
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-pairing of 1-methylthymine mediated by two and three Ag(I) ions: a gas phase study using infrared dissociation spectroscopy and density functional theory†
Yevgeniy
Nosenko
,
Christoph
Riehn
and
Gereon
Niedner-Schatteburg
Fachbereich Chemie and Forschungszentrum OPTIMAS, Technische Universität Kaiserslautern, Erwin-Schrödinger-Str. 52, Kaiserslautern 67663, Germany. E-mail: nosenko@chemie.uni-kl.de; riehn@chemie.uni-kl.de; gns@chemie.uni-kl.de
First published on 16th February 2016
Abstract
Metal base pairs of AgI cations and 1-methylthymine (1MT) or deprotonated 1-methylthymine (1MT-H) are produced and analyzed by electrospray ionization mass spectrometry (ESI-MS). Mass-selected ions of type [Ag2(1MT)(1MT-H)]+ and [Ag3(1MT-H)2]+ are interrogated by infrared multiple-photon dissociation (IRMPD) in an ion trap in the range of 1200–3700 cm−1. Supporting spectroscopic data were obtained from the investigation of the analogous 2′-deoxy-thymidine complexes which exhibit advantageously high fragment yields. By comparison with calculated linear IR spectra (obtained by density functional theory, DFT) we assign the structures and the possible isomeric forms of these metal base pairs and their dependence on the number of mediating AgI ions. Based on the observed Ag+/1MT complexes and related polarizable continuum model DFT calculations we describe the probable formation pathways in aqueous solution. The present findings pave the way for subsequent UV investigations of the multi-metal mediated base pairs.
Introduction
DNA molecules have been recognized for a while as promising templates for atomic scale metal structures due to their ability to bind transition metal ions between their oligonucleotide strands and because of the advanced level of their synthetic structural control.1 The selectivity and binding affinity of natural or artificial nucleobases can be utilized for controlling the metal sequence coordinated to nucleobases inside a DNA double helix, forming a so-called metal–DNA (M–DNA) complex.2 The metal clusters thus formed inside DNAs are interesting targets due to their specific and controllable conductivity, and magnetic, optical or catalytic properties, emerging from metal–ligand and metal–metal interactions. The same motivation fueled numerous studies of various homo- and hetero-nuclear transition metal chain or string structures assembled by means of polymeric ligands.3,4 In particular, silver clusters show luminescence and low toxicity emphasizing their potential use as biosensors with DNA molecules as naturally biocompatible carriers for such applications.5 The structural analysis of such systems is scarce6,7 and challenging due to the size and complexity of the DNA strands so that often the binding topology is only indirectly inferred. Nevertheless, the knowledge of binding energies, geometries and isomeric and tautomeric variability is mandatory for a better understanding of the chemical binding situation, the analysis of possible formation mechanisms in solution and the design of new compounds of this type. We follow here a bottom-up approach using IR spectroscopic and density functional theory (DFT) analyses of small model complexes in the gas phase8 which are comprised of two nucleobases mediated by two or three metal ions.9Thymine (T) containing DNA strands are prone to dimerize and stabilize by deprotonation (T-H) with a parallel selective uptake of HgII ions forming neutral (T-H)–HgII–(T-H) metal base pairs (MBPs), representing one of the best documented examples of specific metal complexation by a nucleobase pair.7,10–12 By vibrational analysis in solution, a Raman “fingerprint” band has been identified for the mercury coordinated (T-H) using a thymidylyl-3′–5′-thymidine (TpT) model compound.13 On the other hand, DNA strands containing cytosine–cytosine (C–C) mispairs show a tendency to preferentially incorporate AgI ions in solution forming cationic C–AgI–C MBPs.14 Moreover, an appropriate selection of the nucleobases even allows for a coordination of two metal ions per base pair.9,15–18 Within DNAs containing 1-3-dideazaadenine (DDA) or 1,N6-ethenoadenine (εA) the formation of the DDA–(Ag)2–T16 or εA–(Ag)2–εA18 metal base pairs (MBPs) has been analyzed using melting temperature measurements, UV absorption and circular dichroism titration techniques. The structure of the related model compounds, i.e. isolated nucleobase pairs, has been interrogated by accompanying density functional theory (DFT) calculations. Formation of the FU–(Ag)2–FU15 (FU = 5-fluorouracil) and ST–(Ag)2–ST17 (ST = 2(4)-thiothymine) MBPs within DNA duplexes has also been observed and supported by results of electrospray ionization mass spectrometry (ESI-MS). Moreover, a silver dimer mediated metal base pair formation mechanism in aqueous solution for uracil has been recently proposed and explored theoretically.19 It is characterized by a rate-limiting step which involves the replacement of the N3H proton by a metal ion in the investigated uracil derivatives.19 In a related comprehensive theoretical study deprotonation of thymine has also been found crucial for the formation of (T-H)–HgII–(T-H) metal base pairs.12
In detail, the gas phase structure of the DDA–(Ag)2–(1MT-H) MBP (1MT = 1-methylthymine) has been recently investigated by one- and two-color IR multiple photon dissociation (1C-/2C-IRMPD) spectroscopy in an electrospray ionization ion trap experiment.9 In that study, vibrational analysis by DFT led to the assignment of a reversed Hoogsteen base pairing topology mediated by two silver coordinative bridges with likely argentophilic interactions due to the close proximity of the metal units.20
The deprotonated,21 protonated22 or metal-complexed single nucleotides23 have been well studied by IR and UV gas phase photodissociation. To the best of our knowledge there are no similar IRMPD reports on metal mediated nucleoside pairs. In order to fill this gap we will present in this work the results of the Ag mediated complexes of 2′-deoxy-thymidine (dT) which also support the here proposed IR spectral assignments of the Ag/1-methylthymine metal base pairs.
In experimental studies derivatives of thymine have already shown their particular ability to form MBPs mediated by two AgI centers.9,15–18 These results have motivated us to perform a study of complexes of thymine (or dT) with AgI ions and explore in detail the structures of their conceivable MBPs. Since the nucleobases reveal tautomerization upon coordination with transition metal ions,24 we have chosen the thymine molecule methylated at its glycosidic nitrogen (1MT) in order to reduce the number of possible tautomers and binding sites. Moreover, the choice of dT for analogous complexes gives rise to higher fragment yields which is beneficial for the applied action spectroscopy.
In the current study we report on the structure of the cationic BP of 1MT mediated by two and three AgI ions investigated using IRMPD spectroscopy. The IR spectra are obtained in an ion trap and assigned by comparison with DFT calculations. Furthermore, these assignments are supported by results obtained for AgI mediated BPs of dT. Eventually, we present the resulting structures and discuss the possible isomeric forms. The thus obtained geometrical parameters and energetics are essential for a deep understanding of the binding topology and the possible formation mechanisms in aqueous solution of these important building blocks of M–DNAs. They are giving evidence for metallophilic interactions and open perspectives to apply this effect to the design of new M–DNA assemblies.
Experimental and calculation details
The ionic AgI complexes of 1MT (or dT) were generated by electrospray ionization from a methanol/water (3/1 v/v) solution of 1MT (dT) and AgNO3 with a stoichiometric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, respectively, at c(1MT/dT) = 0.1 mM. Subsequent ion storage, fragmentation by either collision-induced dissociation (CID) or IRMPD and mass-resolved analysis were carried out in a Paul-type ion trap at ca. 305 K. The silver nitrate compound was of 99.8% purity. Water and methanol solvents of LC-MS grade as well as the 1MT and dT samples were obtained from Sigma-Aldrich.
:2, respectively, at c(1MT/dT) = 0.1 mM. Subsequent ion storage, fragmentation by either collision-induced dissociation (CID) or IRMPD and mass-resolved analysis were carried out in a Paul-type ion trap at ca. 305 K. The silver nitrate compound was of 99.8% purity. Water and methanol solvents of LC-MS grade as well as the 1MT and dT samples were obtained from Sigma-Aldrich.
The experimental setup was described in detail previously.9 Briefly, it consists of a modified ESI quadruple ion trap mass spectrometer (Amazon-SL, Bruker Daltonics) and an injection seeded 10 Hz Nd3+:YAG (PL-8000, Continuum) pumped IR optical parametric oscillator/amplifier (OPO/A) laser system (LaserVision). The sample solution was continuously infused into the ESI chamber by a syringe pump at a flow rate of 2 μL min−1 and sprayed at a nebulizer pressure of 310 mbar and needle voltage of 4.5 kV. Nitrogen as dry gas was supplied with a flow rate of 3.5 L min−1 at 210 °C. The IRMPD spectra in the 3800–2800 cm−1 and 2000–1200 cm−1 ranges were recorded using the idler and signal minus idler difference frequency waves of the OPA, respectively. The IR spectra were evaluated as Fi/(Fi + Pj), with Fi and Pj being the total fragment yield and the parent yield, respectively. Since the IRMPD signal response on the laser fluence is not only species-dependent but also frequency-dependent,9,25 the presented IRMPD spectra were not corrected for laser power unless explicitly stated.
