
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOligothiophene wires: impact of torsional conformation on the electronic structure†
D. A.
Kislitsyn
a,
B. N.
Taber
a,
C. F.
Gervasi
a,
L.
Zhang
b,
S. C. B.
Mannsfeld
c,
J. S.
Prell
a,
A. L.
Briseno
b and
G. V.
Nazin
*a
aDepartment of Chemistry and Biochemistry, Materials Science Institute, Oregon Center for Optical, Molecular and Quantum Science, University of Oregon, 1253 University of Oregon Eugene, OR 97403, USA. E-mail: gnazin@uoregon.edu
bDepartment of Polymer Science and Engineering, University of Massachusetts-Amherst, Silvio O. Conte National Center for Polymer Research, 120 Governors Drive Amherst, MA 01003, USA
cCenter for Advancing Electronics Dresden, Dresden University of Technology, 01062 Dresden, Germany
First published on 18th January 2016
Abstract
Charge transport in polymer- and oligomer-based semiconductor materials depends strongly on the structural ordering of the constituent molecules. Variations in molecular conformations influence the electronic structures of polymers and oligomers, and thus impact their charge-transport properties. In this study, we used Scanning Tunneling Microscopy and Spectroscopy (STM/STS) to investigate the electronic structures of different alkyl-substituted oligothiophenes displaying varied torsional conformations on the Au(111) surface. STM imaging showed that on Au(111), oligothiophenes self-assemble into chain-like structures, binding to each other via interdigitated alkyl ligands. The molecules adopted distinct planar conformations with alkyl ligands forming cis- or trans- mutual orientations. For each molecule, by using STS mapping, we identify a progression of particle-in-a-box-like states corresponding to the LUMO, LUMO+1 and LUMO+2 orbitals. Analysis of STS data revealed very similar unoccupied molecular orbital energies for different possible molecular conformations. By using density functional theory calculations, we show that the lack of variation in molecular orbital energies among the different oligothiophene conformers implies that the effect of the Au–oligothiophene interaction on molecular orbital energies is nearly identical for all studied torsional conformations. Our results suggest that cis–trans torsional disorder may not be a significant source of electronic disorder and charge carrier trapping in organic semiconductor devices based on oligothiophenes.
Introduction
Solution-processable thiophene-based polymers and oligomers represent an important class of organic semiconductor materials1,2 with potential applications in field-effect transistors,3–5 light-emitting diodes,6,7 photodetectors8 and photovoltaic devices.9–11 The electronic structures of polymers and oligomers, and consequently their charge transport properties, are determined, to a significant degree, by the conformations of their π-conjugated backbones.12–14 Molecular conformations not only govern the extent of electronic conjugation, but are also interdependent with molecular packing,15,16 which controls intermolecular charge transport in polymer- and oligomer-based thin films.17 While bulk molecular packing is of central importance in defining the electronic transport characteristics,18 the latter are also often strongly affected by the properties of molecular interfaces with other materials used in electronic devices, where molecular structures can be quite different from those of the bulk.19,20 Examples of such interfaces include the molecule-dielectric interfaces, found in field-induced conduction channels in transistors,21–23 and molecule-electrode interfaces.24–26 Furthermore, the thin film morphology can be strongly impacted by the structure of the molecular layer at the interface.27 The nature of the interface has a profound impact on the molecular morphology and self-organization, which are defined by the competition between the intermolecular and molecule-surface interactions. For this reason, molecular structures at interfaces with solids and other molecular layers have been a subject of numerous studies, with techniques based on scanning tunneling microscopy (STM) being particularly powerful in the absence of long-range order due to their ability to resolve local molecular structures. Many STM studies have focused on molecular self-assembly in the regime of weak molecule-surface interactions simulating molecule-dielectric interfaces, with molecules often deposited on highly-oriented pyrolytic graphite surfaces.28–34 The resulting structures are typically dominated by the intermolecular interactions between the ligands, attached to thiophene backbones in order to enhance solubility and facilitate processing.Several STM studies have addressed the regime of stronger molecule-surface interactions with molecules deposited on metal surfaces simulating molecule-electrode interfaces.30,35–42 A common observation of these studies was the existence of conformational polymorphisms of both polythiophenes40,41 and oligothiophenes,36–39 with the overall degree of disorder being higher than that found for more weakly interacting surfaces.30,40 Understanding the degree of electronic disorder associated with such conformational polymorphisms is important in view of the potentially significant impact of molecular conformation on the electronic structure. Theoretical and optical spectroscopic investigations of organic donor–acceptor molecules incorporating thiophene donors revealed that unoccupied frontier electronic levels were largely unaffected by cis–trans conformational isomerization, finding variations in LUMO energies due to rotational disorder of less than 90 meV.43,44 STM offers the capability to probe the impact of conformational polymorphism on molecular electronic structure directly via scanning tunneling spectroscopy (STS), which has been used to investigate oligothiophene electronic structures on both strongly and weakly interacting surfaces.29,30,45–49 However, the role of conformational effects in defining the molecular electronic structure has so far only been investigated in very short oligothiophenes (incorporating only four thiophene rings), where few electronic states are accessible to STS due to their relatively high energies.50,51
Here we report a conformation-resolved STM/STS study of alkyl-substituted oligothiophenes adsorbed on the Au(111) surface. To obtain a more complete physical picture of the oligothiophene properties on Au(111), we investigated two types of oligothiophenes with similar structures: molecules incorporating eight thiophene rings and four alkyl ligands (we will refer to these molecules as 8T in the following), as well as molecules incorporating seven thiophene rings and three alkyl ligands (7T in the following). Both types of molecules are sufficiently long to be considered as finite-length models for probing conformational effects in application-relevant alkyl-substituted thiophene-based polymers and oligomers. The Au(111) surface serves as a model of Au metal electrodes often used in proof-of-principle organic semiconductor devices. We obtained STS maps of oligothiophene molecules to identify the nature of molecular orbitals, and found that all molecules displayed particle-in-a-box-like progressions of electronic states. Further, we found that different oligothiophene conformations, despite considerable structural differences, result in nearly indistinguishable molecular electronic structures. We use density functional theory (DFT) calculations to show that the electronic structures of oligothiophenes of different torsional conformations (in the gas phase) are quite similar. The lack of conformational sensitivity in our experimental electronic structure data thus suggests that the Au–oligothiophene interaction is relatively insensitive to the specific molecular torsional conformation.
