
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The ultrafast reactions in the photochromic cycle of water-soluble fulgimide photoswitches†
C.
Slavov
a,
C.
Boumrifak
a,
C. A.
Hammer
a,
P.
Trojanowski
a,
X.
Chen
b,
W. J.
Lees
b,
J.
Wachtveitl
a and
M.
Braun
*a
aInstitute of Physical and Theoretical Chemistry, Goethe University, 60438 Frankfurt/Main, Germany. E-mail: braun@theochem.uni-frankfurt.de
bBiomolecular Sciences Institute, School of Integrated Sciences and Humanities and Department of Chemistry and Bio-chemistry, Florida International University, Miami, Florida 33199, USA
First published on 21st March 2016
Abstract
Photochromic switches are essential for the control and manipulation of nanoscale reactions and processes. The expansion of their application to aqueous environments depends strongly on the development of optimized water-soluble photoswitches. Here we present a femtosecond time-resolved investigation of the photochromic reactions (transition between the open and the closed form) of a water-soluble indolylfulgimide. We observe a pronounced effect of the protic nature of water as a solvent on the ultrafast ring-opening reaction. Typically, the excited state of the closed form has a larger dipole moment than the ground state, which leads to stabilization of the excited state in polar solvents and hence a lifetime (3 ps) longer than in non-polar solvents (2 ps). However, in water, despite the increased solvent polarity and the increased excited state dipole moment, the opposite trend for the excited state lifetime is observed (1.8 ps). This effect is caused by the opening of a new excited state deactivation pathway involving proton transfer reactions.
Introduction
Contemporary (bio)chemical science and (bio)technology are focused on the understanding, control and utilization of nanoscale reactions and structures, which necessitates the development of specialized manipulation tools. One promising technique uses molecular photochromic switches (photoswitches). Photoswitches are capable of reversible, light-induced transition between forms with different physicochemical properties.1,2 The utilization of light is advantageous since it permits instantaneous, precise and most importantly non-invasive manipulation.There are several classes of photochromic switches (e.g. azo compounds, diarylethenes, fulgides, spiropyrans, etc.) that are relatively well characterized and actively applied in research and industry.3–8 However, most photochromic compounds have low solubility and stability in water and thus their application is restricted to non-aqueous environments. To date, there is a limited number of studies of photoswitches in aqueous solutions,9–17 of which only a few are focused on the investigation of the ultrafast photochemical reactions (e.g. spiropyran13 and azobenzene18). Thus, despite the importance of water as a solvent environment for chemical and biological processes and the excessive demand for their control and manipulation, the behaviour of photoswitches in aqueous surroundings remains largely uncharacterized. This deficiency represents a major obstacle to a more extensive utilization of photoswitches.
Fulgides are photochromic switches derived from aryl substituted dimethylenesuccinic anhydride (Scheme 1). The substitution leads to the formation of a 1,3,5-hexatriene motif, which permits 6π-electrocyclization to cyclohexadiene. Therefore, these photoswitches exist in three isomeric forms – open Z- and E-forms (colourless hexatriene motif) and closed C-form (coloured cyclohexadiene motif).6 The Z- and the E-forms have similar optical properties and can be interconverted using near UV light.19 However, the photochromic reaction of interest with the strongest change of optical properties involves the electrocyclic ring-closing of the cyclizable open form to the thermally stable C-form (Fig. 1).6 Of particular interest are indolylfulgides, which have a bulkier aryl substituent and thus exhibit a reduced formation of the non-cyclizable open form. Furthermore, they show excellent thermal stability and fatigue resistance in organic solvents.6,20,21 The absorption spectra of their open and closed forms extend into the visible spectral range making them suitable for biochemical and biotechnological applications. Nevertheless, the presence of a succinic anhydride ring in indolylfulgides causes rapid solvolytic degradation in aqueous environments.22 It has been shown that indolylfulgimides, where the succinic anhydride ring is replaced by a succinimide ring, have significantly improved hydrolytic stability6 and certain application examples are available.23,24
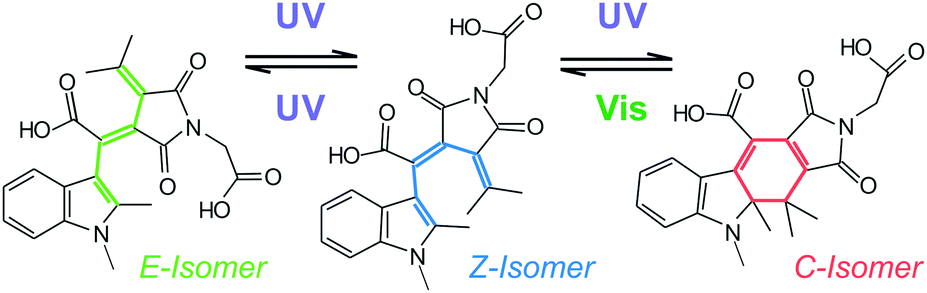 | ||
| Scheme 1 Chemical structures of WF (closed form (C); non-cyclizable open form (E); cyclizable open form (Z)). | ||
 | ||
| Fig. 1 (A) WF Z- and C-form absorption (solid blue and dashed red lines correspondingly) and fluorescence spectra (green dot-dash and yellow dotted lines correspondingly) in sodium phosphate buffer (pH 7.4). Extinction coefficients: εC,588 = 5600 L mol−1 cm−1; εZ,389 = 9600 L mol−1 cm−1;33 (B) Arrhenius plots of the quantum yield for the ring-opening and ring-closing reaction. | ||
The ultrafast photochemical reactions of fulgides and fulgimides are well characterized in organic solvents. The ring-opening proceeds on the picosecond timescale25–28 after overcoming a barrier on the S1 excited state potential energy surface.28,29 This makes the reaction sensitive to external factors like temperature, excitation wavelength and solvent polarity. The ring-closing reaction is barrierless (no dependence on the experimental conditions)28,30 and typically occurs on the sub-picosecond timescale.31,32 Both reactions occur without the involvement of long-lived intermediates, which is an advantage over similar systems (e.g. spiropyrans).