Optionally, the idler wave of an additional IR OPO/A (LaserVision/Continuum) was applied to facilitate fragmentation by two-color IR excitation.9 This probe pulse was typically delayed by 80 ns with respect to the tunable pump IR pulse. Its frequency was fixed in resonance with a selected vibrational transition taking into account the known preexcitation induced broadening and shift of the band (Fig. S7, ESI†). Typically the probe frequency was shifted a few cm−1 off the one-color band maximum but set to a value within the ca. 10 cm−1 bandwidth. The probe laser intensity was attenuated to the one-color fragmentation threshold.
The IRMPD signals were generated using up to 4 laser pulses or pulse pairs per ion cloud. The lasers and the mass-spectrometer were synchronized by a delay generator (SRS DG645). The IR beam path was continuously flushed using a purge gas generator (PG85L, CMC Instruments) to reduce the atmospheric water and CO2 absorption. Typically the IR experiments were performed at 1–2% relative humidity. The dependence of the IRMPD signal on the laser power was recorded at higher relative humidity (ca. 10%), so that a laser intensity modulation up to 50% due to atmospheric water could lead to perturbation of the related IRMPD band shapes (Fig. S5, ESI†).
The minimum energy structures, interaction energies and linear IR spectra were calculated at the DFT/B3LYP/aug-cc-pVDZ level as implemented in the Gaussian 09 program package.26 Related calculations were performed using the dispersion corrected functional B3LYP-D3.27 The Stuttgart–Dresden effective core potential28 was applied to the Ag atoms. All optimized isomeric structures represent true energy minima as they were controlled for the absence of imaginary vibrational frequencies. The harmonic vibrational frequencies were scaled by factors of 0.9869 and 0.954 below and above 2000 cm−1, respectively. The basis set superposition error was calculated using the counterpoise method.29 Reaction free energies in water were estimated using the implicit conductor-like polarizable continuum model (CPCM).30 For these calculations we applied ΔG(Haq+) = −1138 kJ mol−131 for the sake of comparability with the literature.19
Results and discussion
Mass spectra and fragmentation pattern
An ESI mass spectrum obtained in the cationic mode showing the formation of complexes that are comprised of monomers and dimers of 1MT with various numbers of AgI ions is depicted in Fig. 1a. All assigned species are singly charged. We will concentrate on the complexes with two 1MT units in this work. The number of AgI ions involved in the different complexes is readily discernible from their isotope patterns. For the main observed complex cations, the number of AgI ions and the degree of deprotonation of the 1MT self-dimer are related as follows: (AgI)k(1MT)2(–H)(k−1) with k = 1, 2, 3. A similar replacement of protons by AgI ions is reported by Vrkic et al. for cationic silver–adenine heteroclusters.32 Aside from the 1MT complexes containing one AgI ion (m/z 247, 265, 288, 387), the 1MT self-dimers mediated by 2 and 3 metal ions were observed at m/z 495 ([Ag2(1MT-H)(1MT)]+) and m/z 601 ([Ag3(1MT-H)2]+), respectively. The m/z values of the investigated cations are listed in Table 1 along with their major fragment channels.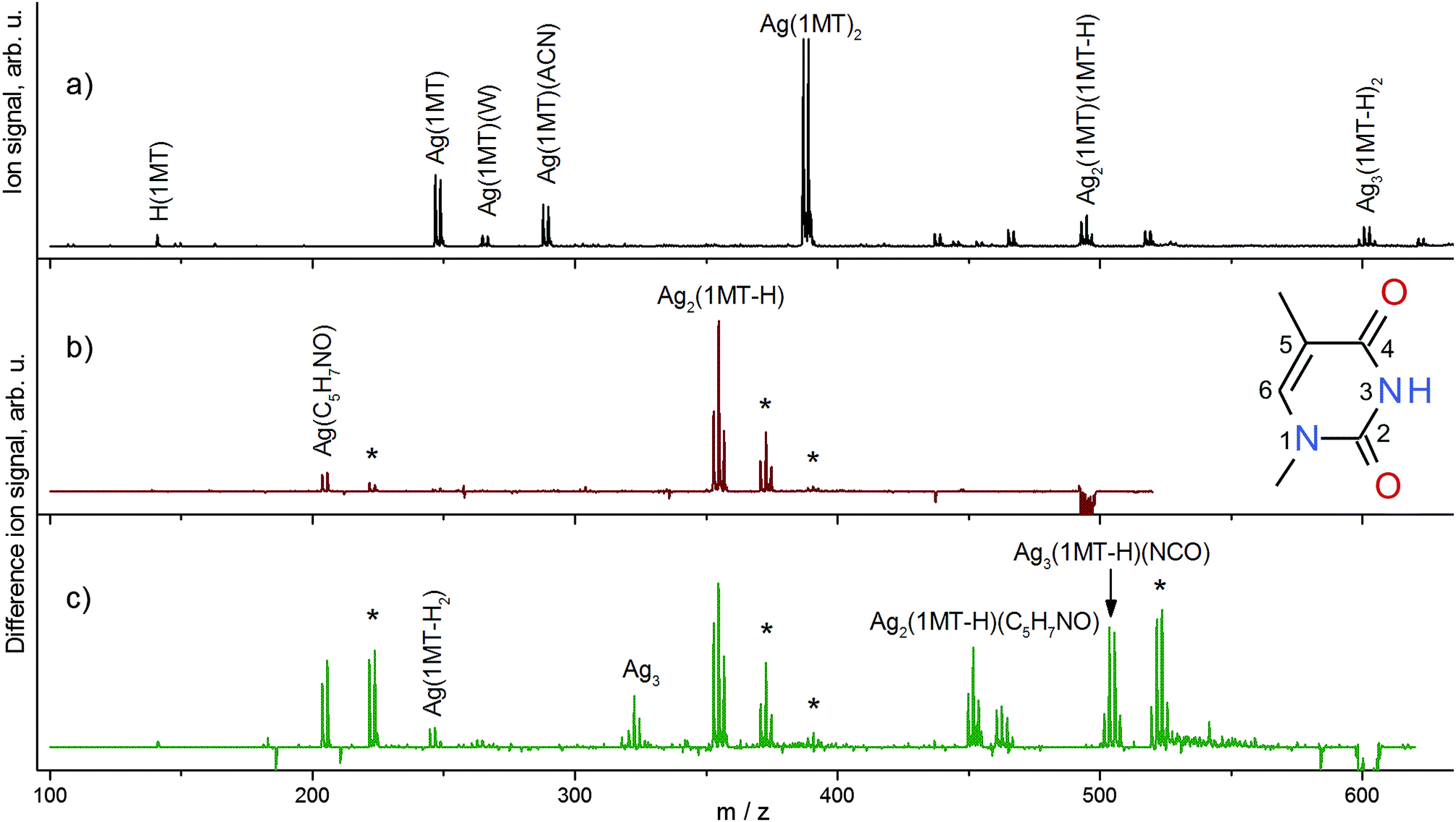 | ||
Fig. 1 (a) ESI mass spectrum in the positive mode of a solution containing 1-methylthymine (1MT) and AgNO3. Difference mass spectra emphasizing IR induced fragmentations measured at the corresponding C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch modes for the (b) [Ag2(1MT-H)(1MT)]+ and (c) [Ag3(1MT-H)2]+ complexes. The negative signals indicate depletion of the parent ions; the positive peaks show an appearance of the related ion fragments. Assignments for the major ionic fragments are given. The gas phase hydration products formed by ion-molecule collisions with traces of water in the ion trap are denoted with asterisks. Inset: a 1MT molecule with the ring atom numbering. O stretch modes for the (b) [Ag2(1MT-H)(1MT)]+ and (c) [Ag3(1MT-H)2]+ complexes. The negative signals indicate depletion of the parent ions; the positive peaks show an appearance of the related ion fragments. Assignments for the major ionic fragments are given. The gas phase hydration products formed by ion-molecule collisions with traces of water in the ion trap are denoted with asterisks. Inset: a 1MT molecule with the ring atom numbering. | ||
| Parent ion/Da | Fragment ion/Da | Neutral loss/Da | ||
|---|---|---|---|---|
| Ag2(1MT-H)(1MT) | ||||
| 495 | 355 | Ag2(1MT-H) | 140 | 1MT |
| 204 | Ag(C5H7NO) | 289 | Ag(1MT)(NCO) | |
| Ag3(1MT-H)2 | ||||
| 601 | 504 | Ag3(1MT-H)(NCO) | 97 | C5H7NO |
| 452 | Ag2(1MT-H)(C5H7NO) | 149 | Ag(NCO) | |
| 355 | Ag2(1MT-H) | 246 | Ag(1MT-H) | |
| 323 | Ag3 | 278 | 2 × (1MT-H) | |
| 245 | Ag(1MT-H2) | 356 | Ag2(1MT) | |
| 204 | Ag(C5H7NO) | 397 | Ag2(1MT-H)(NCO) | |
| Ag3(dT-H)2 | ||||
| 805 | 689 | Ag3(dT-H)(T-H) | 116 | C5H8O3 |
| 573 | Ag3(T-H)2 | 232 | 2 × (C5H8O3) | |