Experimental methods
STM and STS measurements were performed at ∼20 K using a home-built ultra-high vacuum (UHV) cryogenic (closed-cycle cryostat-based) STM system incorporating a STM scanner from RHK Technology.52 Atomically clean Au(111) on mica was prepared in UHV by multiple cycles of Ne-ion bombardment followed by ∼300 °C anneals. 8T and 7T molecules were prepared by Briseno et al.53 The composition was verified with nanoelectrospray ionization mass spectrometry (Fig. S1, ESI†). Sub-monolayers of oligothiophene molecules were deposited on the Au surface via in situ sublimation with the sample held at room temperature and vacuum pressure not exceeding 10−10 Torr. STS spectra were measured in constant-height mode using the lock-in technique, with a modulation frequency of 570 Hz and a bias modulation of 50 mV. This allowed for the direct measurement of the local differential conductance (dI/dV) of the sample, which serves as a measure to the local density of electronic states.54 DFT computations were performed with Gaussian 0955 using B3LYP/6-31G*,56,57 and analyzed with Multiwfn.58Results and discussion
Molecular adsorption configurations from STM imaging
After deposition on the Au(111) surface, STM imaging revealed that 8T and 7T molecules self-assembled into chain-like aggregates comprised of varying numbers of molecules (Fig. 1a, Fig. S2a and S3a, ESI†). High-resolution STM images show that the molecules bind to each other with their alkyl ligands, forming interdigitating patterns (Fig. 1b and Fig. S2b, ESI†).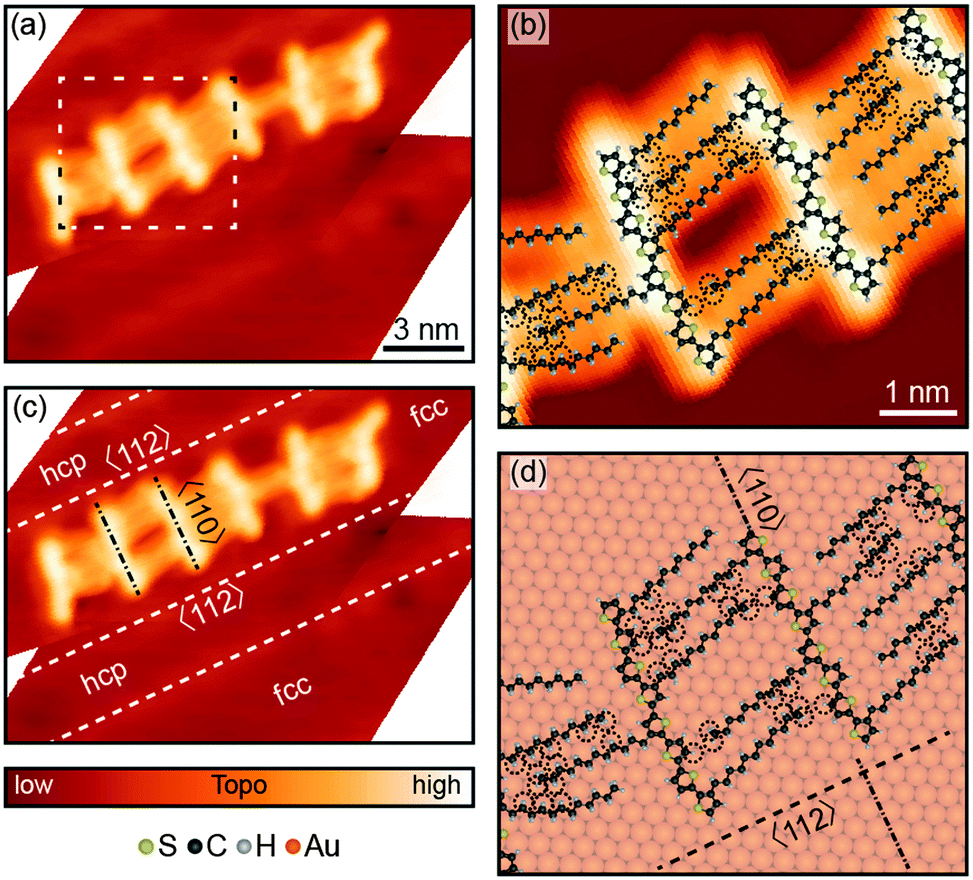 | ||
| Fig. 1 Adsorption of 8T molecules on Au(111). (a) STM image [set point 100 mV, 5 pA] of an aggregate of oligothiophene molecules absorbed on the Au(111) surface. (b) Close-up STM topography of the region confined by the dotted rectangle in (a). Atomic models of 8T molecules are overlaid on the STM image. The atomic models show that molecules are attached to each other via alkyl substituents. The thiophene rings comprising the DDQT backbones are nearly flat on the Au(111) surface, as determined by STM topographies. (c) STM image from (a) with indicated molecular orientations and Au(111) crystallographic directions and highlighted surface-reconstruction ridges. The oligothiophene backbones of 8T molecules are aligned along the 〈110〉 directions of the Au(111) surface, perpendicular to the straight sections of surface-reconstruction ridges which run parallel to the 〈112〉 directions. (d) Model of 8T molecules from (b) matched to the Au(111) surface lattice. Au(111) crystallographic directions are indicated. Dashed circles indicate the van der Waals radii of the hydrogen atoms. | ||
Adsorption of oligothiophenes was highly correlated with the structural features of the Au(111) surface associated with the Au(111) 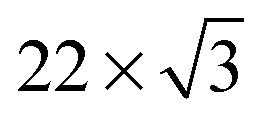 surface reconstruction, which results in the formation of regions with fcc (face-centered cubic) and hcp (hexagonal close-packed) surface structures separated by reconstruction “ridges” visible in STM images (dashed lines in Fig. 1c, Fig. S2c and S3b, ESI†). The molecular aggregates primarily formed in the fcc regions of the Au(111) surface, and were almost entirely absent from the hcp regions (Fig. 1c, Fig. S2c and S3b, ESI†), consistent with results for shorter oligo-thiophenes.45,50 This preferential adsorption has been attributed to different reactivities of the fcc and hcp regions, a result of varied coordination of the surface Au atoms in the two regions.50 STM images of 8T and 7T backbones also showed molecular profiles of ∼2 Å with typical variations of <0.1 Å, consistent with flat molecular backbones.49
surface reconstruction, which results in the formation of regions with fcc (face-centered cubic) and hcp (hexagonal close-packed) surface structures separated by reconstruction “ridges” visible in STM images (dashed lines in Fig. 1c, Fig. S2c and S3b, ESI†). The molecular aggregates primarily formed in the fcc regions of the Au(111) surface, and were almost entirely absent from the hcp regions (Fig. 1c, Fig. S2c and S3b, ESI†), consistent with results for shorter oligo-thiophenes.45,50 This preferential adsorption has been attributed to different reactivities of the fcc and hcp regions, a result of varied coordination of the surface Au atoms in the two regions.50 STM images of 8T and 7T backbones also showed molecular profiles of ∼2 Å with typical variations of <0.1 Å, consistent with flat molecular backbones.49
