
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational design of donor-bridge-acceptor systems exhibiting pronounced quantum interference effects†
Natalie
Gorczak
,
Nicolas
Renaud
,
Elena
Galan‡
,
Rienk
Eelkema
,
Laurens D. A.
Siebbeles
and
Ferdinand C.
Grozema
*
Department of Chemical Engineering, Delft University of Technology, Delft, The Netherlands. E-mail: f.c.grozema@tudelft.nl
First published on 5th February 2016
Abstract
Quantum interference is a well-known phenomenon that dictates charge transport properties of single molecule junctions. However, reports on quantum interference in donor-bridge-acceptor molecules are scarce. This might be due to the difficulties in meeting the conditions for the presence of quantum interference in a donor-bridge-acceptor system. The electronic coupling between the donor, bridge, and acceptor moieties must be weak in order to ensure localised initial and final states for charge transfer. Yet, it must be strong enough to allow all bridge orbitals to mediate charge transfer. We present the computational route to the design of a donor-bridge-acceptor molecule that features the right balance between these contradicting requirements and exhibits pronounced interference effects.
1 Introduction
Photoinduced charge transfer is at the heart of numerous biological processes and technological applications, such as (artificial) photosynthesis,1–3 DNA damage and repair,4–8 organic solar cells,9,10 molecular computation and biosensing.11–13 Synthetic donor-bridge-acceptor (DBA) systems have therefore been used extensively for systematic studies of charge (electron or hole) transfer.14–19 These DBA systems consist of three covalently bound moieties: the charge donor where the transferring charge is generated by absorption of light, the bridge through which the charge passes, and the acceptor where the charge arrives. The vast majority of studies focussed on demonstrating the key parameters that govern charge transfer. Bridge length, energy barrier, and driving force have been identified as the most important ones.20–22 Recently, other parameters like bridge conjugation, and the position at which donor, bridge, and acceptor are connected to each other received attention.23–25The common ground for all these studies is the assumption that the transferring charge is initially localised on the charge donor and, after one or more transfer steps, arrives at the charge acceptor. However, this assumption is only rarely tested. Previously, we have demonstrated that the delocalisation of the initial state strongly affects the distance dependence of electron transfer.26 Notably, the delocalisation was not expected, given that the absorbance spectra of the DBA compounds were virtually identical to the absorbance of the neat donor. The question whether the initial and final state are localised is very important since the distance dependence of the charge transfer rate is often considered as an indication of the charge transfer mechanism that is operative. A weak dependence on distance is usually considered to be indicative of hopping transport, while a strong dependence points to single step tunnelling.27,28 We argue that a careful consideration of the initial state is essential to support such claims. Apart from this fundamental interest, localised initial and final states are also crucial for the design of quantum interference based molecular switches. The phenomenon of quantum interference has been proposed to be a powerful approach to control charge transfer through molecular bridges as large ON/OFF ratios are expected when switching off destructive interference.29–36 Destructive interference is usually met in cross-conjugated molecular bridges.37,38 A switching mechanism could be realised for instance by an electrochemical reduction from a cross-conjugated anthraquinone bridge to a linearly conjugated form,39 or generally by charging a cross-conjugated bridge.40 Quantum interference effects in single molecule junctions have been experimentally demonstrated a number of times.41–45 Recently, also the electrochemical switching of such an anthraquinone molecular switch in a single molecule junction has been realised experimentally.46
Experimental demonstrations of the occurrence of quantum interference in DBA systems are scarce.23,47,48 This could be due to the difficulty in designing suitable DBA systems. The design rules are evident considering that quantum interference originates from interfering tunnelling pathways offered by the bridge orbitals. This implies that charge transfer must follow a coherent superexchange mechanism. Therefore, a suitable DBA system has to fulfil two requirements:
1. Initial and final states must be localised on the charge donor and acceptor, respectively, and
2. All bridge orbitals must take part in the transfer mechanism.
For the first condition to hold, the donor and acceptor state must be essentially decoupled from the bridge states. However, the second condition requires a certain degree of coupling between the donor/acceptor state and all bridge states. Admittedly, the two requirements contradict each other, which makes clear that a suitable DBA system must be in an intermediate regime between too weak and too strong coupling. In our earlier work on hole transfer in DBA systems containing linearly and cross-conjugated biphenyl bridges (Fig. 1) we demonstrated that the second condition demands a certain degree of asymmetry in the DBA system.24 Beforehand, a relatively high hole transfer rate constant was expected for the linearly conjugated DBA molecule (pp in Fig. 1) because of constructive quantum interference. For the two cross-conjugated DBA systems (mp and mm in Fig. 1) slow hole transfer was expected due to destructive quantum interference. Yet we showed, that the symmetry relation between the hole donor (PDI) and the bridge in pp and mp caused the exclusion of certain tunnelling pathways through the bridge. This pathway selection obscured the expected interference effects and led to an equally low rate constant in pp and mp, which was even lower than in mm. This exclusion of certain pathways through the bridge resulting from symmetry considerations is not only important for observing quantum interference effects, but can also explain large differences in the charge transfer rate when an acceptor is coupled to a bridge on different positions. This was shown by Shoer et al.25 for DBA molecules where a PDI acts as the acceptor. Symmetric coupling to the imide nitrogen (similar to the structure in Fig. 1) leads to much lower charge transfer rates than coupling in an asymmetric way at the so-called peri-positions. Therefore, the symmetry arguments that we focus on here are of general importance in determining the efficiency of charge transfer in DBA systems.