Recently, we have reported on the synthesis of a water-soluble dicarboxylic acid indolylfulgimide (WF) (Scheme 1) suitable for applications in aqueous environments.33 WF is thermally very stable in water (phosphate buffer, pH 7.4); after more than 20 days at 37 °C the compound degrades by less than 20%.33 Furthermore, the photochemical stability is also very high for a water-soluble photoswitch. WF is capable of 670 photochemical cycles before reaching 20% degradation.33 For comparison, the recently investigated water-soluble Py-BIPS compound shows 20% degradation in about 35 cycles.13 At present, there are no ultrafast studies on water-soluble fulgides or fulgimides and thus virtually nothing is known about the early photochemistry of those compounds in aqueous solutions. The information on the early photoreaction of fulgides and fulgimides is crucial for the intelligent design of new and improved compounds for extensive application in aqueous environments. In this respect, we have focused our study on the investigation of the ultrafast ring-opening and ring-closing reactions of WF (Scheme 1).
Materials and methods
Sample preparation
The synthesis of the investigated water soluble dicarboxylic acid indolylfulgimide, has been previously described in detail.33 The compound in pure C-form was dissolved for the experiments in 50 mM sodium phosphate buffer (pH 7.4) or in 50 mM sodium carbonate buffer (pH 9.8). The ratio of the different forms, C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z:E, in the photostationary state (PSS) after 405 nm illumination as determined by 1H NMR is 87:6:6.33
:Z:E, in the photostationary state (PSS) after 405 nm illumination as determined by 1H NMR is 87:6:6.33
Steady-state spectroscopy and quantum yield determination
The absorption spectra were recorded on a Specord S600 (Analytik Jena) absorption spectrometer using a fused silica cuvette with 1 cm optical path length. The temperature dependence (10–50 °C) of the quantum yields (QY) for the ring-opening and -closing reactions were determined as described previously in details.28 High power LEDs (ThorLabs, M385L2, M530L2, driver DC4100) were used for the illumination of the Z- and the C-forms. The intensity of the illuminating light was determined using a calibrated light detector (P-9710, Gigahertz-Optik). The PSS530 (530 nm illumination) and the PSS385 (385 nm illumination) were always prepared at 20 °C before transferring the sample to the temperature of interest. The samples were continuously stirred using a magnetic stirrer (Thermo Scientific).The fluorescence spectrum of the WF Z-form was recorded on a FP-8500 fluorimeter (Jasco Analytical Instruments, Tokyo) using a fused silica cuvette with 1 cm optical path length. The fluorescence spectrum of the WF C-form was recorded using a home-built CCD fluorimeter with sensitivity extending up to ∼950 nm using a fused silica with 1 mm optical path length. The fluorescence spectra were corrected for detector sensitivity and self-absorption.
The mid-IR absorption spectra of WF were recorded on a Vertex 80 FTIR-Spectrometer (Bruker, Ettlingen) using 50 μm cuvettes built out of two CaF2 windows. The C → Z (PSSVIS) and the Z → C (PSSUV) conversions were accomplished by illuminating the samples directly in the spectrometer. The PSSVIS was reached by illumination with >520 nm (HgXe arc lamp, Hamamatsu L9588-04, and a colour glass filter OG520, Schott AG), while the PSSUV was reached by illumination with 385 nm (LED, ThorLabs, M385L2).