The molecular aggregates showed reproducible registry with the Au(111) surface lattice, as can be observed by comparing molecular orientations with the ridge-like surface reconstruction features (Fig. 1c for 8T, Fig. S2c and S3b for 7T, ESI†). In particular, in the straight sections of fcc regions, where the Au crystal structure is more regular, the oligothiophene backbones were preferentially oriented along the 〈110〉 directions of Au(111), orthogonal to the growth-directions of molecular aggregates corresponding approximately to the 〈112〉 directions (Fig. 1c, Fig. S2c and S3b, ESI†). This backbone orientation is analogous to that observed for shorter four-thiophene oligomers (4T in the following) on Au(111),50 which is a direct consequence of the similarities in intermolecular and molecule-surface interactions for both systems. In particular, the preferred orientations of 4T molecules along the 〈110〉 directions have been attributed to the charge-transfer interaction involving S atoms of the thiophene units comprising 4T molecules and the Au surface. For individual thiophene units, this interaction results in preferential adsorption on Au top sites,59 with a local energy minimum of 40 meV.60 The tendency of 4T oligothiophenes to align along the 〈110〉 directions on Au(111) has been explained by the fact that this orientation leads to the best matching of all four S-atoms of 4T to the top sites of the Au(111) surface lattice.50 In accordance with this picture, locations of S-atoms in 8T and 7T molecules show similar patterns (Fig. 1d and Fig. S2d, ESI†), even though some deviations from perfect top-site placement are evident. The existence of these deviations is not unexpected, since the longer molecular structures of 7T and 8T molecules are more difficult to match to the Au(111) lattice. For example, while in 4T molecules the distance between the S-atoms of the outer thiophene rings (1.19 nm) was closely matched to four Au atoms along the 〈110〉 direction (1.15 nm), for 8T molecules the distance between the S-atoms of the outer thiophene rings (∼2.8 nm) is a poorer match to Au(111) surface lattice sites, with the best match corresponding to ten lattice constants (2.89 nm). The lack of commensurability between the molecular structures and the Au(111) surface may also be the reason for the commonly observed deviations from linearity of 7T and 8T backbones, in contrast to 4T molecules, which appeared straight in all STM images.50
Another important factor that likely impacted the shapes of 7T and 8T backbones is the intermolecular interactions originating from the alkyl ligands. These include both alkyl–alkyl interactions as well as interactions involving terminal methyl groups of alkyl ligands and thiophene units of neighboring molecules. For example, the strength of the alkyl–alkyl interaction can be expected to be substantial given that for a pair of dodecane molecules the interaction energy is estimated to be 0.4–0.5 eV.61 This interaction tends to affect individual molecules differently due to the widely varied inter-digitation patterns of alkyl substituents connecting neighboring molecules (Fig. 1b and Fig. S2b, ESI†), and may contribute to the observed deviations from linearity for the oligothiophene backbones. Indeed, this argument is supported by the fact that despite the variations in the oligothiophene backbone shapes, the patterns formed by alkyl chains show highly reproducible angles, especially when close alkyl–alkyl contacts are formed (Fig. 1b and Fig. S2b, ESI†). The observed orientations of tightly packed alkyl ligands on the Au(111) lattice form angles of ∼12° with respect to the 〈110〉 direction (Fig. 1d and Fig. S2d, ESI†). This ligand orientation is intermediate between those of the gas-phase molecules (ligand nearly orthogonal to the backbones), and alkanes self-assembled on Au(111), which typically align themselves approximately along the 〈110〉 directions62 (with theoretical calculations giving an angle of ∼7°).63 This observation suggests that orientations of the alkyl ligands are also affected by the local bonding orientation and placement of the oligothiophene backbones.
