 75–525 °C, for clarity. | ||
The TGA-MS analysis of 2 revealed a substantial peak, of m/z 50, evolving during the primary mass-loss event (ca. 225–300 °C); this peak was assigned to chloromethane, [CH3Cl]+˙. In addition, smaller peaks of m/z 99 and m/z 141 were detected during the main period of decomposition, corresponding to N-methylpiperidine [C1pip]+˙ and N-butylpiperidine [C4pip]+˙, respectively (intensities of peaks do not necessarily indicate relative quantities of degradation products, differences in ionisation efficiency/secondary ionisation should be considered). These three decomposition products strongly indicate that SN2 nucleophilic substitution mechanisms occur at the [C4C1pip]+ methyl and butyl substituents (mechanisms A1 and B1, Fig. 7). By contrast, the TGA-MS investigation (Fig. 8) provided no clear evidence to suggest that E2 elimination mechanisms were simultaneously occurring during the thermal degradation of [C4C1pip]Cl, 2.
The TGA-MS investigation of [7+6]Cl, 8, did not provide any clear indication of specific thermal decomposition pathways; mass spectra obtained at regular intervals were far less trivial to interpret compared to salt 2. No peak was observed at m/z 203, corresponding to the total mass of 8, [C11H22NCl]+˙. A parent peak at this mass would suggest the opening of either the six or seven-membered ring by nucleophilic attack of the chloride anion, generating a neutral heterocyclic amine with a terminal chloroalkyl substituent. Furthermore, peaks for piperidine and azepane, which would form from complete detachment of one of the ring alkyl substituents, were absent. However, the absence of the parent MS peak does not necessarily infer that such decomposition species do not form.
The added complexity of the thermal decomposition mechanisms of [7+6]Cl, 8, demanded the use of Thermogravimetric Analysis-Gas Chromatography/Mass Spectrometry (TGA-GCMS). A primary advantage of the additional GC step is the separation of individual components in the product mixture. The difficulties associated with superimposed EI mass spectra (arising from the simultaneous evolution of all decomposition products) in TGA-MS experiments are therefore minimised.
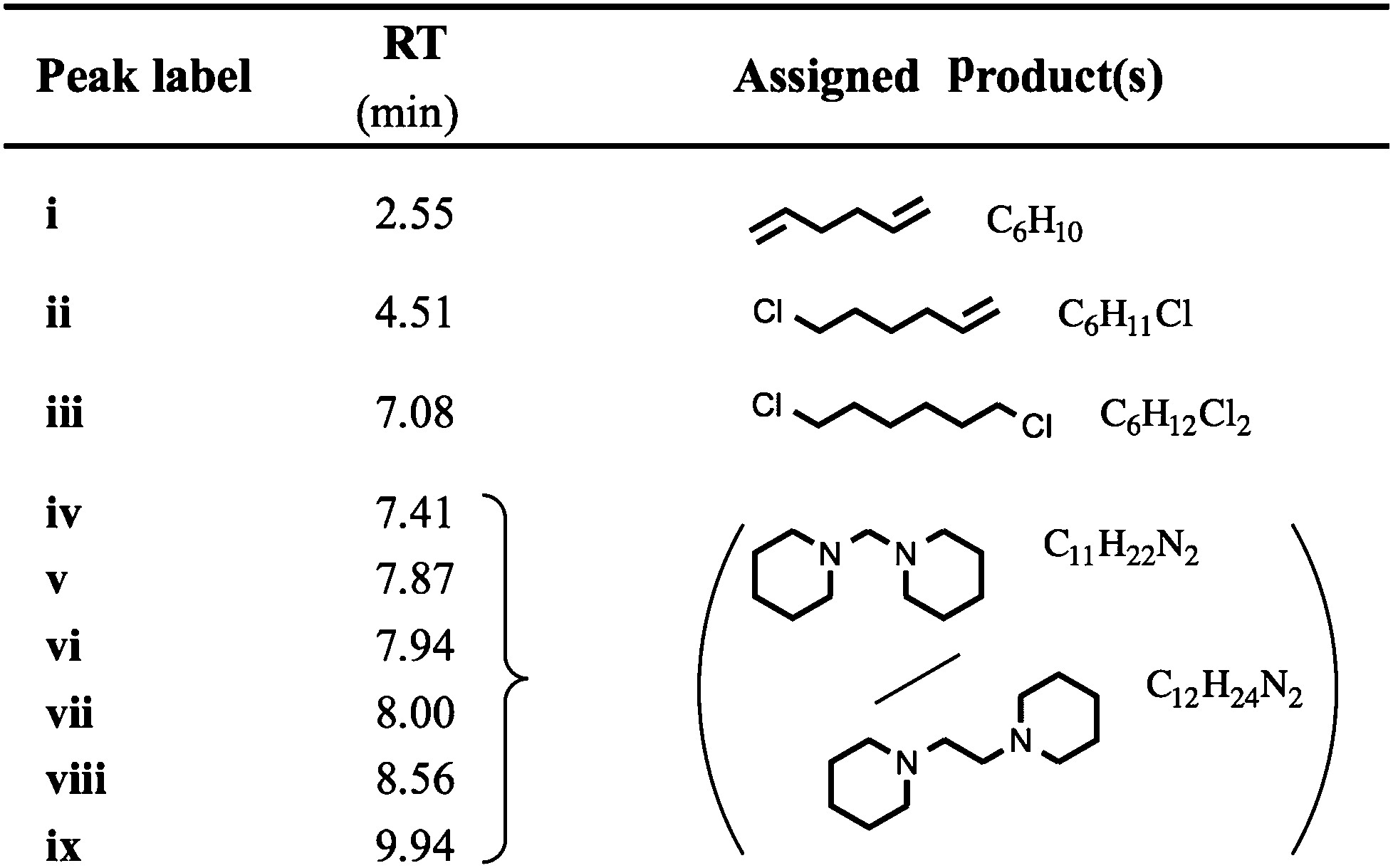
TGA-GCMS experiments were performed on smaller quantities (ca. 2 mg) of [7+6]Cl, 8, using porcelain crucibles, and a heating rate of 10 °C min−1. Thermal decomposition products were transferred, via a heated capillary, and were collected at two temperatures during the ramp (310 and 340 °C) onto activated carbon tubes, for periods of 15 seconds. Subsequently, the tubes were loaded into the GCMS apparatus, and the tubes were heated to evolve the adsorbed decomposition products. Observed GC peaks (‘i–viii’, Fig. 9, Table 4) were cautiously assigned to thermal decomposition products by comparing the MS data against the NIST Chemistry WebBook.46 Full TGA-GCMS procedures are described in the ESI,† including photos of the experimental setup (Fig. E4, ESI†). Proposed thermal degradation mechanisms of 8 are indicated in Fig. 10, based on the assigned products in Table 4.