The fragmentation patterns obtained by IRMPD at various wavelengths were found to be identical to those obtained by collision-induced dissociation (CID). Difference mass spectra (on vibrational resonance minus off-resonance, Fig. 1b and c) depict the IRMPD effect and assist in the analysis of fragmentation patterns which in turn provide insights into the composition and structure of the subunits of the ions under study. A neutral loss of the intact ligand (1MT) is predominant for [Ag2(1MT-H)(1MT)]+ (Fig. 1b). Another fragment channel characterized by chemical disintegration of the ligand becomes discernible by the fragment peak at m/z 204 (formal loss of [Ag(1MT)(NCO)]). In order to rationalize this fragment we assume the formation of the isocyanate anion (NCO−) coordinated to one silver ion. This is in agreement with the observed dominant loss of HNCO from neutral thymine by VUV or electron impact ionization.33 The remainder of the “denaturated” 1MT-H ligand (97 Da, C5H7NO) may form either an oxazole derivative (e.g. dimethyloxazole) or an open unsaturated structure similar to those discussed for the uracil fragmentation.34 It is noteworthy that for [Ag2(1MT-H)(1MT)]+ a dissociation channel prevails in which the two silver ions remain in one fragment, tentatively indicating a conceivable argentophilic interaction, although the DFT calculations of the dissociation energies suggest that splitting of the silver dimer would be feasible (see Table 2). A similar metal–metal interaction is reported for the IRMPD mass spectra of [(DDA)Ag2(1MT-H)]+ MBP where preferentially the loss of neutral 1MT and the formation of [Ag2(DDA-H)]+ are observed.9
| Fragmentation pathway | Parent ion isomer | ΔEff | ΔEdef | ΔE0 | ΔG298 |
|---|---|---|---|---|---|
| [Ag(1MT)2]+ → [Ag(1MT)]+ + 1MT | ag44h3h3 | 176 | 167 | 162 | 119 |
| [Ag2(1MT-H)(1MT)]+ → [Ag2(1MT-H)]+ + 1MT | ag2433h4 | 283 | 262 | 254 | 196 |
| [Ag2(1MT-H)(1MT)]+ → [Ag(1MT)]+ + 3Ag1MT | ag2433h4 | 284 | 251 | 244 | 184 |
| [Ag3(1MT-H)2]+ → [Ag2(1MT-H)]+ + 3Ag1MT | ag243342 | 415 | 367 | 357 | 290 |
The [Ag3(1MT-H)2]+ ion shows a diverse fragment distribution (Fig. 1c). The fragment containing intact ligands (m/z 355) diminishes somewhat in favor of other fragments related to the formation of the Ag coordinated isocyanate. This observation can be understood in terms of higher binding energy and/or higher activation barrier for dissociation of this complex in comparison to [Ag2(1MT-H)(1MT)]+. Also, the observation of a remarkably strong signal corresponding to a fragment containing three Ag centers (m/z 504) and even that of a bare silver trimer cation (m/z 323) imply that the metal–metal interaction is supposedly strong and plays a significant role in the binding and structure of the [Ag3(1MT-H)2]+ ion.
Since the [Ag3(1MT-H)2]+ complex revealed only a small total fragment yield upon IRMPD, even at the highest laser power and also upon 2-color IR irradiation, some computationally predicted weak IR bands could not be recorded by the IRMPD action spectroscopy scheme. As a remedy, we substituted the 1MT ligands by the nucleoside deoxythymidine (dT). The dT molecule forms stoichiometrically the same complexes with the AgI ions as 1MT. The IR induced fragmentation of the [Ag3(dT-H)2]+ ion gave larger fragment yields compared to [Ag3(1MT-H)2]+ under otherwise identical conditions and was clearly dominated by the neutral loss of one or two sugar units accompanied by reprotonation of the glycosidic nitrogen atoms (loss of neutral C5H8O3 or (C5H8O3)2; Fig. S1, ESI†).35 These findings also support the high stability of the multi-metal mediated MBP core.
IRMPD spectra and their assignment
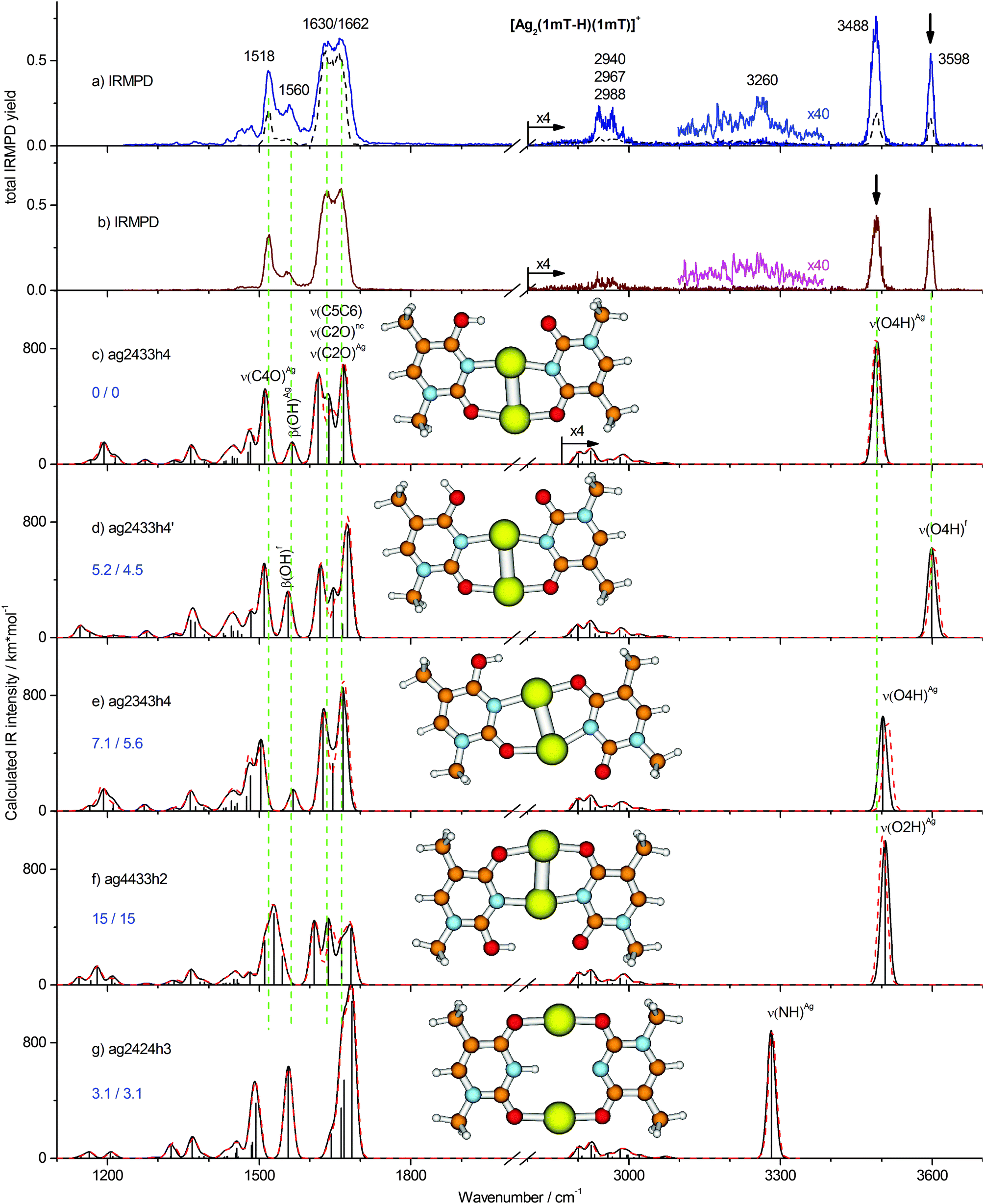 | ||
| Fig. 2 Two-color IRMPD spectra of [Ag2(1MT-H)(1MT)]+ measured with the probe pulse fixed at the (a) 3598 cm−1 and (b) 3488 cm−1 bands as indicated by the arrows. One-color IRMPD spectrum recorded at otherwise same conditions is depicted with dashed line. (c–g) Calculated linear IR absorption spectra of selected isomers of [Ag2(1MT-H)(1MT)]+ (B3LYP/aug-cc-pVDZ/ECP) simulated with the Gaussian profile of fwhm = 20 cm−1. Simulated IR spectra based on B3LYP-D3 calculations are presented with dashed lines. The optimized structures as well as relative Gibbs free energies at 0/298 K are shown. | ||
In contrast to the computationally predicted low energies of the NH, di-keto isomers (Fig. 2g and Fig. S2h, ESI†), the IRMPD spectra of [Ag2(1MT-H)(1MT)]+ indicate no considerable abundance of these species. We would expect IRMPD bands of the NH oscillators at around 3300 cm−1. However, large anharmonicity prevails for the NH stretching mode under the constraints of the structure as given in Fig. 2g, where the proton is likely delocalized in a quadrupole-like NAgNAg cage. Thus, we expect an anharmonicity-induced broadening and a significant red-shift of the ν(NH)Ag band possibly out of the investigated spectral range. On the other hand, we believe that the stability of the enolic tautomers (calculated Ag–Ag distance is typically ≤290 pm, Table S1, ESI†) is underestimated due to the inadequate treatment of the metal–metal interaction at the B3LYP level of theory, which is otherwise well suitable for vibrational analysis.9,23 For instance, an argentophilic term of ca. 64 kJ mol−1 is calculated at the BLYP-D/TZ2P level for the [DDA–Ag2–(T-H)]+ MBP mediated by a silver dimer with the Ag–Ag distance of 288 pm.16 Employing a modified functional with a long-range term B3LYP-D3 we obtained an additional stabilization of ca. 3 kJ mol−1 for the enolic isomers which are characterized by the close metal–metal contact (Table S1, ESI†). The D3 long range correction reduces the estimated Boltzmann population of the di-keto isomers from 27% to 11%. Although this result is not sufficient to rule them out completely, the energy trend upon consideration of metal–metal interactions is not in their favor.