The specific interdigitating structures formed by alkyl ligands of neighboring molecules depend on the particular torsional conformations adopted by the corresponding thiophene backbones. Oligothiophene aggregates studied in our work contained a variety of molecules adopting different torsional conformations (corresponding to the different possible mutual orientations of thiophene units along the oligothiophene backbone, as can be seen in Fig. 1b and Fig. S2b, ESI†), which is one of the reasons for varied intermolecular binding interactions. In addition to controlling the structure of oligothiophene aggregates, torsional conformations can be expected to control the electronic structures of the corresponding molecules, since changing the mutual orientation of individual thiophene units can affect the energies of molecular orbitals. From the point of view of electronic applications based on oligothiophene materials, it is then important to understand the extent of electronic structure variations caused by the existence of different torsional conformers, as well as the physical driving forces responsible for the diversity of torsional conformations in oligothiophene thin films.
The observed tendency of 8T molecules to form relatively ordered aggregates suggests that after deposition, these molecules have sufficient energy (at room temperature) to be able to freely migrate on the surface on the Au(111) surface. Moreover, the well-ordered assembly of interconnecting alkyl ligands suggests that molecular torsional transformations readily occur in these conditions. This is illustrated, for example, by the fact that alkyl ligands unattached to neighbouring molecules were almost never observed in STM imaging, with one particular manifestation of this being the fact that molecules located at the ends of the oligothiophene aggregates typically assumed a conformation with all alkyl ligands facing the rest of the aggregate, whenever possible (a rare exception to this is the left molecule in Fig. S2a (ESI†), where one of the ligands is facing away from the molecular aggregate due to interference from a ligand of a neighbouring molecule).
Scanning tunneling spectroscopy
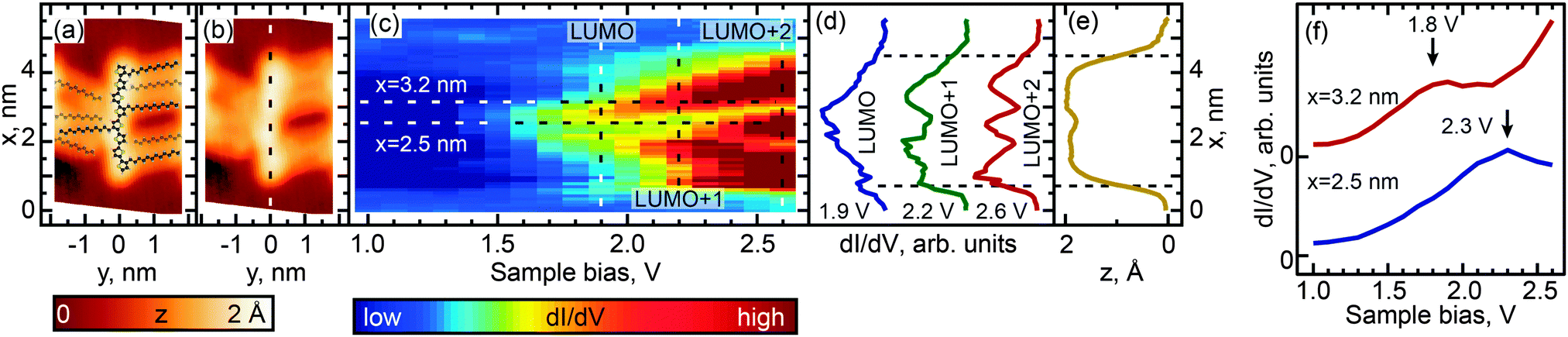
We investigated the effect of length and conformation on oligothiophene electronic structure by using scanning tunneling spectroscopy (STS) to measure the local electronic structure of 8T and 7T molecules adsorbed on the Au(111) surface. Specifically, these measurements were carried out by recording the differential conductance (derivative of the tunneling current, dI/dV), which is representative of the local electronic density of states (DOS). By recording the differential conductance as a function of the applied bias voltage (which serves as the energy scale), we obtain energy-dependent DOS spectra (see Experimental methods for further details).54 In total, 55 straight oligothiophene molecules were studied using this approach (see Fig. S4, ESI†), and STS maps were obtained for 31 molecules, as described below.Among the different possible conformations of 8T molecules, we focus on three representative conformations: CCC (orientation of ligands corresponding to a cis–cis–cis combination, Fig. 2a), TTT (trans–trans–trans ligand orientations, Fig. 3a), and CTT (cis–trans–trans ligand orientations, Fig. 4a). While we are unable to discern the orientations of thiophenes lacking alkyl chains, it is likely that, since the oligothiophene backbones are straight, neighboring thiophenes are preferentially oriented ∼180° relative to each other, as oligothiophene backbones containing neighboring thiophenes oriented ∼0° relative to each other have a tendency to bend due to the steric hindrance between neighboring thiophenes.30 By recording progressions of STS spectra along the oligothiophene backbones (corresponding spatial paths shown in Fig. 2b, 3b and 4b) we obtain DOS maps showing the spatial landscape of molecular electronic states (Fig. 2c, 3c and 4c). Here we focus on the unoccupied states because the occupied molecular orbitals were not clearly distinguishable in the DOS spectra due to the structured background originating from the d-bands of the Au(111)surface. All three DOS patterns (Fig. 2c, 3c and 4c) show a very high degree of similarity. Each map contains features attributable to a progression of three orbitals—LUMO, LUMO+1 and LUMO+2—showing the characteristic particle-in-a-box spatial distributions: the LUMO+1 and LUMO+2 orbitals show one and two spatial nodes respectively, while the LUMOs are relatively featureless along the molecular backbones (Fig. 2d, 3d and 4d). The assignment of LUMO, LUMO+1 and LUMO+2 orbitals in Fig. 2–4 is supported by DFT calculations carried out for 8T molecules in gas phase (Fig. 5–7). Indeed, for CCC, TTT and CTT conformations, DOS patterns similar to those of Fig. 2c, 3c and 4c are found in calculations (Fig. 5a, 6a and 7a). For each molecular conformation, the patterns are formed by three states, LUMO, LUMO+1 and LUMO+2 with particle-in-a-box-like spatial structures (Fig. 5b, 6b and 7b) similar to those in Fig. 2d, 3d and 4d. The particle-in-a-box-nature of these states is evident from the corresponding calculated wavefunctions showing the characteristic nodal patterns (Fig. 5c, 6c and 7c).