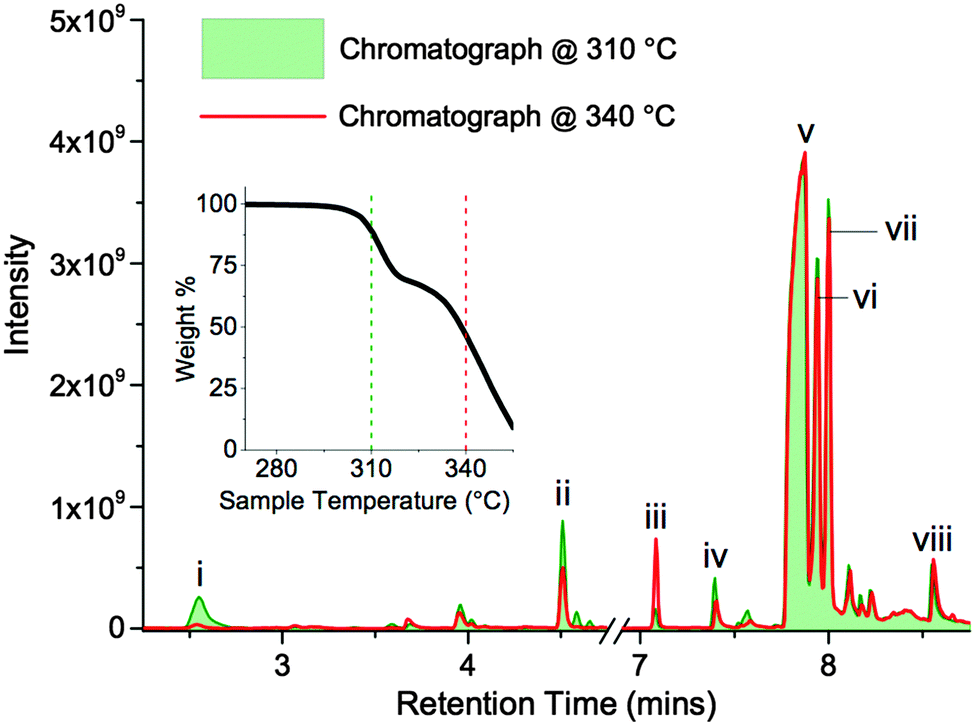 | ||
| Fig. 9 Gas chromatographs of TGA-GCMS experiments, representing thermal decomposition of [7+6]Cl, 8. Selected primary peaks are labelled (i–viii). The two overlaid chromatographs highlight products condensed onto activated carbon tubes, for 15 second periods, at ca. 310 and 340 °C in the TGA thermograph (inset). | ||
|
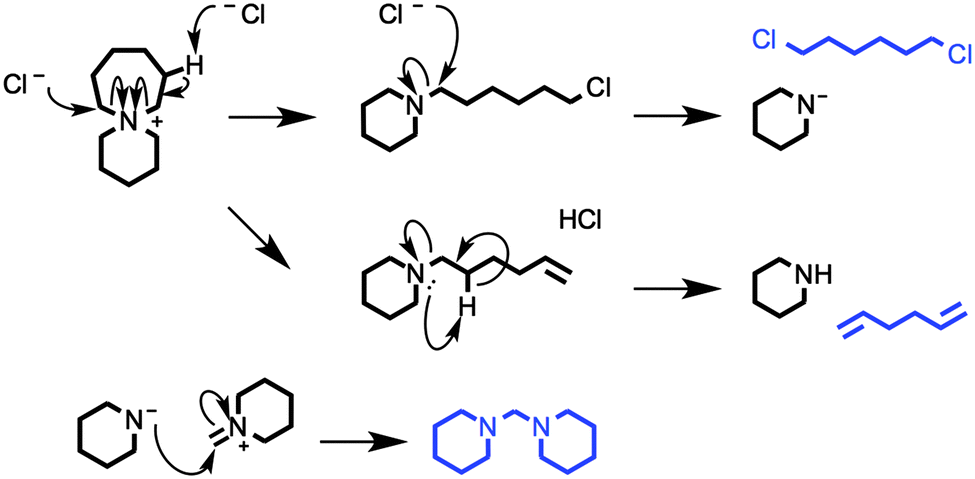 | ||
| Fig. 10 A series of possible thermal decomposition mechanisms for 6-azoniaspiro[6.5]dodecenium chloride, [7+6]Cl 8. Degradation products, tentatively assigned in the TGA-GCMS investigation (Fig. 9, Table 4), are highlighted in blue. | ||
The two chromatographs in Fig. 9, representing thermal decomposition products isolated at 310 and 340 °C, are highly similar; the majority of the significant peaks are present in both spectra. This observation indicates that the two separate mass loss steps in the temperature-ramped TGA thermograph of 8 do not correspond to two distinct chemical mechanisms. Instead, the two step weight loss phenomenon possibly arises due to a heat transfer effect associated with the melting of the salt, or due to multiple crystalline polymorphs of 8 that exhibit marginally differing thermal properties.