Sample conditions for the time-resolved experiments
The Z- and the C-form of the dicarboxylic acid indolylfulgimide were prepared at an OD of ∼0.7–1. Fused silica cuvettes were used for the time-resolved experiments. For the Z-form a flow-through cuvette was used (0.5 or 1 mm optical path length), while for the C-form a normal cuvette (1 mm optical path length) that was continuously moved in the plane perpendicular to the direction of the probe pulse propagation was used. The samples were continuously illuminated to keep the PSSVIS (>520 nm, HgXe arc lamp, Hamamatsu L9588-04, and a colour glass filter OG520, Schott AG) or the PSSUV (385 nm, LED, ThorLabs, M385L2).UV-VIS pump–probe spectroscopy
The femtosecond pump–probe setup used for the transient absorption measurements was described in detail previously.28 In short, the set-up is based on an oscillator–amplifier system (Clark, MXRCPA-iSeries, 1 kHz, 775 nm, 150 fs). The 590 nm pump pulses were generated in a two stage NOPA (non-collinear optical parametric amplifier)34,35 and compressed by a prism compressor to ∼40–50 fs (determined with a PulseCheck USB 15 ShortPulse Autocorrelator, APE). The 390 nm pump pulses (150 fs) were produced from the laser fundamental (775 nm) by second harmonic generation. The pulse energy was adjusted to ensure that <10% of the molecules are excited per pulse. Single filament white light (WL) pulses (300–700 nm) were generated by focusing the laser fundamental in a 5 mm thick CaF2 crystal. The detection of the transient signals was performed in referenced mode using the spectrometers described previously.28 The experiments were performed under magic angle conditions (54.7° pump–probe polarization angle difference) to eliminate anisotropic contributions.Time-resolved fluorescence
The femtosecond fluorescence experiments were conducted on a Kerr shutter set-up36,37 based on an oscillator–amplifier system (Tsunami-Spitfire Pro F, Spectra Physics, 1 kHz, 800 nm, 100 fs). The 400 nm excitation pulses for the Z-form were generated from the laser fundamental via second harmonic generation. The Kerr gate pulses (1300 nm) were generated using a home-built two stage OPA.38 The fluorescence was detected using a Spectrograph (Acton Research, SpektraPro 2358) and a CCD camera (Princeton Instruments, Spec-10:400B/LN). The wavelength-dependent time zero dispersion was corrected by using the Sellmeier equation.39,40Data analysis
Global lifetime analysis (GLA)41,42 was used to analyse the fs transient absorption and time-resolved fluorescence data. In GLA the transients at all detection wavelengths are analysed simultaneously with a single set of exponential functions. The coherent artifact contribution at time zero in the transient absorption data was approximated with a function composed of a Gaussian and/or its first and second derivative43 and fitted within the same routine as the GLA.42 The data analysis was performed using OPTIMUS.42Theoretical calculations
The theoretical calculations were performed with the help of Gaussian 0944 using density functional theory (DFT) and time-dependent density functional theory (TD-DFT)45–47 in combination with the BHandHLYP functional48 and the 6-311G* basis set.49,50 Additionally, a polarizable continuum model (PCM)51 for water was applied and four explicit water molecules located in proximity to the carboxylic groups of the molecule were used. Due to the experimental conditions (phosphate buffer, pH 7.4) both carboxylic acid groups are deprotonated and therefore the deprotonated molecule was treated as doubly negatively charged anion.Results and discussion
Stationary spectroscopy and QY determination
The absorption maximum of the C-form of WF dissolved in sodium phosphate buffer (pH 7.4) is located at 588 nm, while the maximum of the Z-form is at 389 nm (Fig. 1). The position of the absorption bands of indolylfulgimides in different solvents (non-polar or polar) depends strongly on the type of the substituents present in the molecule.33,52,53 Nevertheless, a clear solvent dependence can be observed for the absorption of the C-form which shifts strongly to the red side of the spectrum with increasing solvent polarity.The QY of the ring-opening (C → Z) and the ring-closing (Z → C) reactions were measured as a function of temperature. The QY of the ring-closing reaction is ∼7% and is essentially temperature independent (Fig. 1B). Thus, similarly to other fulgides studied in organic solvents,28,30 in aqueous environment this reaction also proceeds without a barrier on the S1 potential energy surface. In contrast, the QY of the ring-opening reaction shows a strong temperature dependence, going from 0.055% at 10 °C to 0.29% at 50 °C (Fig. 1B). Using the Arrhenius plots of the QY (Fig. 1B) and assuming knon-reactive ≫ kreactive,54 the activation energy, Ea, of the ring-opening reaction of WF in water was calculated to be ∼860 cm−1 (∼10 kJ mol−1). This Ea is significantly larger than previously estimated for an indolylfulgimide in toluene and acetonitrile.53 Overall, the QY of both photochromic reactions of WF are strongly reduced as compared to indolylfulgimides studied in organic solvents.53,55,56 An additional QY measurement at higher pH shows that the ring-opening reaction is sensitive to the proton concentration in the solvent (at 20 °C: QYpH 7.4 = 0.15%; QYpH 9.8 = 0.20%), which indicates that the pathway of the ring-opening reaction of WF is affected by the protic nature of the solvent.
The photoinduced interconversion of WF C- and Z-forms could also be monitored via steady-state FTIR spectroscopy (Fig. 2), which gives detailed insight into the rearrangement of the molecular structure. Previously, we have investigated a similar indolylfulgimide in a non-polar solvent and assigned the IR bands to specific vibrational modes of the molecule.26 Despite the different solvent used here and the differences in the substitution pattern, the majority of the IR bands of WF are preserved with a slight frequency shift. Additionally, we performed theoretical calculations using a polarizable continuum model for water and taking into account that under the experimental conditions used here (phosphate buffer, pD 7.4) the two carboxylic groups (pKa typically < 5) of WF are deprotonated. The model included also 4 explicit water molecules positioned in the vicinity of the carboxylic groups (ESI,† Fig. S1). The calculations are in excellent agreement with the experimental data and confirmed the former assignment of the IR bands (Fig. S2, ESI†).