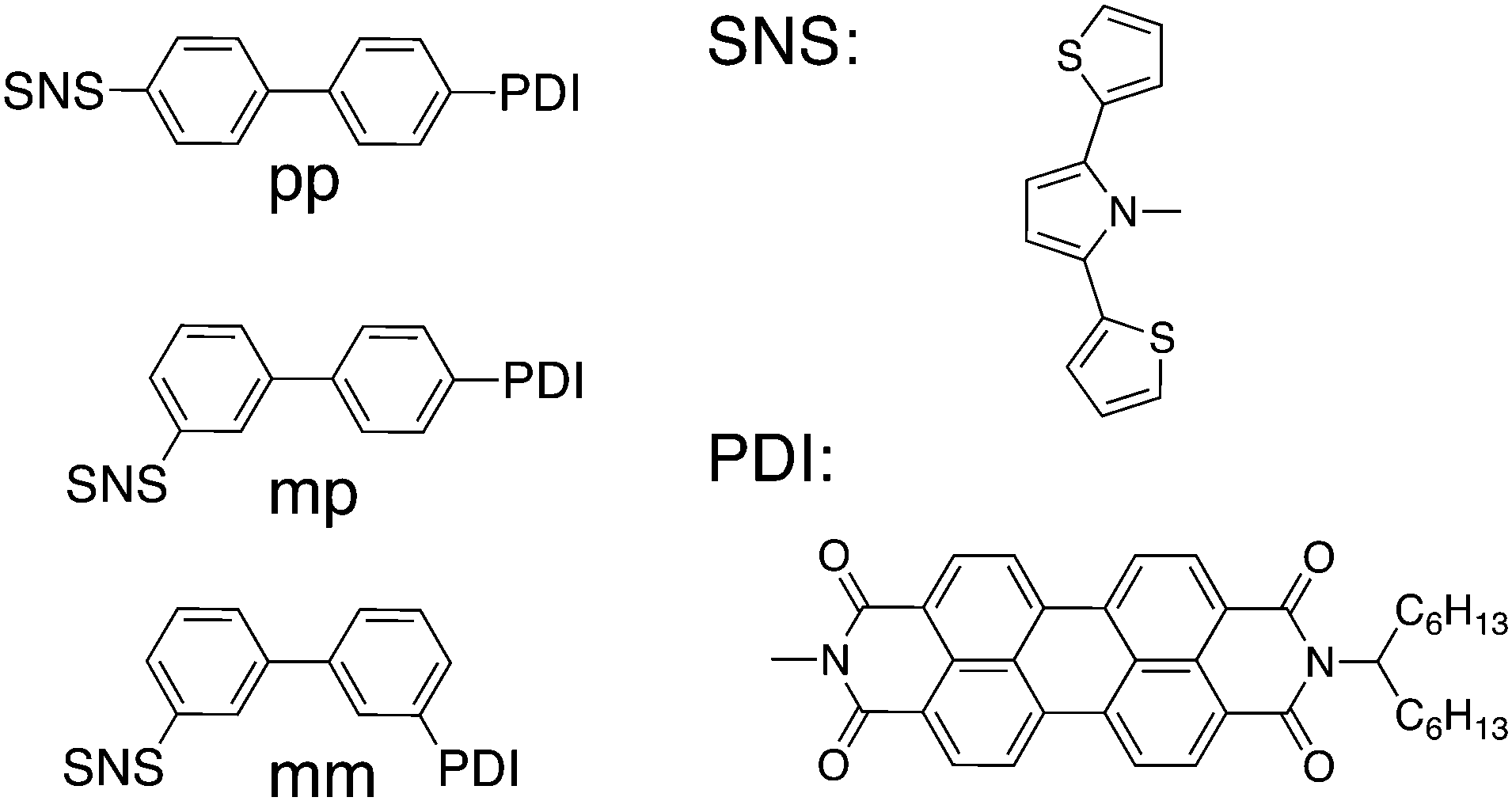 | ||
| Fig. 1 Chemical structures of the linearly and cross-conjugated DBA system previously investigated with the chemical structures of the hole donor and acceptor referred to as PDI and SNS. | ||
In this paper, we present the computational design of a linearly and cross-conjugated DBA system exhibiting prominent interference effects. By using the same biphenyl bridges as in pp and mp and screening for an appropriate hole donor and acceptor, we also emphasise how substantial the thorough choice of initial and final states is. The screening follows the two design rules by examining the initial and final state and comparing the effective electronic coupling for the linearly and the cross-conjugated bridge.