One exception is peak ‘i’, at a retention time (RT) of 2.55 minutes, which is primarily absent from the chromatograph at 340 °C. The mass spectrum of this peak closely matches that of 1,5-hexadiene, [C6H10]+˙.46 This degradation product may form by elimination reactions occurring sequentially at both β carbons of the seven-membered ring of 8 (Fig. 10). Another minor peak, at RT = 7.08 mins, closely matches (the MS data of) 1,6-dichlorohexane, [C6H12Cl2]+˙, likely to be formed from sequential nucleophilic attack of two chloride anions at the two α carbons of the seven-membered ring of 8.46
The most prominent peaks in the TGA-GCMS study of 8 occur in the region RT = 7.8–8.1 mins. Mass spectra associated with these peaks each matched moderately well with two known compounds, bis(1-piperidyl)methane and 1,2-bis(1-pi-peridyl)ethane (Table 4). It is therefore possible that either or both of these piperidine-containing products are present in the thermal decomposition mixture, and correspond to particular peaks in the ensemble at RT ca. 8 mins. However, three major peaks (at RT = 7.87, 7.94 and 8.00 minutes) each display an intense fragment mass peak at m/z 98. This can be assigned to the 1-methylenepiperidinium cation radical [(CH2)5NCH2]+˙. This fragment at m/z 98 would likely be generated from any decomposition product containing the N-methylene-piperidine subunit, including the two open-chain intermediates generated from SN2 and/or E2 ring-opening mechanisms of the seven-membered ring; these intermediates, N-(6-chlorohexyl)piperi-dine and N-(hex-5-enyl)piperidine, are displayed in Fig. 10.
No reference EIMS data for these complex structures exists in the NIST Chemistry WebBook.46 Therefore, it is possible that the significant gas chromatograph peaks (RT = 7.8–8.1 mins) correspond to these decomposition products from the opening of the seven-membered ring.
Although these primary peaks of the gas chromatographs cannot be assigned with certainty, the observations indicate that thermal decomposition mechanisms occur predominantly at the seven-membered ring of [7+6]Cl, 8, leaving the six-membered piperidine ring intact. Increased conformational flexibility (and the unfavourable bond angles) of the seven-membered ring may partially account for this. Regardless of the explanation, azoniaspiro salts must undergo ring-opening upon thermal decomposition in direct contrast to the single-ring systems (e.g. salt 2).
Subsequently, following the thermogravimetric investigation into thermal decomposition of single-ring and azoniaspiro chloride salts, B3LYP-D2/6-311++G(d,p) calculations were employed in order to rationalise observed degradation mechanisms of [C4C1pip]Cl, 2, and [7+6]Cl, 8. Details of the computational method are described in the ESI.†
Initially, five transition state (TS) structures of [C4C1pip]Cl, 2, corresponding to the five mechanisms in Fig. 7, were obtained. These TS structures are displayed below in Fig. 11. ΔE, ΔG, ΔH and TΔS energies for the TSs and products are listed in Table 5, quoted in kJ mol−1 relative to the lowest energy ‘C (Me-Ax) ααα 1’ ion pair conformer (Fig. 3).