 | ||
| Fig. 2 (A) Infrared absorption spectra of the WF C- and Z-forms dissolved in sodium phosphate buffer (pD 7.4); (B) infrared absorption difference spectra resulting from the ring-opening and -closing reaction. | ||
In the C-form, the frequencies of the asymmetric and the symmetric stretch modes of the CO groups from the succinimide ring are located at 1696 cm−1 and 1763 cm−1, respectively (Fig. 2). As a result of the C → Z ring-opening reaction, which changes the electronic configuration of the succinimide ring, the CO oscillation frequencies shift significantly down to 1682 cm−1 (asymmetric stretch) and 1736 cm−1 (symmetric stretch). In the absolute IR spectra (Fig. 2A) a strong and broad band is observed in the 1550–1650 cm−1 region, which does not change considerably during the C → Z conversion. However, in the difference spectra (Fig. 2B) a clear feature appears at ∼1550 cm−1. Based on the theoretical calculations the 1550 cm−1 band in the difference spectra (Fig. 2B and Fig. S2, ESI†) was assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations of the ring involved in the electrocyclic reaction, while the strong IR absorption band (1550–1650 cm−1) in the absolute spectra (Fig. 2A) was assigned to the asymmetric stretch of the –COO− groups. The rest of the pronounced bands (<1450 cm−1) are mostly due to CC (single and double bond) or NC (single bond) stretching modes from the different rings.
C stretching vibrations of the ring involved in the electrocyclic reaction, while the strong IR absorption band (1550–1650 cm−1) in the absolute spectra (Fig. 2A) was assigned to the asymmetric stretch of the –COO− groups. The rest of the pronounced bands (<1450 cm−1) are mostly due to CC (single and double bond) or NC (single bond) stretching modes from the different rings.
Ultrafast ring-opening reaction (C → Z)
The ultrafast ring-opening reaction of the water-soluble indolylfulgimide (WF) was investigated using transient absorption spectroscopy with ∼50 fs time resolution (Fig. 3A). The C → Z reaction was induced by excitation at the absorption maximum (590 nm) of the C-form. The transient absorption data is dominated by the presence of strong excited state absorption (ESA) signal due to the S1 → Sn transitions covering the complete detection window (Fig. 3A). In the range above 500 nm the ESA overlaps with the ground state bleach signal (GSB). The GSB has the same spectral shape as the C-form absorption spectrum (Fig. 1A, maximum at ∼590 nm) but with negative sign and thus partially compensates the ESA in this spectral range. This compensation causes a reduced amplitude in the ESA spectrum around 590 nm. An additional ESA band, associated with another S1 → Sn transition, is present below 470 nm. | ||
| Fig. 3 Results from the investigation of the C → Z ring-opening reaction of WF dissolved in sodium phosphate buffer (pH 7.4). (A) Transient absorption data after 590 nm excitation. (B) Decay-associated spectra (DAS) obtained from the global analysis (see Materials and methods) of the transient absorption data. | ||
The decay-associated spectra (DAS) resulting from the global lifetime analysis (GLA)41,42 is composed of four lifetimes and a non-decay component (inf) (Fig. 3B). The 0.06 ps and the 0.4 ps components show typical positive (600–650 nm) – negative (550–570 nm) amplitude features, which are due to the shift of the corresponding band (in this case ESA around 600 nm) to higher energies. In this respect, those two lifetime components in the ultrafast kinetics of the C-form can be assigned to the relaxation of the excited molecule from the Franck–Condon region and to solvent reorganization. The dominant third lifetime component (1.8 ps) is characterized by an all-positive DAS, reflecting the decay of the ESA bands and the repopulation of the ground state. The fourth lifetime component (∼10 ps), associated with the vibrational cooling of the ground state, shows only a minor amplitude contribution. The non-decaying component (inf) corresponds to the final absorption difference spectrum. This spectrum is nearly flat for the C-form, which reflects the low amount of product formation. This result is explained by the relatively low QY of the ring-opening reaction (∼0.15% at 23 °C), which does not permit the detection of the product formation in the transient absorption experiment.
Overall, the ultrafast C → Z ring-opening reaction of WF in water shows similar dynamics to previously studied indolylfulgimides. Nevertheless, an important solvent effect arises from the comparison with the previous studies. While the change from non-polar (tetrachloroethylene or toluene)27,53 to polar aprotic solvent (acetonitrile)31,35,41 leads to an increase of the excited state lifetime of the C-form (2 ps → 3 ps), an opposite trend is detected in water (polar protic solvent) where the excited state lifetime (1.8 ps) becomes even shorter than in non-polar solvents. Considering the significantly lower ring-opening QY in water and the increased energy barrier along the reaction pathway, the shortening of the excited state lifetime could be attributed to an increased preference of the internal conversion pathway. Our theoretical calculations for the relaxed S0 and S1 structures of the C-form showed that the dipole moment of the molecule in the excited state (∼33.5 D) is larger than in the ground state (∼26.2 D). Hence, the excited state of the C-form is stabilized in polar solvents. However, the short excited state lifetime observed in the present experiments with WF indicates that due to the protic nature of water an additional relaxation pathway may play a role. Previous theoretical calculations indicated that the excited state dynamics of the C-form is governed by the interplay of two conical intersections, where only one has a contribution to the ring-opening reaction, while the other one leads to fast deactivation into the C-form ground state.57,58 Within this conical intersection picture the shortened excited state lifetime of the C-form in protic solvents could be caused either by the opening of a new proton transfer reaction channel or an improved accessibility of the conical intersection leading to the C-form ground state.