The one-color IR induced fragmentation yield even after excitation of the OH stretching frequencies of the [Ag2(1MT-H)(1MT)]+ complex was relatively modest compared to other related systems, e.g. [Ag2(1MT-H)DDA]+,9 under similar experimental conditions. Thus, we applied the 2C-IRMPD scheme in order to improve the signal-to-noise ratio of the related bands. In addition, we planned to discriminate between the two OH rotamers by applying the second laser frequency rotamer selectively which should result in spectral enhancement or depletion of the corresponding bands. The 2C-IRMPD spectra recorded using the laser probe fixed at 3598 and 3488 cm−1 bands are shown with continuous traces in Fig. 2a and b, respectively. Interestingly, they look very similar. All the vibrational bands were enhanced regardless of the probe frequency. However, the 3598 cm−1 probe gave a slightly larger signal enhancement, possibly due to its higher photon energy. This probe scheme also provided a very weak signal near 3260 cm−1 (see magnified trace in Fig. 2a) which is close to the calculated NH stretch of the already discussed di-keto isomer (Fig. 2g). However, since no other evidence of this isomer was observed, its identification under the applied experimental conditions remains uncertain. It is noteworthy that the intensity ratio of the isomer-specific OH stretches in the 2C-IRMPD spectra was affected by a larger enhancement of the non-probed OH stretch. In other words, a ν(OH) preexcitation of one isomer slightly increased the relative two-color signal of the other isomer. These observations can only be explained in terms of the dynamic equilibrium of the two isomers after vibrational excitation, i.e. an IR induced isomerization.37
In order to rationalize this behavior we have explored the isomerization process in more detail by DFT modeling of the energy path along the reaction coordinate. A relaxed potential energy surface scan along the OH torsion coordinate of the ag2433h4 isomer was performed at the B3LYP/aug-cc-pVDZ/ECP level (Fig. 3). It gave a barrier height of ∼36 kJ mol−1 which corresponds to a transition state structure with the OH group located on a plane orthogonal to the ring plane. The zero-point vibrational energy correction reduces the barrier to ∼32 kJ mol−1. Both values for the transition barrier are smaller than the energy of one photon of 3488 cm−1 (41.7 kJ mol−1). So, a one-photon ν(OH) excitation might be sufficient for the OH rotamerization, given the excitation energy is redistributed in favor of the reaction coordinate. Since IRMPD is usually considered as a multiple photon process its application will therefore affect the isomeric equilibrium of [Ag2(1MT-H)(1MT)]+. This process of energy uptake and storage via isomerization partially explains the relatively small 1C-IRMPD signal of [Ag2(1MT-H)(1MT)]+ in the NH/OH stretching region.
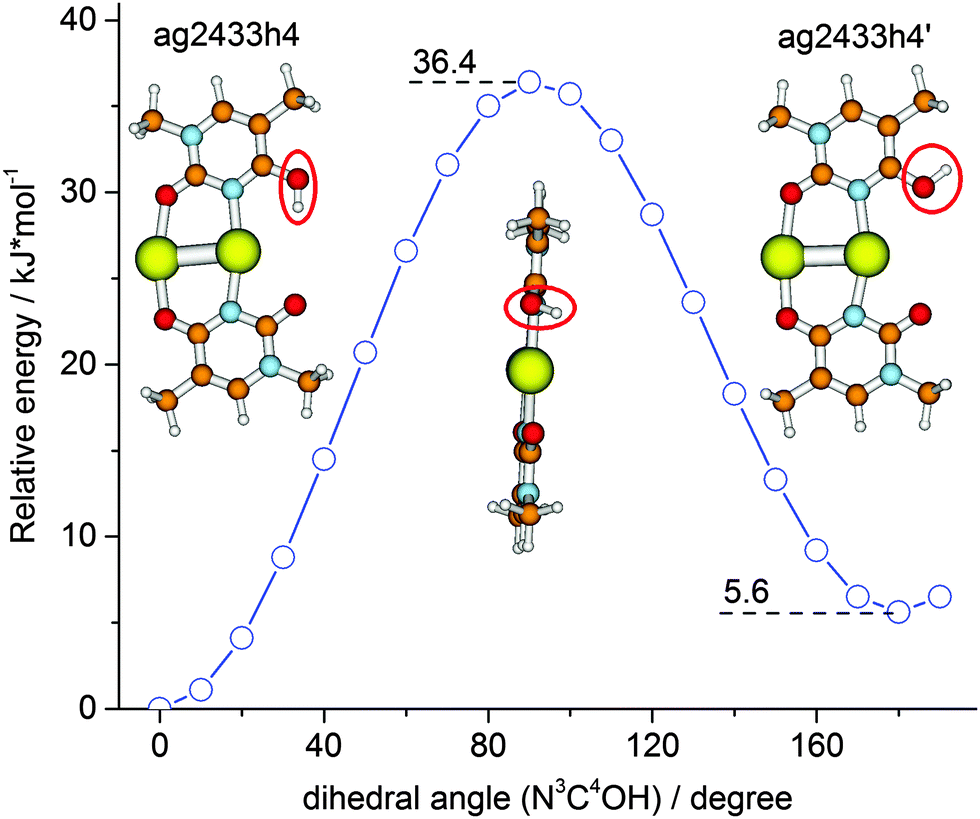 | ||
| Fig. 3 Relaxed potential energy surface scan (B3LYP/aug-cc-pVDZ/ECP) for the energy minimum structure of [Ag2(1MT-H)(1MT)]+ along the hydroxyl group torsion coordinate. | ||
The IR induced ν(OH)Ag ↔ ν(OH)f rotamer interconversion challenges the population analysis for [Ag2(1MT-H)(1MT)]+. The 1C-IRMPD intensities of the ν(OH)Ag and ν(OH)f bands indicate a comparable abundance of the two isomers in contrast to their calculated Gibbs free energy difference of 4.5 kJ mol−1 at 298 K which implies at least 6 times higher abundance of the ag2433h4 isomer. Entropy terms do not significantly affect the isomer stabilities of [Ag2(1MT-H)(1MT)]+ (Fig. 2 and Table S1, ESI†). Therefore, the IR induced isomerization and equilibration are responsible for the observed nearly equal signal intensities on both ν(OH) bands.