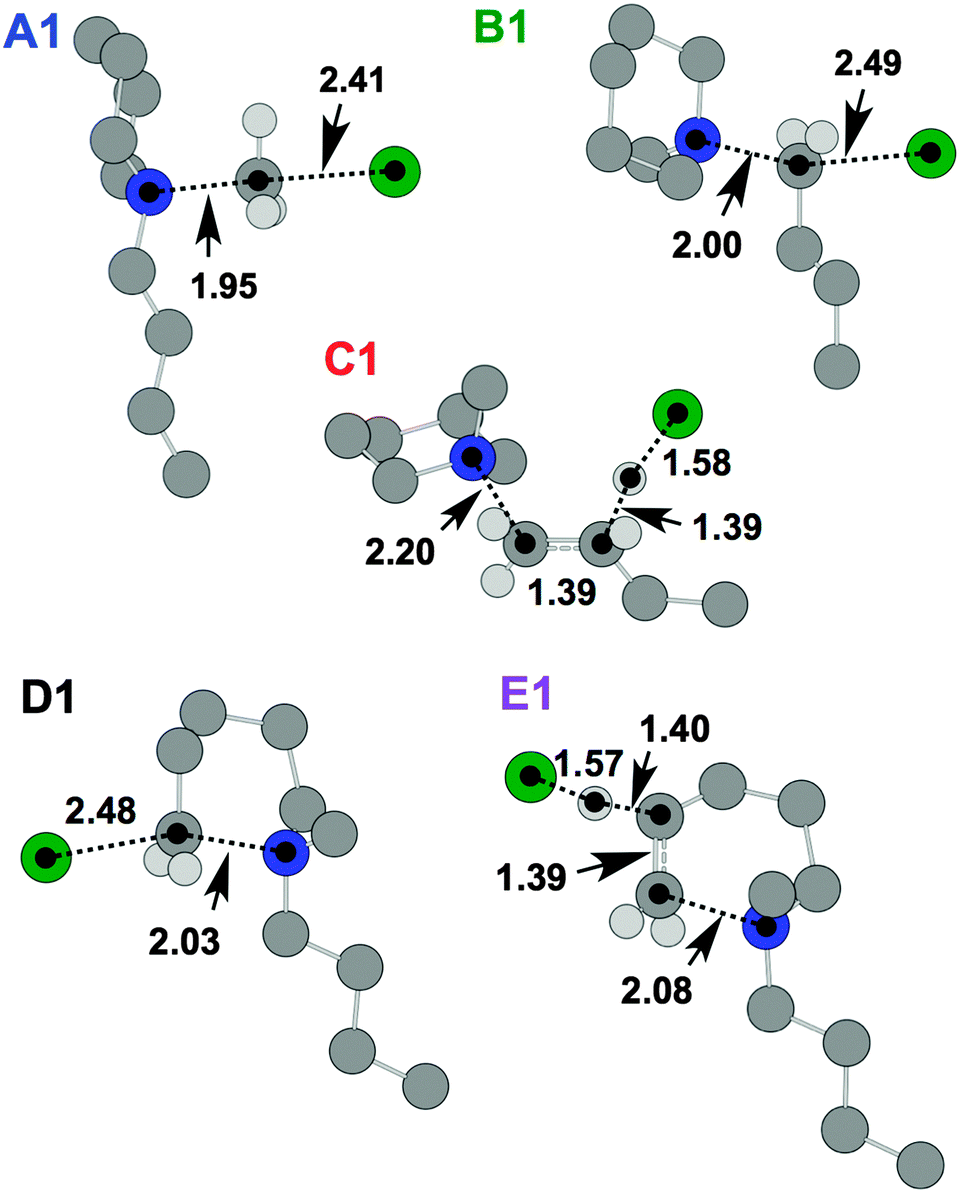 | ||
| Fig. 11 TS structures for [C4C1pip]Cl, 2, representing SN2 (A1, B1 and D1) and E2 (C1 and E1) mechanisms. Atom distances are in angstroms. | ||
| Mechanism | Transition state (kJ mol−1) | Product (kJ mol−1) | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔE | ΔG | ΔH | TΔS | ΔE | ΔG | ΔH | TΔS | |
| A1 | 88.4 | 86.7 | 89.0 | 2.3 | 13.4 | −34.4 | 15.5 | 49.9 |
| B1 | 90.2 | 91.3 | 90.0 | −1.2 | 20.4 | −31.2 | 21.2 | 52.4 |
| C1 | 117.5 | 116.0 | 119.0 | 3.1 | 77.4 | −12.4 | 83.0 | 95.4 |
| D1 | 92.8 | 91.5 | 93.3 | 1.8 | 7.8 | −3.5 | 12.1 | 15.5 |
| E1 | 122.6 | 118.1 | 124.6 | 6.5 | 65.3 | 15.6 | 74.2 | 58.6 |
The SN2 dealkylation (A1, B1) and SN2 ring-opening (D1) mechanisms, for thermal decomposition of [C4C1pip]Cl 2, are 26–36 kJ mol−1 lower in activation energy (ΔHactivation) than the two E2 elimination pathways (C1 and E1), consistent with the nucleophilic character of the chloride anion. By incorporating a more basic anion (e.g. [OAc]−) elimination mechanisms may become favoured over nucleophilic substitution. The calculations indicate only minor disparities (<5 kJ mol−1) in ΔHactivation energy barriers for the multiple SN2 mechanisms (A1, B1 and D1), and thus are of a similar magnitude to the computational error associated with this level of DFT calculation. Gas-phase calculations, therefore, find no difference in activation barrier between SN2 pathways. The TGA-MS analysis suggested that mechanism A1 involving dealkylation at the N-methyl substituent might predominate (indicated by the more intense peak at m/z 50, [CH3Cl]+˙, and the smaller peak at m/z 141, [C4pip]+˙).
Calculations using the SMD solvation methodology47 were subsequently carried out, enabling an approximation of the thermal degradation occurring within a solvent environment (IL). Solvent parameters related to the IL [C4C1pip]Cl were employed, and all structures were re-optimised and confirmed as minima or TS at the B3LYP-D2/6-311++G(d,p) level of theory (further details are provided in the ESI†). Similar to the gas-phase calculations, there are only minor differences in the ΔHactivation/ΔGactivation barriers for the different SN2 mechanisms with SMD (Table E7, ESI†). Although uncertain (considering small TS TΔS values, Table E7, ESI†), it is possible that a subtle entropic cost associated with Cl− attack at the flexible butyl substituent is an explanation for the calculated difference in ΔGactivation for mechanisms A1 and B1.
E2 mechanisms (C1 and E1) involve large positive entropy changes (TΔS = +95.4 and +58.6 kJ mol−1 respectively (gas phase), +89.7 and +53.0 kJ mol−1 respectively (IL), Table 5), associated with formation of two or three neutral molecules from one ion pair.
TS and product geometries were also investigated for the thermal decomposition mechanisms of [7+6]Cl, 8, using DFT (Fig. 12, Table 6). The TGA-GCMS investigation of 8 (Fig. 9 and 10) lent evidence towards preferential opening of the seven-membered ring. In an attempt to rationalise this observation, ΔGactivation barriers for SN2 and E2 ring-opening mechanisms of both the six and seven-membered rings were compared.