 | ||
| Fig. 2 Spatial (STS) DOS mapping across a CCC conformer of 8T molecules. (a) STM image with an overlaid atomic model of the CCC-8T molecule. (b) STM image from (a) showing the path of mapping (dashed line). (c) DOS as a function of the bias voltage and position x along the path shown in (b). (d) LUMO, LUMO+1 and LUMO+2 DOS along the path shown in (b), obtained at voltages corresponding to the vertical dashed lines in (c). These voltages were chosen to maximize the contributions of the corresponding individual orbitals. Curves are shifted and normalized for clarity. (e) Backbone profile (z height vs. x coordinate) along the dashed line from (b). (f) Individual STS spectra from (c) measured at x = 2.1 and 2.7 nm as indicated by horizontal lines in (c). Spectra are shifted for clarity. The LUMO state manifests itself as a peak at 1.85 V in the spectrum measured at x = 2.7 nm, while LUMO+1 is observed as a peak at 2.3 V in the spectrum measured at x = 2.1 nm. Only a shoulder of the LUMO+2 states is observed at 2.6 V (x = 2.7 nm). | ||
 | ||
| Fig. 3 Same as Fig. 2 for a TTT conformer of 8T molecules. The molecule is situated in the center of the molecular aggregate, with alkyl chains on both sides (see Fig. S1d, ESI†). | ||
 | ||
| Fig. 4 Same as Fig. 2 for a CTT conformer of 8T molecules. | ||
 | ||
| Fig. 5 Calculated electronic DOS for a CCC conformer of 8T molecules. (a) DOS (compare to Fig. 2c) as a function of the bias voltage and position x along the path similar to that shown in Fig. 2b. (b) LUMO, LUMO+1 and LUMO+2 DOS (compare to Fig. 2d) along the same path as in (a), obtained at voltages corresponding to the vertical dashed lines in (a). These voltages were chosen to maximize the contributions of the corresponding individual orbitals. Curves are shifted and normalized for clarity. (c) Three-dimensional representations of LUMO, LUMO+1 and LUMO+2 showing nodal patterns (along the molecular backbone) characteristic of the particle-in-a-box nature of these states (no nodes for LUMO, one node for LUMO+1 and two nodes for LUMO+2). (d) Individual DOS spectra from (a) measured at spatial locations indicated by horizontal lines in (a). Spectra are shifted for clarity. The LUMO state manifests itself as a peak at 1.86 V (top curve), while LUMO+1 is observed as a peak at 2.32 V (bottom curve). Electronic structure calculations were performed with density functional theory (DFT) calculations using B3LYP/6-31G*. | ||
 | ||
| Fig. 6 Same as Fig. 5 for a TTT conformer of 8T molecules. | ||
 | ||
| Fig. 7 Same as Fig. 6 for a CTT conformer of 8T molecules. | ||
The LUMO+1 and LUMO+2 orbitals appear more delocalized relative to the LUMO orbitals (compare DOS profiles in Fig. 2d, 3d and 4d to topography profiles in Fig. 2e, 3e and 4e), which is a consequence of several factors. First, due to the longer spatial wavelength of the lowest-energy particle-in-a-box state, the probability density of this state changes on a longer scale and therefore is suppressed over a longer spatial range at the ends of the oligothiophene backbone. In addition, higher-energy states are generally more spatially extended due to the lower effective tunneling barrier. Finally, the fact that the tunneling current (identical for all measured spectra) is composed of contributions from each unoccupied state means that the LUMO contribution is further suppressed via topographic effects when the tip is positioned near the ends of the oligothiophene backbone, where higher-energy states have larger contributions to the tunneling current (the feedback loop was opened at a bias of 2.6 V for all STS maps, so that the tip height was therefore determined by contributions from the first three unoccupied molecular orbitals). A similar relationship between the localization scales of the LUMO, LUMO+1 and LUMO+2 orbitals is reproduced in our theoretical calculations (Fig. 5b, 6b and 7b).
While the unoccupied molecular orbital energies are the same within experimental error for all conformations, there are asymmetries observed in DOS intensity across the molecular backbones (Fig. 2d, 3d and 4d). DFT calculations for the CCC and TTT conformers do not exhibit asymmetry in DOS intensity (Fig. 5b and 6b). It is likely that the observed asymmetric experimental DOS intensity (Fig. 2d and 3d) is due to factors other than conformational variation for these conformers. DFT results for the CCT conformer, however, show uneven LUMO, LUMO+1 and LUMO+2 intensity across the molecular backbone (Fig. 7b), suggesting that asymmetric torsional conformation variations can lead to asymmetry in DOS intensity.