The major features in the lower frequency range are the 1662/1630 cm−1 doublet and the band at 1518 cm−1. The band at 1560 cm−1 as well as a few features below 1500 cm−1 gain strongly in the two-color experiments. The IR band pattern obtained in the 2C-IRMPD experiments shows the best agreement with the calculated spectrum of the lowest energy isomer ag2433h4 and a few other isomers characterized by the coordinated C4O/N3 and C2O/N3 positions of the 1MT-H and 1MT ligands, respectively, (Fig. 2c–e). Hence, these isomers are barely distinguishable in the low frequency spectral range. The band at 1518 cm−1 is then assigned to the ν(C4O)Ag mode of the 1MT-H ligand. The rest of the IR active double bond stretchings, ν(C5C6), ν(C2O)f and ν(C2O)Ag, contribute to the 1662/1630 cm−1 doublet. This assignment is supported by the following published results of related molecular systems. The ν(C2O)f and ν(C4O)Ag modes of the [Ag2(1MT-H)(DDA)]+ MBP are reported at 1648 and 1510 cm−1, respectively,9 in good agreement with the values assigned for [Ag2(1MT-H)(1MT)]+. Moreover, the ν(C5C6) and ν(C2O)f modes are reported to be overlapped within a Raman band at 1654 cm−1 for the mercury complex of TpT containing (T-H)–HgII–(T-H) MBPs.13
The other absorption features below 1500 cm−1 could be assigned to the overlap of several methyl group modes and CH bending modes. The band at 1560 cm−1 is assigned to the in-plane OH bending, β(OH). Interestingly, it is red shifted by ∼5 cm−1 (which is a small but reproducible value) in the 2C-IRMPD spectrum and parallels the already described relative gain of the ν(OH)f band (Fig. 2b). This finding is in agreement with the calculations of the ag2433h4 and ag2433h4′ isomers (Fig. 2c and d) revealing the β(OH)Ag and β(OH)f modes at 1565 and 1557 cm−1, respectively, (δν = 8 cm−1).
In summary, the superposition of two isomers (ag2433h4 and ag2433h4′; Fig. 2c and d) explains the observed IRMPD spectra of [Ag2(1MT-H)(1MT)]+ very well. Their populations cannot be analyzed quantitatively from the band intensities, due to their IR induced interconversion. Moreover, some contribution of the NH di-keto isomers cannot be completely excluded as their strong bands are expected at 1680, 1560 and 1490 cm−1 overlapping the pattern of the ag2433h4 and ag2433h4′ isomers. The long range D3 correction to the B3LYP functional was found to provide only small, rather negligible changes to the calculated vibrational frequencies of [Ag2(1MT-H)(1MT)]+ (Fig. 2 and Fig. S2, ESI†). The root-mean square difference of the OH/NH stretch frequencies for all the considered isomers is 6 cm−1 with a maximum deviation of 14 cm−1.
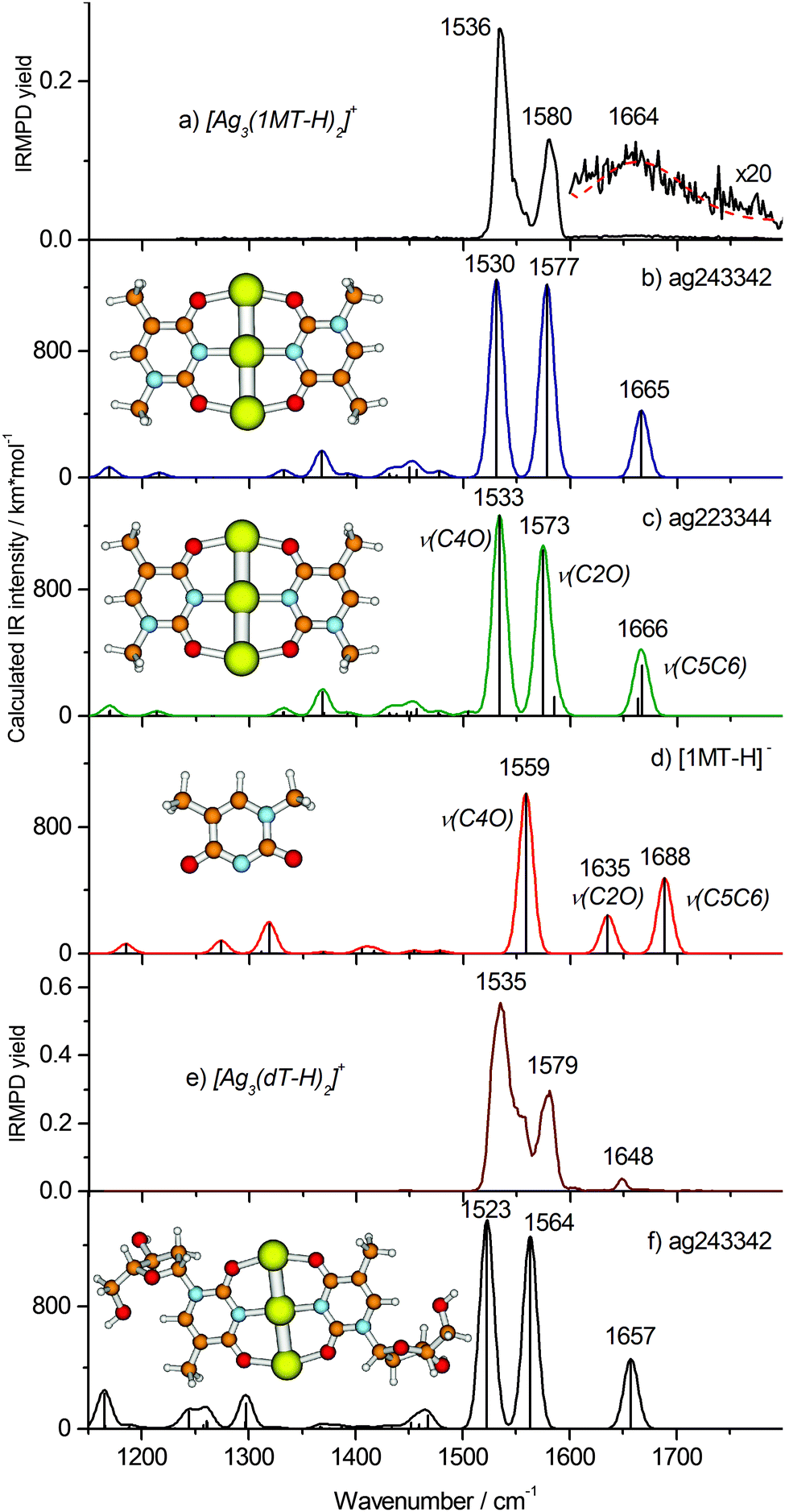 | ||
| Fig. 4 IRMPD spectra of (a) [Ag3(1MT-H)2]+ and (e) [Ag3(dT-H)2]+ and calculated linear IR absorption spectra (B3LYP/aug-cc-pVDZ/ECP) of the relevant complex structures (b, c and f) as well as of the (1MT-H)− ligand (d) simulated using the Gaussian profile of fwhm = 16 cm−1. The optimized structures are shown. | ||
Our calculations reveal two minimum structures with a linear Ag trimer ligated by two (1MT-H) units arranged either in C2H (trans) or C2V (cis) symmetry abbreviated ag243342 and ag223344 forms, respectively, (Fig. 4b and c). In order to explore other possible structures, in particular those with differently arranged metal cores, we carried out a computational geometry optimization of [Ag3(1MT-H)2]+ with a triangular starting structure known to be the minimum for [Ag3]+.38 However, this starting geometry eventually converged to the linear metal core (Fig. 4b and c). The two depicted structures are nearly isoenergetic (calculated energy difference of 0.5 kJ mol−1). Both isomers reveal very good agreement with the experimental vibrational spectrum for the carbonyl stretches, being thus not distinguishable by means of IRMPD spectroscopy. In conclusion, we assign the experimental bands at 1580 and 1536 cm−1 to the ν(C2O)Ag and ν(C4O)Ag modes, respectively. The corresponding calculated normal modes are collective vibrations involving both ligands with most significant contributions located at the denoted bonds (Fig. S4, ESI†). The ν(CO)Ag modes of [Ag3(1MT-H)2]+ exhibit a significant red shift of ca. 190 cm−1 with respect to the corresponding free thymine modes.39 A large fraction of the red shift, 101 and 156 cm−1 (at the B3LYP/aug-cc-pVDZ level, Fig. 4d), is due to the deprotonation of the 1MT ligands, in agreement with the literature.11 The rest stems from the CO–Ag coordination.