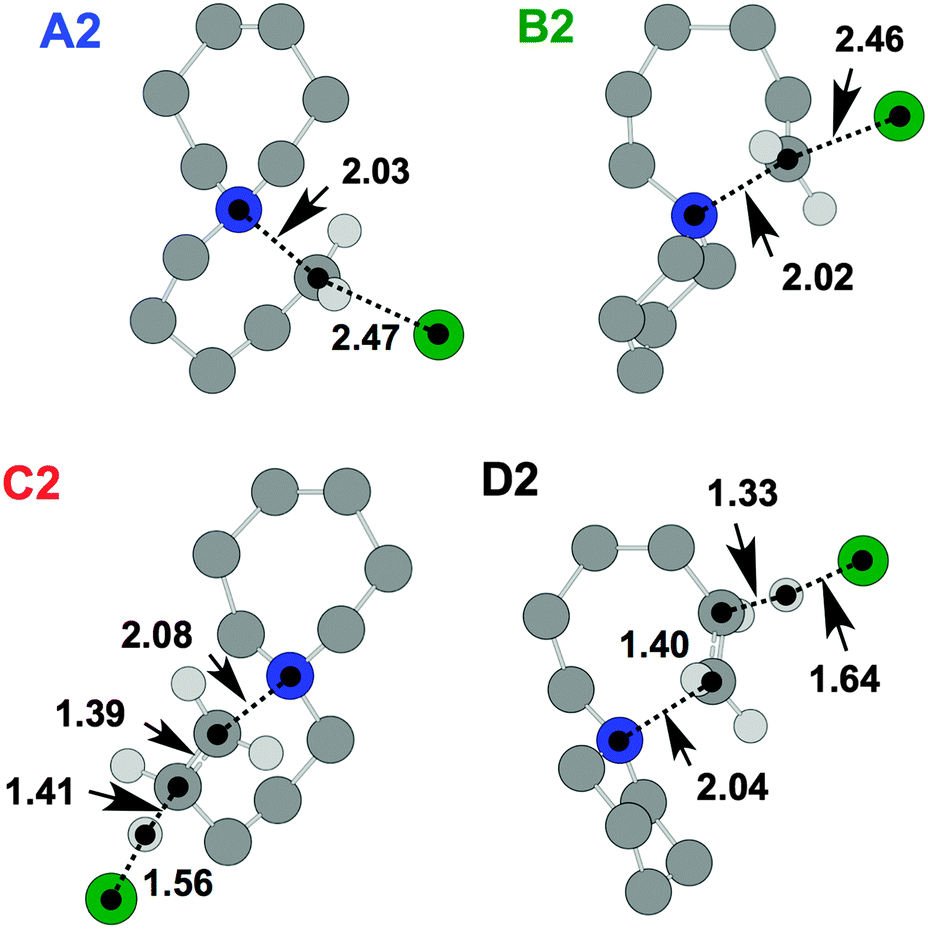 | ||
| Fig. 12 TS structures for [7+6]Cl, 8, representing SN2 (A2 and B2) and E2 (C2 and D2) mechanisms relative to the lowest energy ‘7chair1 6chair ααα’ ion pair conformer (Fig. 4). Atom distances are in angstroms. | ||
| Mechanism | Transition state (kJ mol−1) | Product (kJ mol−1) | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔE | ΔG | ΔH | TΔS | ΔE | ΔG | ΔH | TΔS | |
| A2 | 97.4 | 96.0 | 97.9 | 1.9 | 9.6 | −2.4 | 14.0 | 16.4 |
| B2 | 93.3 | 93.3 | 93.3 | 0.0 | −8.4 | −21.8 | −4.4 | 17.4 |
| C2 | 127.6 | 122.4 | 129.5 | 7.1 | 67.1 | 16.5 | 76.2 | 59.6 |
| D2 | 115.3 | 109.8 | 117.1 | 7.3 | 50.0 | −2.9 | 59.3 | 62.2 |
The calculations of thermal decomposition mechanisms of [C4C1pip]Cl, 2, demonstrated that ΔGactivation barriers for SN2 mechanisms were 24–32 kJ mol−1 (gas phase) or 37–55 kJ mol−1 (IL) lower in energy than for the E2 mechanisms. Similarly, for [7+6]Cl, 8, ΔGactivation energies for SN2 pathways (A2, B2) are approximately 14–29 kJ mol−1 (gas phase) or 6–38 kJ mol−1 (IL) lower than for the corresponding E2 mechanisms (C2, D2).
Moreover, routes involving opening of the seven-membered ring are slightly more facile (ΔΔGactivation = 3–13 kJ mol−1 (gas phase) or 4–28 kJ mol−1 (IL)) than those that involve the opening of the six-membered ring, consistent with the findings of the TGA-GCMS experiments (Fig. 9 and 10). These minor disparities in ΔGactivation suggest that E2 mechanisms could predominate if the chloride anion were to be substituted for other anions. Indeed, the precise thermal decomposition mechanisms that predominate for all single-ring/azoniaspiro tetraalkylammonium salts are likely to be heavily dependant on the nature of the anion, as for the dialkylimidazolium ionic liquids20 and others30 in the chemical literature.
The calculated activation barrier for each of the investigated mechanisms of 8 (ΔHactivation = 93–130 kJ mol−1 (gas phase)) are substantially lower than the experimental activation energy (Ea = 147 kJ mol−1) from isothermal TGA experiments.
An explanation for this disparity may be due, partially, to the limitations of using single ion pairs (stable clusters of one cation and one anion) as the starting point for thermal decomposition reaction profiles. Recent investigations have demonstrated that ion pair dimers are more capable of recovering the structural and bonding features that are exhibited in the bulk ionic liquid (or crystalline phase).43–45 The X-ray structure of azoniaspiro compound [6+6]Cl, 5, Fig. 2 (analogous to [7+6]Cl, 8) reveals that any particular chloride anion is likely to be strongly interacting with α and β hydrogen atoms on as many as four neighbouring cations. Therefore, an additional input of energy will be required to break these favourable interactions between chloride and the near cations, moreover chloride will be less nucleophilic in this more ‘solvated’ environment.