2 Computational method
Starting off with the linearly conjugated biphenyl bridge and the previous hole acceptor SNS, various hole donors were tested with regard to the localisation of the initial state. The degree of localisation was defined as the contribution of the highest occupied fragment orbital (HOFO) of the hole donor to the initial state. To this end, time-dependent density functional theory (TD-DFT) calculations were performed on the ground state geometries of the DBA molecule using the fragment orbital approach of the Amsterdam Density Functional software.49 This means the molecular orbitals of the donor, bridge, and acceptor radical fragment molecules were used as basis set.50 These fragment orbitals were obtained from electronic structure calculations of the isolated radical fragment molecules at the DFT level of theory with the DZP basis set and M06-2X functional.51 The M06-2X functional was chosen since this meta-hybrid functional is known to perform very well for the calculation of excitation energies by TD-DFT, especially for large conjugated molecules and in cases where charge transfer contributions play an important role.52 The latter is here of prime importance since the charge transfer character in the initially excited state is crucial in this work. The calculation of the electronic coupling is relatively insensitive to the functional used. Although there are differences in the magnitude, the differences between the linearly and cross-conjugated DBA molecules that are important for this work are reproduced very well. This is shown in ESI,† where the charge transfer character and the effective electronic couplings are compared for two functionals.The ground state geometries of the different DBA molecules were obtained (DZP/M06-2X) optimising the ground state geometries of the isolated donor-bridge, bridge-acceptor, and the isolated bridge fragments and assembling these fragments to form the DBA molecules. This way of generating the structures because the optimisation of the full DBA molecules is very time-consuming in some cases since the potential energy surfaces can be rather flat where the dihedral angles between the donor and the bridge and the bridge and the acceptor are involved. Additionally, it should be noted that the exact dihedral angles do not alter the symmetry arguments described below, since the cancellation of electronic couplings is independent of the dihedral angles.24
After finding the appropriate hole donor, various hole acceptors were examined with regard to the localisation of the final state. The degree of final state localisation was measured in terms of the contribution of the HOFO of the hole acceptor that makes up the HOMO of the entire DBA system. This contribution was obtained from DFT calculations using the same fragment orbital approach as described above. For the linearly conjugated DBA molecules with localised initial and final states, effective electronic couplings between the HOFO of the hole donor and the HOFO of the hole acceptor were calculated. These couplings should be above 1 meV, which is one order of magnitude larger than in the previous DBA system pp and regarded as the limit of accuracy that can be obtained with DFT. Finally, for the set of donor and acceptor molecules with localised initial and final states and considerable effective electronic coupling, the effective electronic couplings were also calculated using the cross-conjugated bridge. In order to examine whether the effective electronic couplings are dictated by quantum interference, the calculated values were compared between the linearly and the cross-conjugated DBA systems. The effective electronic coupling was calculated according to the method from our previous publication.24 It is expressed as53
 | (1) |
The various direct couplings VXY and the energies of the i-th bridge orbitals HBiBi in eqn (1) were extracted from the Fock matrix of the DBA molecules obtained with DFT (M06-2X) using the fragment orbital approach described above.50 The coupling between the fragment orbitals and their energy is then given by the off-diagonal and the diagonal matrix elements of the Fock matrix: HXY = 〈X|H|Y〉 and HBiBi = 〈Bi|H|Bi〉. Because the fragment orbitals are in general not orthogonal, the finale value for the electronic coupling between the fragment orbitals X and Y was determined as VXY = HXY − 0.5SXY(HXX + HYY), where S is the overlap matrix.
By using eqn (1), we explicitly sum over the contributions of all bridge orbitals as tunnelling pathways, which inherently accounts for the possibility of quantum interference effects. At the same time, performing the summation over only a subset of bridge orbitals provides insight into which orbitals are most relevant for hole transfer in a given DBA system. For instance, including only the π-orbitals of the biphenyl bridge in eqn (1) allows us to examine whether the overall effective coupling is dominated by the π-orbitals. In the fragment orbital method used here it is assumed that small changes in the geometry that occur on charge transfer or photoexcitation do not lead to significant changes in the charge transfer integrals. This approach is very common54 and will certainly be sufficiently accurate to describe the large differences in the effective charge transfer integrals that we are interested in here (roughly an order of magnitude).