The weak and broad feature at ∼1660 cm−1 in the experimental IRMPD spectrum of [Ag3(1MT-H)2]+ (Fig. 4a, inset) is assigned to the ν(C5C6) mode, which is much more intense in the computed, linear IR spectrum (Fig. 4b and c) than in the experiment. A Gaussian profile fit (Fig. 4a, dashed line) yielded 100 cm−1 width (fwhm) and 1664 ± 13 cm−1 peak frequency. This value is close to the frequency of the experimental ν(C5C6) stretch in [Ag2(1MT-H)(1MT)]+ (1662 cm−1) where one of the two ligands is deprotonated as well. There are no other vibrational bands (in particular within the CH/CH3 stretch region) by IRMPD under the applied experimental conditions. This observation can be rationalized in terms of the non-linear character of the IRMPD process and by a high dissociation threshold.40 In the case of the ν(CO)Ag modes of [Ag3(1MT-H)2]+ the IRMPD yield scales cubically with the laser fluence (see ESI†). The weak intensity of the ν(C5C6) mode indicates that only a small fraction of the [Ag3(1MT-H)2]+ ions, likely the highest energy tail of the thermal distribution, is able to fragment upon excitation of this vibrational transition. We have no explanation for the unusual broadening of this band. It cannot be ascribed to isomeric differences since the calculated variation in band positions is very small. From this result we estimate a minimum line strength for obtaining fragment ions from [Ag3(1MT-H)2]+ by IRMPD under the applied conditions of 400–800 km mol−1. In contrast, the ν(CO)Ag modes couple more effectively to the dissociation coordinates and thus their IRMPD intensities are relatively large. Along such lines of thought we would consider energy transfer from the ν(C5C6) mode less effective. It is noteworthy to emphasize that in this particular case the otherwise successful two-color IRMPD enhancement technique fails to overcome the seemingly prevailing bottleneck in fragmentation. This strongly non-ergodic behavior is certainly related to a dynamic light-heavy-light structure of the [Ag3(1MT-H)2]+ complex where the line of three heavy Ag centers seem to block effective energy randomization.
In order to analyze and overcome this fragmentation threshold problem we recorded an IRMPD spectrum of the [Ag3(dT-H)2]+ ion (Fig. 4e) which allows with higher yield for a low energy cleavage of its glycosidic bond. Remarkably, this spectrum resembles that of the [Ag3(1MT-H)2]+ complex suggesting a close similarity of the metal coordination patterns in both complexes. The frequencies of the carbonyl stretches at 1579 and 1535 cm−1 reproduce those of [Ag3(1MT-H)2]+ within one wavenumber (Fig. 4a and e). Additionally, there is a well-defined band at 1648 cm−1 which we assign to the ν(C5C6) mode. In this way, we observed all of the characteristic double bond stretching modes of the nucleoside complex, except for the weak bands below 1500 cm−1. The experimental IRMPD positions and intensities above 1500 cm−1 showed an acceptable agreement with the calculated ones for the linear IR spectrum of the ag243342 isomer of the [Ag3(dT-H)2]+ complex (Fig. 4f).
Comparison of different complexes and consequences for MDNA formation in solution
We note that a stepwise increase of the number of AgI ions per 1MT self-pair leads to a decrease of their IRMPD fragment ion yield under otherwise identical experimental conditions. These findings correlate with the laser fluence dependence of the IRMPD band intensities. While the IRMPD signals for [Ag2(1MT-H)(1MT)]+ scale linearly or quadratically with the laser fluence, [Ag3(1MT-H)2]+ showed clearly cubic dependence (Fig. S5, ESI†). We thus conclude that the stability of the silver mediated 1MT self-pair grows with increasing number of AgI ions per dimer. The stabilization energies of these cations support this statement (calculated for the most significant fragmentation pathways and listed in Table 2). Introduction of the third AgI ion leads to a reduction of the calculated Ag–Ag distance from 282 pm (ag2433h4 isomer) to 274 pm (ag243342 isomer) suggesting their partial reduction and enhancement of the metal–metal interaction, i.e. possibly through an enthalpic three-body effect.In order to estimate the energetics of this intermetallic interaction we followed the computational approach applied by Bickelhaupt and co-workers to the [DDA–Ag2–(T-H)]+ MBP.16 The (1MT-H) and (4H1MT) ligands of the ag2433h4 species were thus rotated by 90° about the N3–Ag bond and by 180° about the O2–Ag bond, respectively, while keeping other structural parameters frozen. For the ag243342 complex, the (1MT-H) ligands were accordingly rotated around the O4–Ag bonds. In the resulting structures (see ESI†) the ligands are connected via a dimeric or trimeric silver string. The interaction energy was then calculated for the frozen fragments as formed by the cleavage of a suitable Ag–Ag bond. The obtained B3LYP interaction energies for the silver dimer and silver trimer mediated complexes were −48 and −56 kJ mol−1, respectively. This interaction strengthening correlates well with the Ag–Ag bond shortening when the third metal center is introduced. At the dispersion corrected B3LYP-D3 level of theory these interaction energy values increased to −71 and −70 kJ mol−1, respectively. Although the long-range term contributes considerably to the metal–metal interaction, it seems to cancel the 8 kJ mol−1 non-additive interaction gain at the B3LYP level. A possible rationalization for this result could be the closer spacing of the interacting metal–ligand subsystems in the [Ag2(1MT-H)(1MT)]+ complex. The distances between the closest ring atoms of the two ligands for the silver dimer and trimer mediated model structures were 589 and 760 pm, respectively. From these results we tentatively conclude that the metal–metal interaction can be mainly traced back to a dominant electrostatic and/or charge transfer contribution and likely a small contribution from the dispersion interaction.
The preceding IRMPD/DFT analysis of the [Ag2(1MT-H)(1MT)]+ and [Ag3(1MT-H)2]+ complexes indicates that a multi-metal mediated T–T pairing is accomplished by the ligand tautomerisation and/or deprotonation. A concomitant change in the vibrational fingerprints is characterized by the red shift of the carbonyl stretch modes (Table S2, ESI†).
The studied complexes were prepared by electrospray ionization from aqueous solution. It is an obvious question of concern whether these species are formed or preformed in the solution or whether they result from the spraying and transfer processes. We cannot definitely answer this question by our described experimental means. However, we can explore the thermodynamic possibilities of formation of the observed and related species in solution by calculation of the related energetics and conceivable reaction mechanisms. The available computational models of formation of the thymine MBPs involve a metal ion for the N3H proton substitution as the most energy demanding step.12,16,19 Thus, the formation of (T-H)–HgII–(T-H) involves proton transfer steps from the bases to the metal ion solvate with transition state energies of up to 69 kJ mol−1.12 A formation pathway is also proposed for the neutral (T-H)–Ag2–(T-H) complex in water with the deprotonation barrier of ∼60 kJ mol−1 at the CPCM-B3LYP/aug-cc-pVDZ level.19
Using a similar methodology as in ref. 19 we have examined the formation of the cationic multi-metal mediated 1MT dimers as found in the present work. The Gibbs free energies of selected reaction pathways are summarized in Fig. 5 with the cation–cation reactions being excluded from the consideration. The pathway proposed by Matsui et al.19 is marked by the dotted arrow A in Fig. 5. The free energy of reaction for the AgI/proton exchange via the [Ag(1MT)W]+ complex was found to be endergonic with ΔG = 7 kJ mol−1, which is lower than the previously reported value of 24 kJ mol−1.19 The difference should be attributed to the different basis sets of the Ag atom since the methodology was otherwise the same. Alternatively (Fig. 5, dotted arrow B), the AgI/H+ exchange can proceed exergonically via a doubly H-bonded dimer of 1MT (ΔG = −12 kJ mol−1). A similar pathway was calculated to be favorable for the DDA–(Ag)2–(T-H) MBP.16 Formation of the H-bonded 1MT dimer seems to be not probable (ΔG = 17 kJ mol−1). Once the crucial species of either 3Ag1MT or the [Ag(1MT-H)(1MT)] complex are formed all the multi-metal mediated 1MT dimers investigated here become accessible in aqueous solution. This includes the formation of [Ag3(1MT-H)2]+via the neutral [Ag2(1MT-H)2]. In summary, the formation of the neutral and monocationic multi-Ag mediated MBPs is possible in solution and should proceed via the same rate limiting steps, i.e. the formation of the neutral 3Ag1MT or [Ag(1MT-H)(1MT)] intermediates. The overcoming of the barriers along both the paths (A) and (B) could be facilitated by the ESI-inherent droplet fission process during which the concentration of the partially cationized solute precursors increases up to the solvent free limit. However, as a note of caution, we like to emphasize at this point that the here suggested formation pathways in solution are of explorative character, since they are mainly based on free energy calculations of possible key intermediates. More investigations (in particular in solution) are necessary in order to verify the proposed reaction routes.