We investigated the changes in the partial charge distributions in the C-form of the WF molecule occurring after the transition to the excited state to identify potential positions for proton transfer reactions (Table S1, ESI†). Interestingly, the typical candidate for excited state proton transfer reactions (the O atoms) showed only minor photoinduced changes in their partial charges. The largest effects were observed for the N atoms of the indolyl group and the succinimide ring (positions 9 and 15) and the C atoms of the photochromic ring III (positions 8, 10, and 11) and at the succinimide ring (position 16) (Fig. S3, ESI†).
Coherent oscillations in the ultrafast transient absorption signals of the C-form of indolylfulgides28,30 and indolylfulgimides53 have been detected previously. Such oscillations are present also in the transient absorption data from WF in water (clearly visible in the red edge of the ESA, Fig. 3A and in the single transients, Fig. 4A). The Fourier analysis of the oscillatory pattern shows that the dominant frequency is ∼80 cm−1 (Fig. 4B). Several other higher frequencies could also be resolved (∼170, ∼290, ∼410, ∼530 cm−1). Recently, for an indolylfulgide, we have showed that the observed coherent oscillation (∼80 cm−1) is linked to vibrational wavepacket motion on the excited state potential energy surface.28 The theoretical analysis for this indolylfulgide indicated that the motion is due to a vibrational mode contributing significantly to the ring-opening reaction coordinates.28 The same dominant frequency appears here also for the WF. Consequently, this vibrational mode is universally involved in the reaction dynamics of the C-form of fulgides.
 | ||
| Fig. 4 (A) Coherent oscillations in the single absorption difference transients recorded after excitation (590 nm) of the C-form of WF in sodium phosphate buffer (pH 7.4). (B) Frequency map obtained from the Fourier analysis of the residuals from the multi-exponential fit of the transient absorption data. | ||
Ultrafast ring-closing reaction (Z → C)
Transient absorption (Fig. 5A, 390 nm excitation) as well as time-resolved fluorescence spectroscopy (Fig. 5C, 400 nm excitation) were employed to investigate the ultrafast ring-closing reaction of the water-soluble fulgimides (WF). The excitation wavelength in both experiments was tuned near the absorption maximum of the open Z-form (see Materials and methods for the Experimental details). The transient absorption data (Fig. 5A) is dominated by the strong GSB of the Z-form (∼400 nm), as well as by two strong ESA bands (<370 nm and >440 nm). In addition, on the early timescale (<500 fs) and above 570 nm, also stimulated emission (SE) contribution is discernible. The formation of the C-form product as a result of the Z-form photoinduced cyclization is evident on the timescale >10 ps, where both the remaining GSB band (∼400 nm) due to the non-recovered Z-form, as well as the newly formed C-form absorption band (∼590 nm) are present.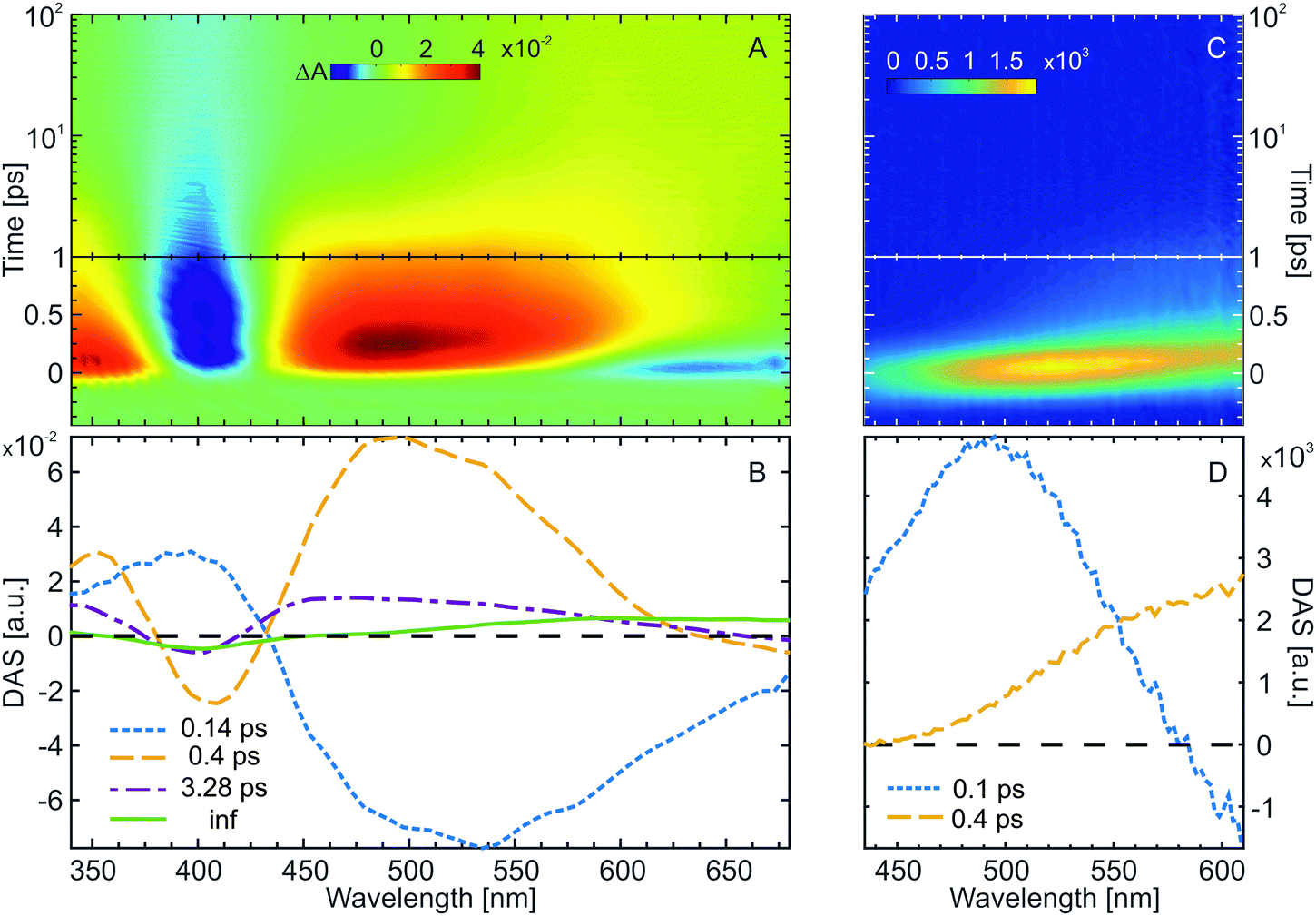 | ||
| Fig. 5 Results from the investigation of the Z → C ring-closing reaction of WF dissolved in sodium phosphate buffer (pH 7.4). (A) Transient absorption data after excitation at 390 nm. (B) DAS obtained from the GLA of the transient absorption data. (C) Time-resolved fluorescence data after excitation at 400 nm. (D) DAS obtained from the GLA of the time-resolved fluorescence data. | ||