Calculations using the SMD model47 were carried out for [7+6]Cl, 8 (Table E8, ESI†), similarly to the calculations performed for 2. Incorporation of the SMD solvation model appears to recover a large proportion of the energy disparity between the calculated gas-phase ΔHactivation values and the experimental Ea from isothermal TGA (147 kJ mol−1). With the addition of SMD, ΔHactivation was calculated as +140.0 and +135.7 kJ mol−1 for SN2 mechanisms A2 and B2 respectively. Similar to gas-phase calculations, solvated E2 pathways exhibited ΔHactivation values significantly higher (ΔΔHactivation = 13–45 kJ mol−1) than for the corresponding SN2 pathways. The SMD results therefore indicate that it is critical to account for solution effects when calculating transition state energy barriers for thermal decomposition routes.
Comparing Tonset values for azoniaspiro chloride salts 4–10 (Table 3), it is sensible to suggest that six and seven-membered rings impart improved thermal stability over five or eight-membered rings. Furthermore, TGA-GCMS and DFT investigations into 8 reveal that decomposition occurs more readily at the seven-membered ring. On this basis, [6+6]Cl, 5, should exhibit the greatest thermal stability of the compounds in the series. However, 5 was found to be among the most unstable, degrading via two distinct thermal events (Tstart = 189 °C). To minimise the possibility that an impurity is responsible for the poor observed stability of 5, repeat temperature-ramped TGA experiments were performed, and a separate batch of the salt was prepared (omitting MgSO4 in the purification); multi-step thermographs were obtained in each circumstance. Moreover, spectroscopic analysis of 5 confirmed high purity. Therefore, the low thermal stability of [6+6]Cl, 5, cannot reasonably be attributed to the presence of an impurity.
Nevertheless the focal azoniaspiro salt in the investigation, [7+6]Cl, 8, exhibited significantly improved thermal stability relative to single-ring analogues 1–3; in terms of the Tonset and Tstart parameters, 8 outperformed [C4C1pip]Cl, 2, by 91 and 76 °C, respectively. The prohibitively high melting temperatures of the azoniaspiro chlorides presents a problem for employing these salts in ionic liquid applications; no salt in the series (5–10) exhibits a significant stable-liquid range. Nevertheless, substituting chloride for a larger, charge-diffuse molecular anion will lower the melting point substantially, whilst retaining the inherent stability imparted by the cation. To this end, two additional azoniaspiro salts were prepared, by the reaction of 8 with either Li[OTf] or Li[NTf2] ([OTf]− = trifluoromethane-sulfonate, [NTf2]− = bis(trifluoromethanesulfonyl)imide). The resultant salts, [7+6][OTf] and [7+6][NTf2], exhibited melting temperatures of 115.5–117 °C and 104.5–106 °C, respectively (full synthesis/characterisation is listed in the ESI†). The Tonset value of [7+6][OTf] was measured as 412 °C, comparing favourably or similarly with analogous heterocyclic triflate salts (e.g. [C4py][OTf] Tonset = 362 °C;30 [(HO)2C2C1im][OTf] Tonset = 391 °C;30 [C1C1C14pz][OTf] Tonset = 395 °C48 or; [C4C1im][OTf] Tonset = 393 °C30 or 410 °C48). The Tonset value of [7+6][NTf2] was measured as 431 °C, approximately equal to other ionic compounds incorporating this anion28,30 (degradation is known to proceed via an anion-centred degradation mechanism27). Nevertheless, in contrast to the commonly-encountered dialkylimidazolium species, azoniaspiro salts are incapable of undergoing N-Heterocyclic Carbene (NHC) formation/metal chelation in the presence of reactive (dissolved) species. This may render them a more suitable choice of solvent media for organic synthesis and catalysis.
Preparation of binary/reciprocal binary mixtures48–52 of azo-niaspiro species is likely to afford still lower melting points. Processes that exploit the specific interactions of a particular anion, therefore, may be undertaken, whilst problems associated with high reactivity between the cation and anion (leading towards decomposition) are minimised. Azoniaspiro species therefore represent worthy candidates for future investigation.
Conclusions
In summary, a broad ensemble of single-ring and azoniaspiro-type chloride organic salts were prepared, in both high purity and acceptable yields. Investigations were undertaken in order to gain insights into the structural and bonding properties, and to ascertain information on the trends, thermodynamics and underlying mechanisms of thermal decomposition.X-ray crystallography and DFT studies demonstrated that, regardless of whether the cation incorporates a single ring, or two cyclic motifs, the chloride anion is preferentially located in close proximity to the cation quaternary nitrogen atom, interacting strongly with tripod-like ensembles of α and β protons. Interestingly, for the monohydrate of 4, [5+5]Cl·H2O, the water molecule appears to compete with and displace the chloride anion, occupying the position closest to the nitrogen.
The relative thermal stabilities of the chloride species were compared by employing temperature-ramped TGA and trends were elucidated. Single-ring salts were susceptible to thermal degradation at markedly lower temperatures than their azoniaspiro analogues. Possible explanations include higher rigidity, and the absence of flexible alkyl chains for azoniaspiro salts; this is supported by the observation that thermal decomposition of 1-butyl-1-methylpiperidinium chloride, [C4C1pip]Cl, 2, principally involves SN2 dealkylation at the pendant N-methyl and N-butyl chains. The absence of these alkyl chains for azoniaspiro salts implies that thermal degradation must proceed via a less optimal ring-opening transition state; TGA-GCMS analysis of [7+6]Cl, 8, indicated that primary decomposition products retain an intact six-membered piperidine ring. To a minor extent full detachment of chloroalkyl/alkenyl fragments (e.g. 1-chlorohex-5-ene, 1,5-hexadiene) occurs. DFT calculations supported these experimental findings, highlighting that mechanisms to open the seven-membered ring of 8 proceed through a lower-energy transition state than for opening of the six-membered ring.