3 Results and discussion
As mentioned in the introduction, we have previously demonstrated a linearly and cross-conjugated DBA system that did not feature the commonly expected trend in hole transfer rate due to quantum interference effects.24 This was attributed to the orbital symmetry of the previously used hole donor and hole acceptor (PDI and SNS) in relation to the biphenyl bridge. Therefore, we start off our search for a suitable hole donor and hole acceptor by breaking the symmetry of PDI and SNS.3.1 Hole donor candidates
There are two asymmetric ways to connect a PDI to a molecular bridge, namely at the bay or the headland position (see PDIbay and PDIhead in Fig. 2). The calculated excitation spectra are shown in Fig. 3(a). According to the results of the TD-DFT calculations, the lowest excitation in PDIbay is described by a one electron transition from the HOMO−1 of the DBA molecule to the LUMO (Fig. 3(b)). This has also been obtained in the case of PDIim. However, by contrast to PDIim the HOMO−1 of PDIbay is delocalised over PDI and biphenyl as depicted in Fig. 3(b): the highest occupied fragment orbital (HOFO) of PDI contributes to the HOMO–1 with 78% (see Table 1). This delocalisation occurs despite the fact that the energy difference between the HOFOs of PDI and biphenyl is similar for PDIbay and PDIim (Fig. 4). The difference could be explained by the direct coupling VDBHOFO, which is 0 eV for PDIim due to symmetry but is 0.24 eV for PDIbay. When PDI is connected at the headland position, the initial state is more delocalised than when connected at the bay position with a contribution of the HOFO of PDI of 66%. The stronger delocalisation can not be explained by the direct coupling VDBHOFO since the coupling is reduced by a factor of 5 (while the energy difference is only reduced by a factor of 1.4). In this case the lowest excitation is characterised by an electron transition from the HOMO−1 of the DBA molecule to the LUMO and a transition from the HOMO−2 to the LUMO. As shown in Fig. 3(b), the HOMO−2 of PDIhead is delocalised over biphenyl and SNS. The example of the imide, bay, and headland substitution of PDI to biphenyl makes clear that although PDI is a widely used hole donor, it might be unsuitable for some applications because of delocalisation of the initial state. The reason lies partially in the differences in the electronic coupling but are mostly caused by subtle differences in the relative energies of the different electronic states on the PDI and the bridge. It is hard to predict these differences based on the electronic properties of the isolated molecular fragments since they are significantly influenced by the coupling to neighbouring parts in the DBA system. A perylene derivative that has similar electron accepting properties as PDI but exhibits additional possibilities for asymmetric attachment to a biphenyl bridge is perylene-monoimide connected to the biphenyl as shown in PMI in Fig. 2. When analysing the initial excited state in the same way as above, it turns out that PMI fulfils the requirement of a localised initial state while maintaining a relatively large electronic coupling to the biphenyl. The latter is caused by the direct asymmetric coupling between the perylene core and the biphenyl. The localisation is also shown to be relatively robust under variations of the dihedral angle between the hole donor and bridge (see Table 1). The contribution of the HOFO of PMI to the initial state merely decreases from 94% in the ground state geometry (69°) to 81% at a fictional dihedral angle of 0°. PMI is therefore an excellent example of a hole donor that fulfils the requirements that we have defined in the introduction and it will be used in the DBA systems that are discussed below. | ||