The GLA of the Z-form transient absorption data yielded three lifetimes and a non-decaying component (inf) (Fig. 5B). The shortest lifetime component (0.14 ps) is associated with the relaxation of the excited Z-form from the Franck–Condon region and towards the conical intersection to the ground state. The DAS of this lifetime is a complex mixture reflecting spectral shifts of the ESA and the SE bands. The main decay of the ESA and recovery of the GSB occurs with 0.4 ps lifetime. With this lifetime, also the rise of the C-form product absorption can be observed. Relaxation of the excited state is followed by vibrational cooling in the ground state (3.3 ps lifetime). The hot ground state band (broad absorption peaking at 450 nm) shifts with the 3.3 ps lifetime to 400 nm to partially repopulate the relaxed ground state and further reveals on the red side of the spectral range (>500 nm) the absorption of the C-form product.
The assignment of the dynamics in the transient absorption data is confirmed by time-resolved fluorescence (Fig. 5C). The spectrally very broad and ultrashort fluorescence has an initial maximum at ∼530 nm, which quickly shifts to the red and decays on the timescale <2 ps. The spectral properties of the Z-form time-resolved fluorescence match the stationary fluorescence spectrum (Fig. 1A). Two lifetimes were sufficient to fit the time-resolved fluorescence of the Z-form. The first one, ∼100 fs, accounts for the ultrafast red-shift of the fluorescence due to the departure of the excited Z-form from the Franck–Condon region. The second one, ∼400 fs, represents the decay of the fluorescence and thus the decay of the excited state. Therefore, the conclusion drawn from the time-resolved fluorescence and the transient absorption data of the Z-form are in excellent agreement. Furthermore, the excited state reaction dynamics of the Z-form of the water-soluble indolylfulgimide is very similar to what was previously observed for a related compound dissolved in acetonitrile.31,59 The main difference for those two compounds and solvents is found in the vibrational cooling lifetime, which is nearly two times faster for WF.
Conclusions
In the current work, we present the first, to our knowledge, investigation of the ultrafast photoconversion dynamics of a water-soluble fulgimide (WF). Despite the reduced QY of this indolylfulgimide in water, as compared to similar compounds dissolved in organic solvents, WF shows very high thermal- and photostability. The kinetics of the ring-closing Z → C reaction is mostly similar to the kinetics of indolylfulgimides in polar solvents. The conical intersection to the ground state for this reaction is readily accessible as there is no energetic barrier on the relaxation pathway from the Franck–Condon region (Scheme 2). Thus the reaction is generally ultrafast (∼0.4 ps) and is not affected significantly by changes in the environment. The C → Z ring-opening reaction in water, on the other hand, is faster than in both polar and non-polar organic solvents. Surprisingly, this is an opposite trend as typically the reaction is slowed down in polar solvents due to stabilization of the polar excited state. Evidently, in protic solvents like water, excited state proton transfer reactions play a role in the relaxation kinetics of photoexcited fulgimides. Such reactions effectively quench the excited state, which reduces the QY of the photoconversion. In this respect, a major solvent effect on the ultrafast kinetics of indolylfulgimides emerges from our study, which should serve as a basis for future optimizations of the WF structure.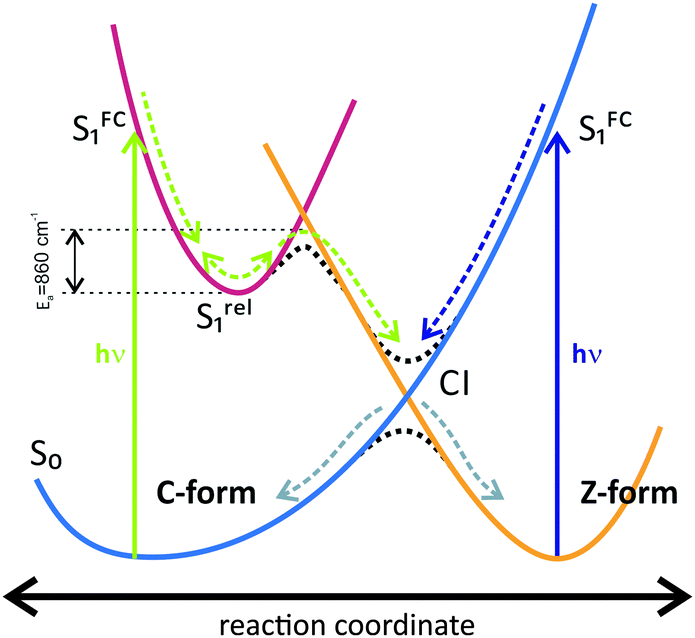 | ||
| Scheme 2 Reaction scheme for the ring-opening and -closing reactions of the water-soluble WF. | ||
Acknowledgements
Chavdar Slavov and Josef Wachtveitl acknowledge DFG (Grant WA 1850/4-1).References
- G. H. Brown, Photochromism, Wiley, New York, 1971 Search PubMed.