Despite the completely different torsional conformations of the molecules in Fig. 2–4, their orbital energies are essentially the same (1.8 V for LUMO and 2.3 V for LUMO+1, as determined from Fig. 2f, 3f, and 4f) within the experimental error (∼50 mV). These observations are mirrored by the calculated state energies (obtained from Fig. 5d, 6d and 7d), which are very similar for all torsional conformations, as summarized in Fig. 8. Because our calculations were carried out for molecules in the gas phase, the similarity of the experimentally determined and calculated orbital energies suggests that the oligothiophene interaction with the Au(111) surface is relatively insensitive to the molecular conformation, with any differences between conformers being less than 50 mV. To further test this conclusion, we studied 7T molecules with analogous conformations to those of 8T (Fig. S5–S7, ESI†). Because of their different structure and reduced symmetry, the interaction of 7T molecules with Au(111) may be different from that of 8T molecules, which could potentially lead to a different magnitude of electronic structure variations for different 7T conformers. Similarly to 8T molecules, we identify three representative conformations: CC (orientation of ligands corresponding to a cis–cis combination, Fig. S5a, ESI†), TT (trans–trans ligand orientations, Fig. S6a, ESI†), and CT (cis–trans ligand orientations following an unsubstituted section of the backbone, Fig. S7a, ESI†). Analogously to the case of 8T molecules, DOS maps of different conformers of 7T oligothiophenes (Fig. S5c, S6c and S7c, ESI†) show patterns attributable to progressions of three particle-in-a-box-like orbitals, LUMO, LUMO+1 and LUMO+2, consistent with theoretical calculations (Fig. S8–S10, ESI†). As with the 8T molecules, all studied conformers of the 7T molecules showed, within the 50 mV experimental error, nearly the same orbital energies (1.9 V for LUMO and 2.5 V for LUMO+1), which are close to the theoretically predicted values summarized in Fig. 8.
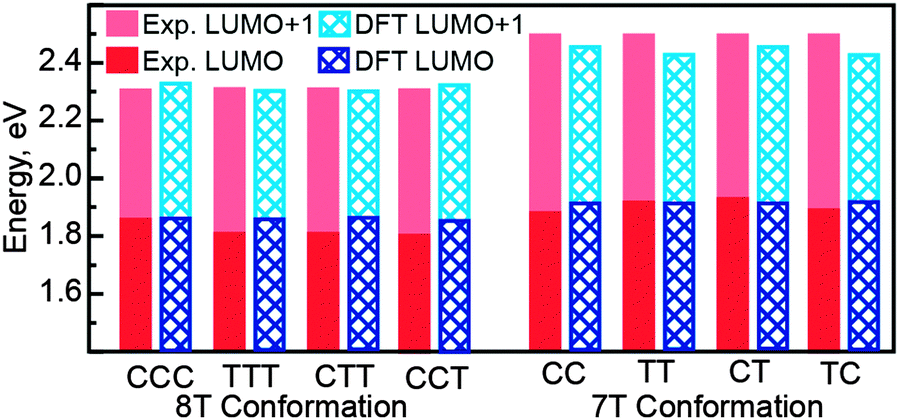 | ||
| Fig. 8 Experimental (red solid bars) and theoretical (blue textured bars) LUMO and LUMO+1 energies for different torsional conformers of alkyl-substituted 8T and 7T oligothiophenes. Electronic orbitals for 7T molecules are upshifted as compared to the 8T molecules due to their shorter thiophene backbone lengths. | ||
Another example of the lack of sensitivity of the oligothiophene electronic structure to molecular conformation is provided by our recent work on 4T molecules, where similar orbital energies were found for both cis and trans conformations, and the variation in molecular orbital energies were found to be attributable to the local variations in the Au(111) surface reactivity caused by the  surface reconstruction.50 In the present study, similar orbital energy variations with standard deviations of 40–60 mV (depending on the molecular structure and conformation), comparable to the experimental error (∼50 mV) were found. Thus, because the oligothiophene unoccupied molecular orbital energies were found to be insensitive to the torsional conformation of molecules with a wide range of structures, it is likely that conformational differences in the oligothiophene interaction with the Au(111) surface are on the scale of a few tens of meV.
surface reconstruction.50 In the present study, similar orbital energy variations with standard deviations of 40–60 mV (depending on the molecular structure and conformation), comparable to the experimental error (∼50 mV) were found. Thus, because the oligothiophene unoccupied molecular orbital energies were found to be insensitive to the torsional conformation of molecules with a wide range of structures, it is likely that conformational differences in the oligothiophene interaction with the Au(111) surface are on the scale of a few tens of meV.
Conclusions
Our results, obtained using STS spectroscopy, demonstrate that different torsional conformers of oligothiophene molecules with different lengths and (alkyl) substitutional patterns show molecular orbital energies nearly independent of the molecular conformations. While these results are consistent with theoretical simulations for oligothiophenes in the gas phase, the experimental demonstration of similar unoccupied molecular orbital energies (within 50 meV) across multiple planar molecular conformation, as demonstrated in our work, suggests that the oligothiophene interaction with the Au(111) surface on molecular energy levels in general is relatively insensitive to the molecular conformation. This is an important result for applications utilizing charge transport through thin films based on longer substituted oligothiophenes and poly-thiophenes, where significant local variations in conformational structure are often found.30,32,40,41 The insensitivity of the oligothiophene molecular electronic structure to torsional conformation presented here may mean a low degree of electronic disorder, and consequently, lower probability of charge carrier trapping within molecular backbones in poly- and oligo-thiophene materials. Indeed, molecular orbital energy variations found in our STS measurements are comparable to the energetic disorder (20–40 meV) characteristic of high-mobility (∼0.1 cm2 V−1 s−1) polythiophene samples.64 Thus, while the molecular conformational and packing structures do affect the trap density in bulk poly- and oligo-thiophene materials,65 the present work suggests that in molecular films with predominating planar molecular conformations, the electronic disorder may be reduced.Acknowledgements
The STM instrument used in this work was constructed with support from the National Science Foundation under Grant DMR-0960211. Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund for support of this research (Grant 52732-DNI6). B. N. T. and G. V. N. gratefully acknowledge support from the Center for Sustainable Materials Chemistry through the NSF CCI Grant CHE-1102637. L. Z. and A. L. B. gratefully acknowledge the Center for Hierarchical Manufacturing (CMMI-0531171). S. C. B. M. acknowledges support by the Deutsche Forschungsgemeinschaft (DFG) within the Cluster of Excellence “cfaed”.References
- L. Zhang, N. S. Colella, B. P. Cherniawski, S. C. B. Mannsfeld and A. L. Briseno, ACS Appl. Mater. Interfaces, 2014, 6, 5327–5343 CAS.