| Fig. 2 Chemical structures of the linearly conjugated DBA system with hole donor candidates. | ||
 | ||
| Fig. 3 (a) Optical excitation of PDIbay, PDIhead, and PMI using TD-DFT (DZP/M06-2X) and (b) the orbitals that are involved in the lowest excitation. | ||
 | ||
| Fig. 4 Energy levels of the HOFO of the hole donor candidates and the HOFO of the linearly conjugated biphenyl bridge in the DBA molecules (solid line) compared to the isolated fragments (dashed line). | ||
3.2 Hole acceptor candidates
The requirements for the hole acceptor in the DBA system are the same as those for the hole donor: a reasonably large electronic coupling to the bridge combined with a localised final state. Just as the PDI, the SNS moiety used in our previous work lacks the electronic coupling to the bridge due to its symmetry with respect to the bond axis. In order to overcome this we introduced asymmetry in the SNS by exchanging one thiophene with a phenyl in SNPh, or by connecting the SNS in an asymmetric fashion to the biphenyl at the thiophene ring in SNSas (Fig. 5). Both variations exhibit a localised character of the final state; the hole acceptor HOFO contributes with 97% to the final state for SNPh and with 84% for SNSas (Table 2). However, when considering the electronic coupling, SNPh is not suitable because the calculated effective coupling for hole transfer Jeff is only 0.4 meV. This is essentially no improvement to 0.1 meV that was obtained for the previous hole acceptor SNS. Therefore, the exchange of one thiophene by a phenyl does not sufficiently break the symmetry. In the case of SNSas, a look at the energy level alignment in Fig. 6 explains why this hole acceptor is not suitable: upon connection of the SNS at the thiophene to the biphenyl the energy of the HOFO of the hole acceptor shifts below the energy of the HOFO of the hole donor. This essentially reverses the roles of donor and acceptor. Again, this arrangement of the different energy levels in the DBA molecule are almost impossible to predict on basis of the properties of the individual fragments and calculation such as the ones described here are essential in this regard.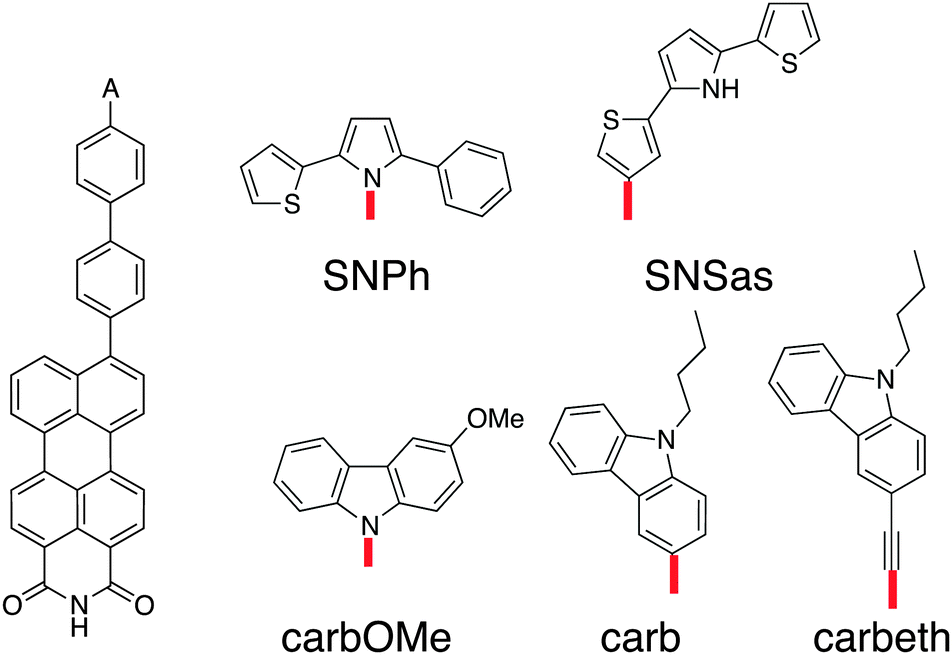 | ||
| Fig. 5 Chemical structures of the linearly conjugated DBA system with the hole donor perylene-monoimide and the hole acceptor candidates. | ||
| χ AB (°) | HOFO contribution (%) | J eff (meV) | |
|---|---|---|---|
| a HOFO of hole acceptor shifted below HOFO of hole donor perylene-monoimide. | |||
| SNS | 60 | 98 | 0.1 |
| SNPh | 58 | 97 | 0.4 |
| SNSas | 42 | 84 | |
| carbOMe | 53 | 90 | 27 |
| carb | 46 | 78 | 29 |
| carbeth | 8 | 83 | 39 |
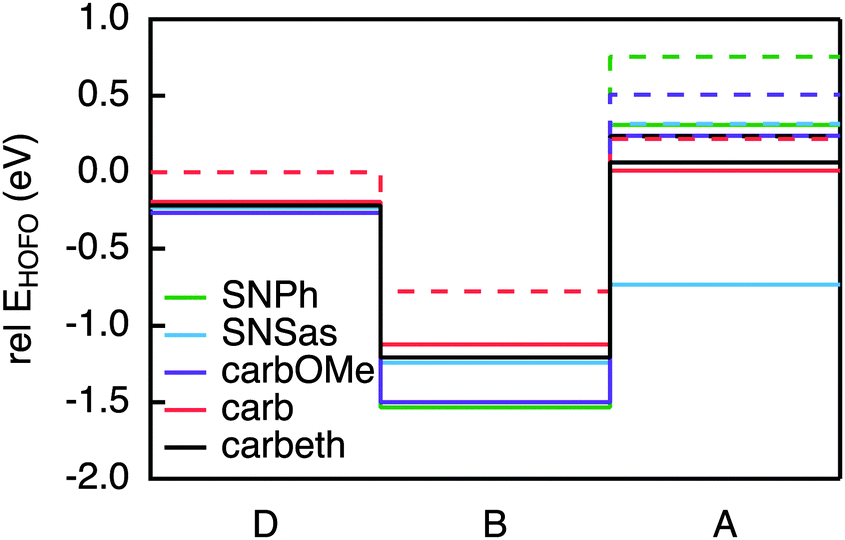 | ||
| Fig. 6 Energy levels of the HOFO of the hole acceptor candidates and the HOFOs of the linearly conjugated biphenyl bridge and PMI in the DBA molecules (solid line) compared to the isolated fragments (dashed line). | ||
In the next step, three derivatives of carbazole, a well-known electron donor, were considered as hole acceptors. These derivatives are shown in Fig. 5. According to the DFT calculations, the final state is localised on the hole acceptor for carbOMe while exhibiting substantial Jeff of 27 meV; two orders of magnitude larger than for SNS. For carb the final state is rather localised with a hole acceptor HOFO contribution of 78%. This contribution could be further increased to 83% by inserting an ethynyl spacer in carbeth. This degree of localisation is quite satisfying considering the dihedral angle between hole acceptor and bridge is almost 0°. With Jeff of 39 meV for the linearly conjugated DBA system, carbeth compares well with carbOMe.