- Photochromism: Molecules and Systems, ed. H. Dürr and H. Bouas-Laurent, Elsevier, Amsterdam, 2003 Search PubMed.
- N. Tamai and H. Miyasaka, Chem. Rev., 2000, 100, 1875–1890 CrossRef CAS PubMed.
- R. H. El Halabieh, O. Mermut and C. J. Barrett, Pure Appl. Chem., 2004, 76, 1445–1465 CrossRef CAS.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- Y. Yokoyama, Chem. Rev., 2000, 100, 1717–1739 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- J. Garcia-Amorós, M. Díaz-Lobo, S. Nonell and D. Velasco, Angew. Chem., Int. Ed., 2012, 51, 12820–12823 CrossRef PubMed.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS PubMed.
- R. Matsushima and H. Sakaguchi, J. Photochem. Photobiol., A, 1997, 108, 239–245 CrossRef CAS.
- T. Stafforst and D. Hilvert, Chem. Commun., 2009, 287–288 RSC.
- X. Chen, N. I. Islamova, R. V. Robles and W. J. Lees, Photochem. Photobiol. Sci., 2011, 10, 1023–1029 CAS.
- J. Kohl-Landgraf, M. Braun, C. Özçoban, D. P. N. Gonçalves, A. Heckel and J. Wachtveitl, J. Am. Chem. Soc., 2012, 134, 14070–14077 CrossRef CAS PubMed.
- O. Sadovski, A. A. Beharry, F. Z. Zhang and G. A. Woolley, Angew. Chem., Int. Ed., 2009, 48, 1484–1486 CrossRef CAS PubMed.
- M. A. Cardona and D. C. Magri, Tetrahedron Lett., 2014, 55, 4559–4563 CrossRef CAS.
- C. Özçoban, T. Halbritter, S. Steinwand, L.-M. Herzig, J. Kohl-Landgraf, N. Askari, F. Groher, B. Fürtig, C. Richter, H. Schwalbe, B. Suess, J. Wachtveitl and A. Heckel, Org. Lett., 2015, 17, 1517–1520 CrossRef PubMed.
- S. Heng, C. A. McDevitt, D. B. Stubing, J. J. Whittall, J. G. Thompson, T. K. Engler, A. D. Abell and T. M. Monro, Biomacromolecules, 2013, 14, 3376–3379 CrossRef CAS PubMed.
- S. Steinwand, T. Halbritter, D. Rastädter, J. M. Ortiz-Sánchez, I. Burghardt, A. Heckel and J. Wachtveitl, Chem. – Eur. J., 2015, 21, 15720–15731 CrossRef CAS PubMed.
- F. Renth, R. Siewertsen and F. Temps, Int. Rev. Phys. Chem., 2012, 32, 1–38 CrossRef.
- Y. Yokoyama and K. Takahashi, Chem. Lett., 1996, 1037–1038 CrossRef CAS.
- N. I. Islamova, X. Chen, S. P. Garcia, G. Guez, Y. Silva and W. J. Lees, J. Photochem. Photobiol., A, 2008, 195, 228–234 CrossRef CAS.
- A. Kaneko, A. Tomoda, M. Ishizuka, H. Suzuki and R. Matsushima, Bull. Chem. Soc. Jpn., 1988, 61, 3569–3573 CrossRef CAS.
- M. W. Berns, T. Krasieva, C. H. Sun, A. Dvornikov and P. M. Rentzepis, J. Photochem. Photobiol., B, 2004, 75, 51–56 CrossRef CAS PubMed.
- F. D. Jochum, F. R. Forst and P. Theato, Macromol. Rapid Commun., 2010, 31, 1456–1461 CrossRef CAS PubMed.
- H. Port, P. Gärtner, M. Hennrich, I. Ramsteiner and T. Schock, Mol. Cryst. Liq. Cryst., 2005, 430, 15–21 CrossRef CAS.