Within the sequence of azoniaspiro chloride salts, thermal stability trends may be established, on the basis of the sizes of the incorporated rings. Six- and seven-membered rings impart greater thermal stability than five- and eight-membered rings. Foremost, compounds based on five-membered rings should be avoided on account of the poor thermal stabilities and their observed hygroscopic nature. Salts derived from morpholine and thiomorpholine should similarly be avoided, because of their potential to evolve toxic mustard species upon thermal decomposition. Future research is necessary, to determine the impact of ring-substitution and branching on thermal stability.
A relative lack of conformational flexibility of the azonia-spiro-cation salts has an unwanted side effect; melting temperatures are significantly raised. The reactive chloride anion was selected, in this investigation, for the deliberate purposes of incurring thermal decomposition (to compare relative stabilities of the different cation structures), to minimise the computational complexity/expense, and for the ease of laboratory preparation. For the examined azoniaspiro chloride salts, the onset of melting appears closely associated with thermal de-composition. However, pairing an azoniaspiro cation with a large, charge-diffuse and bulky anion (e.g. [OTf]− or [NTf2]−) yields compounds that have much lower melting points, albeit considerably above room temperature.
We propose that it is more sensible to consider the concept of a ‘stable-liquid range’; a temperature-range in which the species is both in the liquid state and is thermally stable for a prolonged period of time. ‘Room-Temperature Ionic Liquids’ (RTILs), though (by definition) molten at ambient conditions, often suffer from poor thermal stability.36,37 By contrast all-in-organic ‘Molten Salts’ are highly thermally-robust, yet their prohibitively high melting points preclude their use in a broad range of applications. Simple azoniaspiro salts, or more likely binary/reciprocal binary azoniaspiro mixtures,48,49 have the potential to partially bridge this ‘gap’ in stable-liquid ranges; difficulties associated with the high melting temperatures are therefore minimised, whilst retaining benefits associated with the high thermal stability of the azoniaspiro cation. Certain azoniaspiro chloride salts have been shown, in this investigation, to offer substantially higher operational temperature limits than single-ring (e.g. dialkylpiperidinium) equivalents.
Further investigation is required in order to gain a comprehensive understanding into thermal properties of azoniaspiro salts. Nevertheless, they represent promising candidate solvent systems, offering the beneficial properties of high-thermal stability, wide electrochemical window,18 and trivial synthesis and purification. Furthermore, a broad range of structures and properties are achievable via the modification of the size and substituents of the two cyclic moieties, in analogy to the reputation of ionic liquids as ‘designer solvents’.
Acknowledgements
The authors thank Dr Richard L. Thompson and W. Douglas Carswell of the University of Durham, for assistance with the TGA-MS investigations, and Dr Francis Clegg of Sheffield-Hallam University for assistance with TGA-GCMS. Matthew T. Clough wishes to acknowledge Sven Holzmann of BASF SE for assistance with the azoniaspiro salt syntheses, to Claire Ashworth of Imperial College London for help with the DFT calculations, and to BASF SE for generous sponsorship.References
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- F. H. Hurley and T. P. WIer, J. Electrochem. Soc., 1951, 98, 207–212 CrossRef CAS.
- H. L. Chum, V. R. Koch, L. L. Miller and R. A. Osteryoung, J. Am. Chem. Soc., 1975, 97, 3264–3265 CrossRef CAS.
- J. S. Wilkes, J. A. Levisky, R. A. Wilson and C. L. Hussey, Inorg. Chem., 1982, 21, 1263–1264 CrossRef CAS.
- A. a. K. Abdul-Sada, A. M. Greenway, K. R. Seddon and T. Welton, Org. Mass Spectrom., 1989, 24, 917–918 CrossRef CAS.
- A. a. K. Abdul-Sada, A. M. Greenway, K. R. Seddon and T. Welton, Org. Mass Spectrom., 1992, 27, 648–649 CrossRef CAS.
- A. a. K. Abdul-Sada, A. M. Greenway, K. R. Seddon and T. Welton, Org. Mass Spectrom., 1993, 28, 759–765 CrossRef CAS.
- J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1992, 965–967, 10.1039/c39920000965.
- I. Anugwom, P. Maki-Arvela, P. Virtanen, P. Damlin, R. Sjoholm and J.-P. Mikkola, RSC Adv., 2011, 1, 452–457 RSC.
- D. R. MacFarlane, P. Meakin, J. Sun, N. Amini and M. Forsyth, J. Phys. Chem. B, 1999, 103, 4164–4170 CrossRef CAS.
- H. Sakaebe and H. Matsumoto, Electrochem. Commun., 2003, 5, 594–598 CrossRef CAS.
- T. Belhocine, S. A. Forsyth, H. Q. N. Gunaratne, M. Nieuwenhuyzen, P. Nockemann, A. V. Puga, K. R. Seddon, G. Srinivasan and K. Whiston, Green Chem., 2011, 13, 3137–3155 RSC.
- F. F. Blicke and E. B. Hotelling, J. Am. Chem. Soc., 1954, 5099–5103 CrossRef CAS.
- A. Losurdo, W. Y. Wen and C. Jolicoeur, J. Solution Chem., 1979, 8, 449–460 CrossRef CAS.
- R. Millini, L. Carluccio, F. Frigerio, W. O. Parker and G. Bellussi, Microporous Mesoporous Mater., 1998, 24, 199–211 CrossRef CAS.
- F. Frigerio, L. Carluccio, W. O. Parker and R. Millini, J. Mol. Catal. A: Chem., 2001, 166, 167–174 CrossRef CAS.