3.3 Linear vs. cross-conjugation
The two linearly conjugated DBA systems carbOMe and carbeth show localised initial and final states while still exhibiting a substantial effective electronic coupling for hole transfer. Therefore, both appear to be promising designs for DBA systems exhibiting pronounced quantum interference effects. In the following, the two DBA systems are referred to as lin_carbOMe and lin_carbeth to point out the linear conjugation of these molecules. To test whether hole transfer in these systems is dominated by quantum interference, we have also calculated Jeff for their cross-conjugated versions cross_carbOMe and cross_carbeth and compared these values to Jeff for lin_carbOMe and lin_carbeth in Fig. 7. This comparison reveals that Jeff for cross_carbOMe is merely by a factor of 2 smaller than for lin_carbOMe. A thorough examination of the different contributions to Jeff reveals that in addition to the π-orbitals of the biphenyl bridge the σ-network carries a large extent of Jeff. The strong effect of the σ-network in this case becomes clear when only the contributions due to the π-orbitals to Jeff are considered (values in parentheses in Fig. 7). In this case the difference would be almost a factor of 5. By contrast, the σ-network plays a subordinate role in Jeff for lin_carbeth and cross_carbeth and does consequently not obscure the effects of quantum interference. Note once again, that the exchange of the hole acceptor alone alters the contributions of the respective bridge orbitals. lin_carbeth and cross_carbeth vary in Jeff by one order of magnitude which is due to constructive and destructive interference, respectively. On the basis of these calculations, the hole transfer rate constants for lin_carbeth and cross_carbeth are expected to differ by two orders of magnitude since the hole transfer rate constant scales with the square of the effective electronic coupling in the context of non-adiabatic charge transfer theory. This illustrates that it is rather hard to predict the differences in the electronic coupling for different DBA systems, even if strong effects, such as quantum interference, play a role. Therefore, the type of calculations described here is a very important tool for the design of DBA model systems for specific purposes—also when no quantum interference effects are expected. | ||
| Fig. 7 Chemical structures of the linearly and cross-conjugated DBA systems with localised initial and final states and non-negligible Jeff. The linearly and cross-conjugated bridges are highlighted in red. Jeff were calculated taking into account all bridge fragment orbitals or only the π bridge fragment orbitals (in parentheses). | ||
4 Conclusions
We have shown a computational route to the design of a linearly and a cross-conjugated DBA molecule containing a biphenyl bridge with hole transfer characteristics dominated by quantum interference effects. The screening of several hole donor and hole acceptor candidates demonstrated how difficult it is to find the right balance between a too weak and too strong coupling between the donor, bridge, and acceptor moieties in order to assure localised initial and final states while still allowing for all bridge orbitals to provide hole transfer pathways. This condition needs to be especially met in quantum interference based DBA systems, but should generally be examined in the interpretation of experimental results on electron or hole transfer in all DBA systems. A false assumption of localised initial and final states might for instance lead to wrong conclusions concerning the transfer mechanism.By consciously using asymmetric donor and acceptor molecules, as opposed to the previously investigated symmetric ones presented in the introduction, we ensure that all bridge orbitals can mediate hole transfer. This is clearly reflected in the effective electronic coupling for the linearly conjugated DBA systems, which is two orders of magnitude larger for the asymmetric version than for the symmetric one. However, we have also shown that the participation of all bridge orbitals does not necessarily guarantee a pronounced effect of quantum interference as for instance a dominant contribution of the σ-network of the bridge might conceal interference effects.
Finally, we have seen that the orbital energy of the fragments shift unpredictably upon connection. This furthermore stresses the importance of a holistic approach to the rational design of DBA systems in which the donor, bridge, and acceptor moieties cannot be seen as separate isolated fragments with inherent properties.
Acknowledgements
This work is supported by the Netherlands Organization for Scientific Research (NWO) through a VIDI grant. The research leading to these results has received funding from the European Research Council FP7 ERC Grant Agreement no. 240299 and Horizon 2020 ERC Grant Agreement no. 648433.References
- A. V. Vooren, V. Lemaur, A. Ye, D. Beljonne and J. Cornil, ChemPhysChem, 2007, 8, 1240–1249 CrossRef PubMed.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- D. B. Hall, R. E. Holmlin and J. K. Barton, Nature, 1996, 382, 731–735 CrossRef CAS PubMed.