- F. O. Koller, W. J. Schreier, T. E. Schrader, A. Sieg, S. Malkmus, C. Schulz, S. Dietrich, K. Rück-Braun, W. Zinth and M. Braun, J. Phys. Chem. A, 2006, 110, 12769–12776 CrossRef CAS PubMed.
- S. Malkmus, F. O. Koller, B. Heinz, W. J. Schreier, T. E. Schrader, W. Zinth, C. Schulz, S. Dietrich, K. Rück-Braun and M. Braun, Chem. Phys. Lett., 2006, 417, 266–271 CrossRef CAS.
- C. Slavov, N. Bellakbil, J. Wahl, K. Mayer, K. Rück-Braun, I. Burghardt, J. Wachtveitl and M. Braun, Phys. Chem. Chem. Phys., 2015, 17, 14045–14053 RSC.
- S. Draxler, T. Brust, S. Malkmus, J. A. DiGirolamo, W. J. Lees, W. Zinth and M. Braun, Phys. Chem. Chem. Phys., 2009, 11, 5019–5027 RSC.
- T. Brust, S. Draxler, A. Popp, X. Chen, W. J. Lees, W. Zinth and M. Braun, Chem. Phys. Lett., 2009, 477, 298–303 CrossRef CAS PubMed.
- B. Heinz, S. Malkmus, S. Laimgruber, S. Dietrich, C. Schulz, K. Rück-Braun, M. Braun, W. Zinth and P. Gilch, J. Am. Chem. Soc., 2007, 129, 8577–8584 CrossRef CAS PubMed.
- M. Handschuh, M. Seibold, H. Port and H. C. Wolf, J. Phys. Chem. A, 1997, 101, 502–506 CrossRef CAS.
- X. Chen, N. I. Islamova, S. P. Garcia, J. A. DiGirolamo and W. J. Lees, J. Org. Chem., 2009, 74, 6777–6783 CrossRef CAS PubMed.
- T. Wilhelm, J. Piel and E. Riedle, Opt. Lett., 1997, 22, 1494–1496 CrossRef CAS PubMed.
- E. Riedle, M. Beutter, S. Lochbrunner, J. Piel, S. Schenkl, S. Spörlein and W. Zinth, Appl. Phys. B: Lasers Opt., 2000, 71, 457–465 CrossRef CAS.
- B. Schmidt, S. Laimgruber, W. Zinth and P. Gilch, Appl. Phys. B: Lasers Opt., 2003, 76, 809–814 CrossRef CAS.
- P. Trojanowski, PhD dissertation, Goethe University, 2015.
- G. Cerullo and S. De Silvestri, Rev. Sci. Instrum., 2003, 74, 1–18 CrossRef CAS.
- W. Sellmeier, Ann. Phys., 1871, 219, 272–282 CrossRef.
- D. Meschede, Optics, Light and Lasers, Wiley-VCH, 2007 Search PubMed.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- C. Slavov, H. Hartmann and J. Wachtveitl, Anal. Chem., 2015, 87, 2328–2336 CrossRef CAS PubMed.
- S. A. Kovalenko, A. L. Dobryakov, J. Ruthmann and N. P. Ernsting, Phys. Rev. A: At., Mol., Opt. Phys., 1999, 59, 2369–2384 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- M. E. Casida, Recent Advances in Density Functional Methods, World Scientific, 1995 Search PubMed.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- N. I. Islamova, X. Chen, J. A. DiGirolamo, Y. Silva and W. J. Lees, J. Photochem. Photobiol., A, 2008, 199, 85–91 CrossRef CAS PubMed.
- T. Brust, S. Malkmus, S. Draxler, S. A. Ahmed, K. Rück-Braun, W. Zinth and M. Braun, J. Photochem. Photobiol., A, 2009, 207, 209–216 CrossRef CAS.
- T. Brust, S. Draxler, S. Malkmus, C. Schulz, M. Zastrow, K. Rück-Braun, W. Zinth and M. Braun, J. Mol. Liq., 2008, 141, 137–139 CrossRef CAS.
- F. O. Koller, W. J. Schreier, T. E. Schrader, S. Malkmus, C. Schulz, S. Dietrich, K. Rück-Braun and M. Braun, J. Phys. Chem. A, 2008, 112, 210–214 CrossRef CAS PubMed.
- M. A. Wolak, N. B. Gillespie, C. J. Thomas, R. R. Birge and W. J. Lees, J. Photochem. Photobiol., A, 2001, 144, 83–91 CrossRef CAS.
- D. Geppert, L. Seyfarth and R. de Vivie-Riedle, Appl. Phys. B: Lasers Opt., 2004, 79, 987–992 CrossRef CAS.
- A. Nenov, W. J. Schreier, F. O. Koller, M. Braun, R. de Vivie-Riedle, W. Zinth and I. Pugliesi, J. Phys. Chem. A, 2012, 116, 10518–10528 CrossRef CAS PubMed.
- S. Draxler, T. Brust, S. Malkmus, F. O. Koller, B. Heinz, S. Laimgruber, C. Schulz, S. Dietrich, K. Rück-Braun, W. Zinth and M. Braun, J. Mol. Liq., 2008, 141, 130–136 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures. See DOI: 10.1039/c5cp06866e |
| This journal is © the Owner Societies 2016 |