- S. I. Zones, A. W. Burton, G. S. Lee and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 9066–9079 CrossRef CAS PubMed.
- Y. Abu-Lebdeh, E. Austin and I. J. Davidson, Chem. Lett., 2009, 38, 782–783 CrossRef CAS.
- M. R. R. Prasad and V. N. Krishnamurthy, Thermochim. Acta, 1991, 185, 1–10 CrossRef.
- H. L. Ngo, K. LeCompte, L. Hargens and A. B. McEwen, Thermochim. Acta, 2000, 357–358, 97–102 CrossRef CAS.
- C. P. Fredlake, J. M. Crosthwaite, D. G. Hert, S. Aki and J. F. Brennecke, J. Chem. Eng. Data, 2004, 49, 954–964 CrossRef CAS.
- T. J. Wooster, K. M. Johanson, K. J. Fraser, D. R. MacFarlane and J. L. Scott, Green Chem., 2006, 8, 691–696 RSC.
- A. Seeberger, A.-K. Andresen and A. Jess, Phys. Chem. Chem. Phys., 2009, 11, 9375–9381 RSC.
- F. Heym, B. J. M. Etzold, C. Kern and A. Jess, Phys. Chem. Chem. Phys., 2010, 12, 12089–12100 RSC.
- N. Meine, F. Benedito and R. Rinaldi, Green Chem., 2010, 12, 1711–1714 RSC.
- A. W. T. King, A. Parviainen, P. Karhunen, J. Matikainen, L. K. J. Hauru, H. Sixta and I. Kilpelainen, RSC Adv., 2012, 2, 8020–8026 RSC.
- Y. Chen, Y. Cao, Y. Shi, Z. Xue and T. Mu, Ind. Eng. Chem. Res., 2012, 51, 7418–7427 CrossRef CAS.
- M. T. Clough, K. Geyer, P. A. Hunt, J. Mertes and T. Welton, Phys. Chem. Chem. Phys., 2013, 47, 20480–20495 RSC.
- N. E. A. Cousens, L. J. Taylor Kearney, M. T. Clough, K. R. J. Lovelock, R. G. Palgrave and S. Perkin, Dalton Trans., 2014, 43, 10910–10919 RSC.
- Y. Cao and T. Mu, Ind. Eng. Chem. Res., 2014, 53, 8651–8664 CrossRef CAS.
- J. H. Flynn and L. A. Wall, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 323–328 CrossRef CAS.
- B. Chan, N. Chang and M. Grimmett, Aust. J. Chem., 1977, 30, 2005–2013 CrossRef CAS.
- M. C. Kroon, W. Buijs, C. J. Peters and G.-J. Witkamp, Thermochim. Acta, 2007, 465, 40–47 CrossRef CAS.
- H. Ohtani, S. Ishimura and M. Kumai, Anal. Sci., 2008, 24, 1335–1340 CrossRef CAS PubMed.
- S. D. Chambreau, J. A. Boatz, G. L. Vaghjiani, C. Koh, O. Kostko, A. Golan and S. R. Leone, J. Phys. Chem. A, 2012, 116, 5867–5876 CrossRef CAS PubMed.
- C. Maton, N. De Vos and C. V. Stevens, Chem. Soc. Rev., 2013, 42, 5963–5977 RSC.
- M. T. Clough, Ionic Liquid Thermal Stability: implications for cellulose regeneration, PhD thesis, Imperial College London, 2015 Search PubMed.
- W. Eschweiler, Ber. Dtsch. Chem. Ges., 1905, 38, 880–882 CrossRef.
- P. A. Hunt and I. R. Gould, J. Phys. Chem. A, 2006, 110, 2269–2282 CrossRef CAS PubMed.
- P. A. Hunt, B. Kirchner and T. Welton, Chem. – Eur. J., 2006, 12, 6762–6775 CrossRef CAS PubMed.
- P. A. Hunt, J. Phys. Chem. B, 2007, 111, 4844–4853 CrossRef CAS PubMed.
- P. A. Hunt, I. R. Gould and B. Kirchner, Aust. J. Chem., 2007, 60, 9–14 CrossRef CAS.
- R. P. Matthews, C. Ashworth, T. Welton and P. A. Hunt, J. Phys.: Condens. Matter, 2014, 26, 284112 CrossRef PubMed.
- R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2014, 16, 3238–3253 RSC.
- R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2015, 17, 14437–14453 RSC.
- National Institute of Standards and Technology (NIST) Chemistry WebBook, http://webbook.nist.gov/chemistry.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. T. Clough, C. R. Crick, J. Grasvik, P. A. Hunt, H. Niedermeyer, T. Welton and O. P. Whitaker, Chem. Sci., 2015, 6, 1101–1114 RSC.
- H. Niedermeyer, J. P. Hallett, I. J. Villar-Garcia, P. A. Hunt and T. Welton, Chem. Soc. Rev., 2012, 41, 7780–7802 RSC.
- J. N. Canongia Lopes, T. C. Cordeiro, J. M. S. S. Esperança, H. J. R. Guedes, S. Huq, L. P. N. Rebelo and K. R. Seddon, J. Phys. Chem. B, 2005, 109, 3519–3525 CrossRef PubMed.
- M. Y. Lui, L. Crowhurst, J. P. Hallett, P. A. Hunt, H. Niedermeyer and T. Welton, Chem. Sci., 2011, 2, 1491–1496 RSC.
- M. Brussel, M. Brehm, A. S. Pensado, F. Malberg, M. Ramzan, A. Stark and B. Kirchner, Phys. Chem. Chem. Phys., 2012, 14, 13204–13215 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 978072 and 1434098. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cp07209c |
| This journal is © the Owner Societies 2016 |