- S. Kanvah, J. Joseph, G. B. Schuster, R. N. Barnett, C. L. Cleveland and U. Landman, Acc. Chem. Res., 2010, 43, 280–287 CrossRef CAS PubMed.
- Z. Liu, C. Tan, X. Guo, Y. T. Kao, J. Li, L. Wang, A. Sancar and D. Zhong, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14831–14836 CrossRef CAS PubMed.
- J. C. Genereux and J. K. Barton, Chem. Rev., 2010, 110, 1642–1662 CrossRef CAS PubMed.
- F. D. Lewis, H. Zhu, P. Daublain, B. Cohen and M. R. Wasielewski, Angew. Chem., Int. Ed., 2006, 45, 7982–7985 CrossRef CAS PubMed.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- C. Bauer, J. Teuscher, J. C. Brauer, A. Punzi, A. Marchioro, E. Ghadiri, J. D. Jonghe, M. Wielopolski, N. Banerji and J. E. Moser, Chimia, 2011, 65, 704–709 CrossRef CAS PubMed.
- J. Andreasson and U. Pischel, Chem. Soc. Rev., 2010, 39, 174–188 RSC.
- A. P. de Silva and S. Uchiyama, Nat. Nanotechnol., 2007, 2, 399–410 CrossRef CAS PubMed.
- A. P. de Silva, J. Phys. Chem. Lett., 2011, 2, 2865–2871 CrossRef CAS.
- R. H. Goldsmith, L. E. Sinks, R. F. Kelley, L. J. Betzen, W. H. Liu, E. A. Weiss, M. A. Ratner and M. R. Wasielewski, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3540–3545 CrossRef CAS PubMed.
- W. B. Davis, W. A. Svec, M. A. Ratner and M. R. Wasielewski, Nature, 1998, 396, 60–63 CrossRef CAS.
- E. A. Weiss, M. J. Tauber, R. F. Kelley, M. J. Ahrens, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2005, 127, 11842–11850 CrossRef CAS PubMed.
- G. L. Closs and J. R. Miller, Science, 1988, 240, 440–447 CAS.
- O. S. Wenger, Inorg. Chim. Acta, 2011, 374, 3–9 CrossRef CAS.
- J. Sukegawa, C. Schubert, X. Zhu, H. Tsuji, D. M. Guldi and E. Nakamura, Nat. Chem., 2014, 6, 899–905 CrossRef CAS PubMed.
- O. S. Wenger, Acc. Chem. Res., 2011, 44, 25–35 CrossRef CAS PubMed.
- B. Albinsson and J. Martensson, J. Photochem. Photobiol., C, 2008, 9, 138–155 CrossRef CAS.
- E. A. Weiss, M. R. Wasielewski and M. A. Ratner, Top. Curr. Chem., 2005, 257, 103–133 CrossRef PubMed.
- A. B. Ricks, G. C. Solomon, M. T. Colvin, A. M. Scott, K. Chen, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2010, 132, 15427–15434 CrossRef CAS PubMed.
- N. Gorczak, N. Renaud, S. Tarkuc, A. J. Houtepen, R. Eelkema, L. D. A. Siebbeles and F. C. Grozema, Chem. Sci., 2015, 6, 4196–4206 RSC.
- L. E. Shoer, S. W. Eaton, E. A. Margulies and M. R. Wasielewski, J. Phys. Chem. B, 2014, 119, 7635–7643 CrossRef PubMed.
- N. Gorczak, S. Tarkuc, N. Renaud, A. J. Houtepen, R. Eelkema, L. D. A. Siebbeles and F. C. Grozema, J. Phys. Chem. A, 2014, 118, 3891–3898 CrossRef CAS PubMed.
- M. G. Gatty, A. Kahnt, L. J. Esdaile, M. Hutin, H. L. Anderson and B. Albinsson, J. Phys. Chem. B, 2015, 119, 7598–7611 CrossRef PubMed.
- E. A. Weiss, M. J. Ahrens, L. E. Sinks, A. V. Gusev, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 5577–5584 CrossRef CAS PubMed.
- P. Sautet and C. Joachim, Chem. Phys. Lett., 1988, 153, 511–516 CrossRef CAS.
- G. C. Solomon, D. Q. Andrews, T. Hansen, R. H. Goldsmith, M. R. Wasielewski, R. P. V. Duyne and M. A. Ratner, J. Chem. Phys., 2008, 129, 054701 CrossRef PubMed.
- T. Hansen, G. C. Solomon, D. Q. Andrews and M. A. Ratner, J. Chem. Phys., 2009, 131, 194704 CrossRef PubMed.