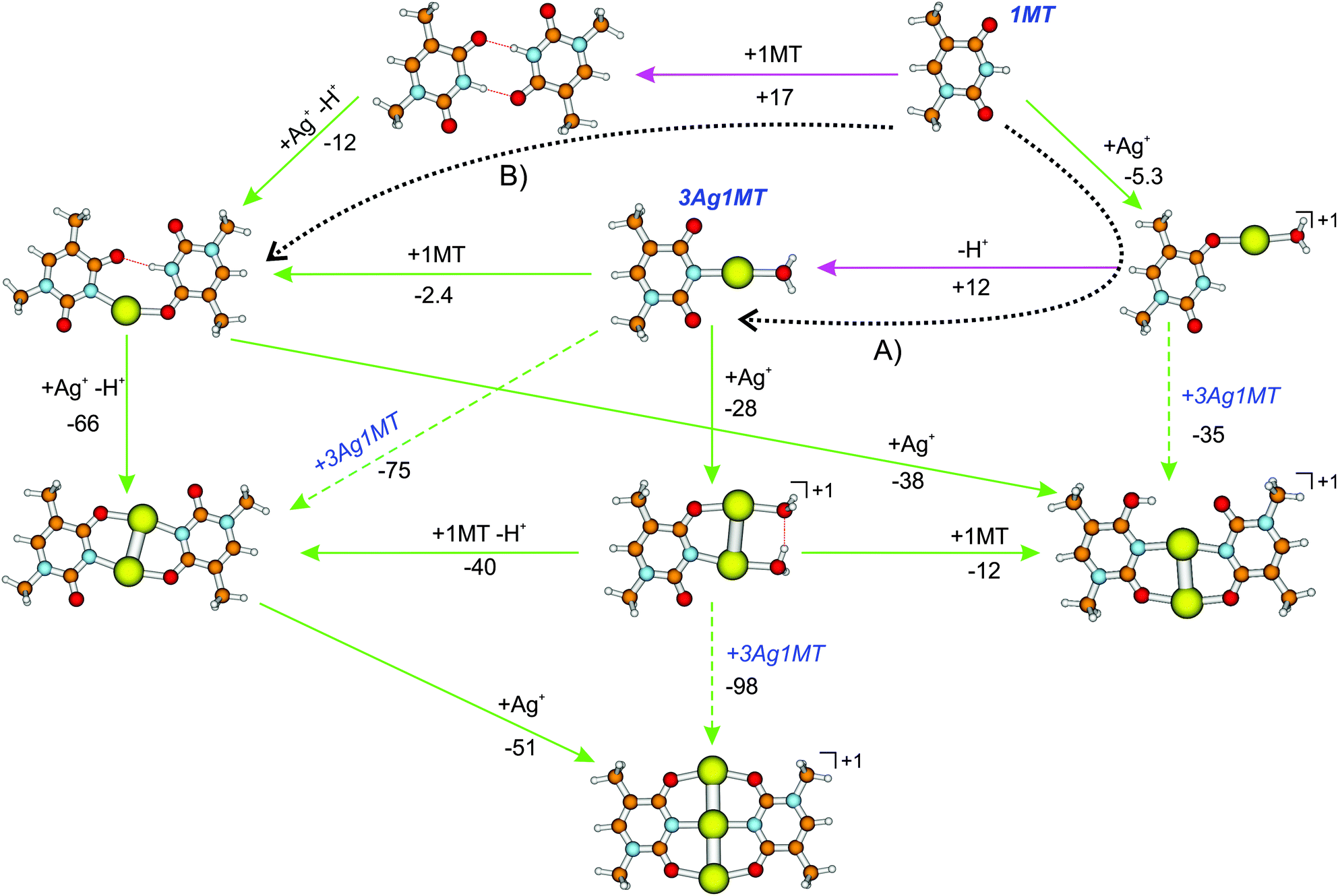 | ||
| Fig. 5 Reaction free Gibbs energies of formation (ΔG298, in kJ mol−1) of multi-metal mediated 1MT dimers in aqueous AgNO3 solution calculated at the CPCM-B3LYP/aug-cc-pVDZ/ECP level. The green and pink arrows indicate exergonic and endergonic reaction steps, respectively. The reaction pathways according to ref. 19 and 16 are denoted by the dotted arrows (A) and (B) respectively. Possible cation–cation reaction pathways are omitted. All cationic species were observed in this work by ion-trap ESI-MS. | ||
Interestingly, solvent-corrected calculations of the [Ag2(1MT-H)(1MT)]+ isomers predict higher relative Gibbs free energies for their NH (diketo) forms than in the gas phase. For the ag2424h3 isomer we thus obtained 8.5(14) kJ mol−1 at the CPCM-B3LYP (CPCM-B3LYP-D3) level relative to the global minimum enolic form (Fig. 5 and Table S1, ESI†). The corresponding population of the NH isomers in solution can therefore be estimated to be below 1%. This finding could in turn explain the lack of observation of the NH isomers in the IRMPD spectra (Fig. 2) with the additional assumption that the ag2433h4 → ag2424h3 gas phase conversion barrier is too high under the applied ion trap conditions. The latter assumption gains support by the fact that we could not detect a considerable signal of the ag2424h3 isomer even under resonant IR heating (2C-IRMPD) of the ag2433h4 and ag2433h4′ species (Fig. S7, ESI†).
As a consequence of these mechanistic considerations, the multi-silver mediated T–T mispairing within a DNA duplex would require tautomerization and a metal ion for the proton replacement. This might explain why it is observed that thymine oligonucleotides do not form silver-mediated homo-duplexes, in contrast to e.g. cytosine polynucleotides, at neutral pH.4 At the considered degrees of metalation we have experimentally identified the MBPs suitable for the MDNA duplex, e.g. ag2433h4 and ag243342. Although even a dicationic MBP, like εA–(Ag)2–εA,18 is known to stabilize the duplex, the charged nature of these MBPs might actually destabilize the MDNA especially in a contiguous MBP arrangement. A design of a silver-rich MDNA based on thymidine is still a challenge and must take this into account. One should also consider that it is energetically more favorable to form a reverse Ag-mediated T–(Ag)2,3–T pairing, where the glycosidic sites are arranged in a transoid-configuration with respect to the N3–N3 axis.
Conclusions
We have investigated the energetic stability of silver mediated 1-methylthymine (1MT) homo-dimers using binding energy calculations and IRMPD experiments. The obtained fragment yields decrease with the number of AgI ions involved. We have observed and analyzed using DFT and IR dissociation spectroscopy species with two and three AgI ions, i.e. [Ag2(1MT-H)(1MT)]+ and [Ag3(1MT-H)2]+.The recorded vibrational spectra are reproduced well by DFT spectra of optimized structures. The long-range D3 correction to the B3LYP functional does not considerably affect the predicted vibrational spectra of the considered metal base pairs. The identified structure of the [Ag2(1MT-H)(1MT)]+ ion features the N3–Ag–N3 and O2–Ag–O4 coordinative bridges and a clearly identified O4H rotamerism. No measurable amount of the NH isomers (di-keto forms) of this complex was detected which could be traced back to their calculated negligible population in solution. Moreover, we found by CPCM-DFT calculation of the Gibbs energies of intermediates that the formation of the neutral and monocationic multi-Ag mediated homogeneous MBPs of 1MT is possible in solution and should proceed via the same rate limiting steps, i.e. the formation of the neutral 3Ag1MT or [Ag(1MT-H)(1MT)] species.
For the [Ag3(1MT-H)2]+ ion we find a remarkable linear arrangement of the three silver ions, bridging all the oxygen and the N3 atoms. This molecular structure proved to be very strongly bound, and we assign it by comparison with results of the corresponding 2′-deoxy-thymidine complex [Ag3(dT-H)2]+. With this result we also hope to stimulate preparative chemists to approach the targeted syntheses of nucleobase complexes of high metal content. The gas phase UV spectroscopy and ultrafast dynamics of this species are subject to further investigation.41
Acknowledgements
We would like to thank Dr Alisa Krishtal (Rutgers University, Newark, USA) for DFT-D3 calculations in the early stage of this research project. We gratefully acknowledge financial support from the SFB/TRR 88 (3MET, project C4 and project A5) and the “Stiftung Rheinland-Pfalz für Innovation” (Project 965). YN thanks the “Carl-Zeiss-Stiftung” for financial support.References
- H. A. Wagenknecht, Angew. Chem., Int. Ed., 2003, 42, 3204–3206 CrossRef CAS PubMed; G. H. Clever and T. Carell, Angew. Chem., Int. Ed., 2007, 46, 250–253 CrossRef PubMed.