- L. Xu, L. Yang and S. Lei, Nanoscale, 2012, 4, 4399–4415 RSC.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS PubMed.
- A. R. Murphy, J. M. J. Fréchet, P. Chang, J. Lee and V. Subramanian, J. Am. Chem. Soc., 2004, 126, 1596–1597 CrossRef CAS PubMed.
- C. Videlot-Ackermann, J. Ackermann, H. Brisset, K. Kawamura, N. Yoshimoto, P. Raynal, A. El Kassmi and F. Fages, J. Am. Chem. Soc., 2005, 127, 16346–16347 CrossRef CAS PubMed.
- M. Mazzeo, V. Vitale, F. Della Sala, M. Anni, G. Barbarella, L. Favaretto, G. Sotgiu, R. Cingolani and G. Gigli, Adv. Mater., 2005, 17, 34–39 CrossRef CAS.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. D. Santos, J. L. Bredas, M. Logdlund and W. R. Salaneck, Nature, 1999, 397, 121–128 CrossRef CAS.
- L. Tong, C. Li, F. E. Chen, H. Bai, L. Zhao and G. Q. Shi, J. Phys. Chem. C, 2009, 113, 7411–7415 CAS.
- R. Fitzner, E. Mena-Osteritz, A. Mishra, G. Schulz, E. Reinold, M. Weil, C. Körner, H. Ziehlke, C. Elschner, K. Leo, M. Riede, M. Pfeiffer, C. Uhrich and P. Bäuerle, J. Am. Chem. Soc., 2012, 134, 11064–11067 CrossRef CAS PubMed.
- L. Zhang, F. Liu, Y. Diao, H. S. Marsh, N. S. Colella, A. Jayaraman, T. P. Russell, S. C. B. Mannsfeld and A. L. Briseno, J. Am. Chem. Soc., 2014, 136, 18120–18130 CrossRef CAS PubMed.
- K. Cnops, B. P. Rand, D. Cheyns, B. Verreet, M. A. Empl and P. Heremans, Nat. Commun., 2014, 5, 3406 Search PubMed.
- C. Schubert, J. T. Margraf, T. Clark and D. M. Guldi, Chem. Soc. Rev., 2015, 44, 988–998 RSC.
- B. Q. Xu, X. L. Li, X. Y. Xiao, H. Sakaguchi and N. J. Tao, Nano Lett., 2005, 5, 1491–1495 CrossRef CAS PubMed.
- B. Capozzi, E. J. Dell, T. C. Berkelbach, D. R. Reichman, L. Venkataraman and L. M. Campos, J. Am. Chem. Soc., 2014, 136, 10486–10492 CrossRef CAS PubMed.
- N. Koch, J. Phys.: Condens. Matter, 2008, 20, 184008 CrossRef.
- A. Höfer, K. Duncker, M. Kiel, S. Wedekind and W. Widdra, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 075414 CrossRef.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS.
- Y. Wu, P. Liu, B. S. Ong, T. Srikumar, N. Zhao, G. Botton and S. Zhu, Appl. Phys. Lett., 2005, 86, 142102 CrossRef.
- S. Duhm, G. Heimel, I. Salzmann, H. Glowatzki, R. L. Johnson, A. Vollmer, J. P. Rabe and N. Koch, Nat. Mater., 2008, 7, 326–332 CrossRef CAS PubMed.
- R. Joseph Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 222–228 CrossRef CAS.
- A. Dodabalapur, L. Torsi and H. E. Katz, Science, 1995, 268, 270–271 CAS.
- J. Huang, J. Sun and H. E. Katz, Adv. Mater., 2008, 20, 2567 CrossRef CAS.
- S. Fabiano, C. Musumeci, Z. Chen, A. Scandurra, H. Wang, Y.-L. Loo, A. Facchetti and B. Pignataro, Adv. Mater., 2012, 24, 951 CrossRef CAS PubMed.
- G. R. Dholakia, M. Meyyappan, A. Facchetti and T. J. Marks, Nano Lett., 2006, 6, 2447–2455 CrossRef CAS PubMed.
- J. E. Lyon, A. J. Cascio, M. M. Beerbom, R. Schlaf, Y. Zhu and S. A. Jenekhe, Appl. Phys. Lett., 2006, 88, 222109 CrossRef.
- H. Ishii, K. Sugiyama, E. Ito and K. Seki, Adv. Mater., 1999, 11, 605 CrossRef CAS.
- S. Wang, A. Kiersnowski, W. Pisula and K. Müllen, J. Am. Chem. Soc., 2012, 134, 4015–4018 CrossRef CAS PubMed.
- E. Mena-Osteritz, A. Meyer, B. M. W. Langeveld-Voss, R. A. J. Janssen, E. W. Meijer and P. Bäuerle, Angew. Chem., Int. Ed., 2000, 39, 2679–2684 CrossRef.
- Z.-Y. Yang, H.-M. Zhang, G.-B. Pan and L.-J. Wan, ACS Nano, 2008, 2, 743–749 CrossRef CAS PubMed.
- Z.-Y. Yang, H.-M. Zhang, C.-J. Yan, S.-S. Li, H.-J. Yan, W.-G. Song and L.-J. Wan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3707–3712 CrossRef CAS PubMed.