- S. Chen, Y. Zhang, S. Koo, H. Tian, C. Yam, G. Chen and M. A. Ratner, J. Phys. Chem. Lett., 2014, 5, 2748–2752 CrossRef CAS PubMed.
- N. Renaud, M. A. Ratner and C. Joachim, J. Phys. Chem. B, 2011, 115, 5582–5592 CrossRef CAS PubMed.
- T. Markussen, R. Stadler and K. S. Thygesen, Nano Lett., 2010, 10, 4260–4265 CrossRef CAS PubMed.
- A. A. Kocherzhenko, F. C. Grozema and L. D. A. Siebbeles, J. Phys. Chem. C, 2010, 114, 7973–7979 CAS.
- R. Härtle, M. Butzin, O. Rubio-Pons and M. Thoss, Phys. Rev. Lett., 2011, 107, 046802 CrossRef PubMed.
- D. Q. Andrews, G. C. Solomon, R. P. V. Duyne and M. A. Ratner, J. Am. Chem. Soc., 2008, 130, 17309–17319 CrossRef CAS PubMed.
- G. C. Solomon, D. Q. Andrews, R. H. Goldsmith, T. Hansen, M. R. Wasielewski, R. P. V. Duyne and M. A. Ratner, J. Am. Chem. Soc., 2008, 130, 17301–17308 CrossRef CAS PubMed.
- E. H. van Dijk, D. J. T. Myles, M. H. van der Veen and J. C. Hummelen, Org. Lett., 2006, 8, 2333–2336 CrossRef CAS PubMed.
- A. A. Kocherzhenko, K. B. Whaley, G. Sforazzini, H. L. Anderson, M. Wykes, D. Beljonne, F. C. Grozema and L. D. A. Siebbeles, J. Phys. Chem. C, 2012, 116, 25213–25225 CAS.
- M. Mayor, H. B. Weber, J. Reichert, M. Elbing, C. V. Hanisch, D. Beckmann and M. Fischer, Angew. Chem., Int. Ed. Engl., 2003, 42, 5834–5838 CrossRef CAS PubMed.
- C. R. Arroyo, S. Tarkuc, R. Frisenda, J. S. Seldenthuis, C. H. Woerde, R. Eelkema, F. C. Grozema and H. S. van der Zant, Angew. Chem., Int. Ed. Engl., 2013, 52, 3152–3155 CrossRef CAS PubMed.
- C. M. Guedon, H. Valkenier, T. Markussen, K. S. Thygesen, J. C. Hummelen and S. J. van der Molen, Nat. Nanotechnol., 2012, 7, 304–308 CrossRef PubMed.
- S. V. Aradhya, J. S. Meisner, M. Krikorian, S. Ahn, R. Parameswaran, M. L. Steigerwald, C. Nuckolls and L. Venkataraman, Nano Lett., 2012, 12, 1643–1647 CrossRef CAS PubMed.
- S. Ballmann, R. Hartle, P. B. Coto, M. Elbing, M. Mayor, M. R. Bryce, M. Thoss and H. B. Weber, Phys. Rev. Lett., 2012, 109, 056801 CrossRef PubMed.
- M. Baghernejad, X. T. Zhao, K. B. Ornso, M. Fueg, P. Moreno-Garcia, A. V. Rudnev, V. Kaliginedi, S. Vesztergom, C. C. Huang, W. J. Hong, P. Broekmann, T. Wandlowski, K. S. Thygesen and M. R. Bryce, J. Am. Chem. Soc., 2014, 136, 17922–17925 CrossRef CAS PubMed.
- C. Patoux, C. Coudret, J. P. Launay, C. Joachim and A. Gourdon, Inorg. Chem., 1997, 36, 5037–5049 CrossRef CAS.
- M. L. Kirk, D. A. Shultz, D. E. Stasiw, D. Habel-Rodriguez, B. Stein and P. D. Boyle, J. Am. Chem. Soc., 2013, 135, 14713–14725 CrossRef CAS PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. V. Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- K. Senthilkumar, F. C. Grozema, F. M. Bickelhaupt and L. D. A. Siebbeles, J. Chem. Phys., 2003, 119, 9809–9817 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- D. Jacquemin, E. A. Perpete, I. Ciofini, C. Adamo, R. Valero, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2071–2085 CrossRef CAS PubMed.
- J. W. Evenson and M. Karplus, J. Chem. Phys., 1992, 96, 5272–5278 CrossRef CAS.
- A. Kubas, F. Hoffmann, A. Heck, H. Oberhofer, M. Elstner and J. Blumberger, J. Chem. Phys., 2014, 140, 104105 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Comparison of the charge transfer character and the effective electronic couplings for two functionals. See DOI: 10.1039/c5cp06728f |
| ‡ Present address: Novaled GmbH, Dresden, Germany |
| This journal is © the Owner Societies 2016 |