- G. H. Clever and M. Shionoya, Met. Ions Life Sci., 2012, 10, 269–294 CAS; D. A. Megger, N. Megger and J. Müller, Met. Ions Life Sci., 2012, 10, 295–317 Search PubMed.
- G. S. M. Tong, S. C. F. Kui, H.-Y. Chao, N. Zhu and C.-M. Che, Chem. – Eur. J., 2009, 15, 10777–10789 CrossRef CAS PubMed.
- S.-A. Hua, M.-C. Cheng, C.-h. Chen and S.-M. Peng, Eur. J. Inorg. Chem., 2015, 2510–2523 CrossRef CAS.
- C. I. Richards, S. Choi, J.-C. Hsiang, Y. Antoku, T. Vosch, A. Bongiorno, Y.-L. Tzeng and R. M. Dickson, J. Am. Chem. Soc., 2008, 130, 5038–5039 CrossRef CAS PubMed; J. M. Obliosca, L. Cong, R. A. Batson, M. C. Babin, J. H. Werner and Y. Hsin-Chih, Biosensors, 2013, 3, 185–200 CrossRef PubMed.
- S. Johannsen, N. Megger, D. Böhme, R. K. O. Sigel and J. Müller, Nat. Chem., 2010, 2, 229–234 CrossRef CAS PubMed; T. Dairaku, K. Furuita, H. Sato, J. Sebera, D. Yamanaka, H. Otaki, S. Kikkawa, Y. Kondo, R. Katahira, F. Matthias Bickelhaupt, C. Fonseca Guerra, A. Ono, V. Sychrovsky, C. Kojima and Y. Tanaka, Chem. Commun., 2015, 51, 8488–8491 RSC.
- J. Kondo, T. Yamada, C. Hirose, I. Okamoto, Y. Tanaka and A. Ono, Angew. Chem., 2014, 126, 2417–2420 CrossRef.
- ed. V. Gabelica, Nucleic Acids in the Gas Phase, Springer-Verlag, Heidelberg, 2014 Search PubMed.
- Y. Nosenko, F. Menges, C. Riehn and G. Niedner-Schatteburg, Phys. Chem. Chem. Phys., 2013, 15, 8171–8178 RSC.
- Y. Miyake, H. Togashi, M. Tashiro, H. Yamaguchi, S. Oda, M. Kudo, Y. Tanaka, Y. Kondo, R. Sawa, T. Fujimoto, T. Machinami and A. Ono, J. Am. Chem. Soc., 2006, 128, 2172–2173 CrossRef CAS PubMed; L. D. Kosturko, C. Folzer and R. F. Stewart, Biochemistry, 1974, 13, 3949–3952 CrossRef PubMed; A. Ono and H. Togashi, Angew. Chem., Int. Ed., 2004, 43, 4300–4302 CrossRef PubMed.
- B. Morzyk-Ociepa and D. Michalska, J. Mol. Struct., 2001, 598, 133–144 CrossRef CAS.
- J. Šebera, J. Burda, M. Straka, A. Ono, C. Kojima, Y. Tanaka and V. Sychrovský, Chem. – Eur. J., 2013, 19, 9884–9894 CrossRef PubMed.
- T. Uchiyama, T. Miura, H. Takeuchi, T. Dairaku, T. Komuro, T. Kawamura, Y. Kondo, L. Benda, V. Sychrovsky, P. Bour, I. Okamoto, A. Ono and Y. Tanaka, Nucleic Acids Res., 2012, 40, 5766–5774 CrossRef CAS PubMed.
- A. Ono, S. Cao, H. Togashi, M. Tashiro, T. Fujimoto, T. Machinami, S. Oda, Y. Miyake, I. Okamoto and Y. Tanaka, Chem. Commun., 2008, 4825–4827 RSC; M. Berdakin, V. Steinmetz, P. Maître and G. A. Pino, J. Phys. Chem. A, 2014, 118, 3804–3809 CrossRef CAS PubMed.
- I. Okamoto, K. Iwamoto, Y. Watanabe, Y. Miyake and A. Ono, Angew. Chem., Int. Ed., 2009, 48, 1648–1651 CrossRef CAS PubMed.
- D. A. Megger, C. F. Guerra, J. Hoffmann, B. Brutschy, F. M. Bickelhaupt and J. Müller, Chem. – Eur. J., 2011, 17, 6533–6544 CrossRef CAS PubMed.
- I. Okamoto, T. Ono, R. Sameshima and A. Ono, Chem. Commun., 2012, 48, 4347–4349 RSC.
- S. Mandal, A. Hepp and J. Müller, Dalton Trans., 2015, 44, 3540–3543 RSC.
- T. Matsui, H. Miyachi, T. Baba and Y. Shigeta, J. Phys. Chem. A, 2011, 115, 8504–8510 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- J. C. Marcum, A. Halevi and J. M. Weber, Phys. Chem. Chem. Phys., 2009, 11, 1740–1751 RSC; Y.-w. Nei, N. Hallowita, J. D. Steill, J. Oomens and M. T. Rodgers, J. Phys. Chem. A, 2013, 117, 1319–1335 CrossRef CAS PubMed.
- F. Lanucara, M. E. Crestoni, B. Chiavarino, S. Fornarini, O. Hernandez, D. Scuderi and P. Maitre, RSC Adv., 2013, 3, 12711–12720 RSC.
- J.-Y. Salpin, L. MacAleese, F. Chirot and P. Dugourd, Phys. Chem. Chem. Phys., 2014, 16, 14127–14138 RSC.
- T. van der Wijst, C. F. Guerra, M. Swart, F. M. Bickelhaupt and B. Lippert, Chem. – Eur. J., 2009, 15, 209–218 CrossRef CAS PubMed; O. Y. Ali and T. D. Fridgen, Int. J. Mass Spectrom., 2011, 308, 167–174 CrossRef; J.-Y. Salpin, S. Guillaumont, D. Ortiz, J. Tortajada and P. Maître, Inorg. Chem., 2011, 50, 7769–7778 CrossRef PubMed; A. K. Vrkic, T. Taverner, P. F. James and R. A. J. O'Hair, Dalton Trans., 2004, 197–208 RSC.
- T. Pankewitz, A. Lagutschenkov, G. Niedner-Schatteburg, S. S. Xantheas and Y.-T. Lee, J. Chem. Phys., 2007, 126, 074307 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Oglario, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision D.01), Gaussian Inc., Wallingford, CT, 2013 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- T. Matsui, A. Oshiyama and Y. Shigeta, Chem. Phys. Lett., 2011, 502, 248–252 CrossRef CAS.
- A. K. Vrkic, T. Taverner and R. A. J. O'Hair, J. Chem. Soc., Dalton Trans., 2002, 4024–4034 RSC.
- H. W. Jochims, M. Schwell, H. Baumgärtel and S. Leach, Chem. Phys., 2005, 314, 263–282 CrossRef CAS.
- D. G. Beach and W. Gabryelski, J. Am. Soc. Mass Spectrom., 2012, 23, 858–868 CrossRef CAS PubMed.
- J. Liu, S. Cao, B. Jia, D. Wei, X. Liao, J. Lu and Y. Zhao, Int. J. Mass Spectrom., 2009, 282, 1–5 CrossRef CAS.
- J. K. Bera and K. R. Dunbar, Angew. Chem., Int. Ed., 2002, 41, 4453–4457 CrossRef CAS.
- B. C. Dian, A. Longarte, P. R. Winter and T. S. Zwier, J. Chem. Phys., 2004, 120, 133–147 CrossRef CAS PubMed; J. Lang, M. Gaffga, F. Menges and G. Niedner-Schatteburg, Phys. Chem. Chem. Phys., 2014, 16, 17417–17421 RSC.
- M. J. Huang and J. D. Watts, Phys. Chem. Chem. Phys., 2012, 14, 6849–6855 RSC.
- P. Colarusso, K. Q. Zhang, B. J. Guo and P. F. Bernath, Chem. Phys. Lett., 1997, 269, 39–48 CrossRef CAS.
- A. Simon, C. Joblin, N. Polfer and J. Oomens, J. Phys. Chem. A, 2008, 112, 8551–8560 CrossRef CAS PubMed.
- Y. Nosenko, C. Riehn and W. Klopper, 2016, in preparation.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp07016c |
| This journal is © the Owner Societies 2016 |