- E. Mena-Osteritz, M. Urdanpilleta, E. El-Hosseiny, B. Koslowski, P. Ziemann and P. Bauerle, Beilstein J. Nanotechnol., 2011, 2, 802–808 CrossRef CAS PubMed.
- X. J. Ma, Y. Guo, T. Wang and Z. H. Su, J. Chem. Phys., 2013, 139, 014701 CrossRef PubMed.
- T. Jaroch, M. Knor, R. Nowakowski, M. Zagorska and A. Pron, Phys. Chem. Chem. Phys., 2008, 10, 6182–6189 RSC.
- B. Grévin, P. Rannou, R. Payerne, A. Pron and J. P. Travers, J. Chem. Phys., 2003, 118, 7097–7102 CrossRef.
- L. Wang, B. J. V. Tongol, S.-L. Yau, T. Otsubo and K. Itaya, Langmuir, 2010, 26, 7148–7152 CrossRef CAS PubMed.
- F. Nishiyama, K. Ogawa, S. Tanaka and T. Yokoyama, J. Phys. Chem. B, 2008, 112, 5272–5275 CrossRef CAS PubMed.
- K. Duncker, M. Kiel, A. Hoefer and W. Widdra, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155423 CrossRef.
- T. Yokoyama, Y. Kogure, M. Kawasaki, S. Tanaka and K. Aoshima, J. Phys. Chem. C, 2013, 117, 18484–18487 CAS.
- B. J. V. Tongol, L. Wang, S.-L. Yau, T. Otsubo and K. Itaya, Langmuir, 2010, 26, 982–989 CrossRef CAS PubMed.
- Y. F. Liu, K. Krug and Y. L. Lee, Nanoscale, 2013, 5, 7936–7941 RSC.
- S. Förster, E. Kohl, M. Ivanov, J. Gross, W. Widdra and W. Janke, J. Chem. Phys., 2014, 141, 164701 CrossRef PubMed.
- D. Wang, L.-J. Wan and C.-L. Bai, Mater. Sci. Eng., R, 2010, 70, 169–187 CrossRef.
- L. V. Brownell, K. A. Robins, Y. Jeong, Y. Lee and D. C. Lee, J. Phys. Chem. C, 2013, 117, 25236–25247 CAS.
- B. He, A. B. Pun, D. Zherebetskyy, Y. Liu, F. Liu, L. M. Klivansky, A. M. McGough, B. A. Zhang, K. Lo, T. P. Russell and L. W. Wang, J. Am. Chem. Soc., 2014, 136, 15093–15101 CrossRef CAS PubMed.
- B. Koslowski, A. Tschetschetkin, N. Maurer, E. Mena-Osteritz, P. Bauerle and P. Ziemann, Beilstein J. Nanotechnol., 2011, 2, 561–568 CrossRef CAS PubMed.
- G. Reecht, H. Bulou, F. Scheurer, V. Speisser, B. Carrière, F. Mathevet and G. Schull, Phys. Rev. Lett., 2013, 110, 056802 CrossRef PubMed.
- J. Repp, P. Liljeroth and G. Meyer, Nat. Phys., 2010, 6, 975–979 CrossRef CAS.
- F. Jäckel, U. G. E. Perera, V. Iancu, K. F. Braun, N. Koch, J. P. Rabe and S. W. Hla, Phys. Rev. Lett., 2008, 100, 126102 CrossRef PubMed.
- E. Varene, Y. Pennec and P. Tegeder, Chem. Phys. Lett., 2011, 515, 141–145 CrossRef CAS.
- B. N. Taber, D. A. Kislitsyn, C. F. Gervasi, S. C. B. Mannsfeld, L. Zhang, A. L. Briseno and G. V. Nazin, ACS Appl. Mater. Interfaces, 2015, 19732–19742 Search PubMed.
- D. A. Kislitsyn, B. N. Taber, C. F. Gervasi, S. C. B. Mannsfeld, L. Zhang, A. L. Briseno and G. V. Nazin, J. Phys. Chem. C, 2015, 26959–26967 CAS.
- J. D. Hackley, D. A. Kislitsyn, D. K. Beaman, S. Ulrich and G. V. Nazin, Rev. Sci. Instrum., 2014, 85, 103704 CrossRef PubMed.
- L. Zhang, N. S. Colella, F. Liu, S. Trahan, J. K. Baral, H. H. Winter, S. C. B. Mannsfeld and A. L. Briseno, J. Am. Chem. Soc., 2012, 135, 844–854 CrossRef PubMed.
- C. J. Chen, Introduction to Scanning Tunneling Microscopy, Oxford University Press, New York, 2nd edn, 2008 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision C.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. Zhou, Y. X. Yang, P. Liu, N. Camillone, III and M. G. White, J. Phys. Chem. C, 2010, 114, 13670–13677 CAS.
- K. Tonigold and A. Gross, J. Chem. Phys., 2010, 132, 224701 CrossRef PubMed.
- A. Goursot, T. Mineva, R. Kevorkyants and D. Talbi, J. Chem. Theory Comput., 2007, 3, 755–763 CrossRef CAS PubMed.
- H. M. Zhang, Z. X. Xie, B. W. Mao and X. Xu, Chem. – Eur. J., 2004, 10, 1415–1422 CrossRef CAS PubMed.
- R. J. Baxter, G. Teobaldi and F. Zerbetto, Langmuir, 2003, 19, 7335–7340 CrossRef CAS.
- H. Sirringhaus, Adv. Mater., 2005, 17, 2411–2425 CrossRef CAS.
- A. Salleo, T. W. Chen, A. R. Völkel, Y. Wu, P. Liu, B. S. Ong and R. A. Street, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 115311 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp07092a |
| This journal is © the Owner Societies 2016 |