
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1,1,2,2-Tetracyanocyclopropane (TCCP) as supramolecular synthon†
Antonio
Bauzá
a,
Antonio
Frontera
 *a and
Tiddo J.
Mooibroek
*b
*a and
Tiddo J.
Mooibroek
*b
aDepartment of Chemistry Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma (Baleares), Spain. E-mail: toni.frontera@uib.es; Fax: +34 971 173426
bSchool of Chemistry of the University of Bristol, Cantock's Close, BS8 1TS, Bristol, UK. E-mail: tiddomooibroek@gmail.com
First published on 9th December 2015
Abstract
The 1,1,2,2-tetracyanocyclopropane (TCCP) unit presents a synthetically accessible and versatile synthon that can interact with lone-pair or π-electrons by ‘non-covalent carbon bonding’. Complexes of TCCP with common small molecules, anions, aromatics like fullerenes, amino acids and nucleobases were computed at the DFT BP86-D3/def2-TZVP level of theory. Binding energies vary between about −10 kcal mol−1 for neutral guests and −15 to −50 kcal mol−1 for anionic species. This is comparable to strong and very strong hydrogen bonding respectively. Thus, in addition to synthons that contain polarized hydrogen or halogen atoms, TCCP presents a new supramolecular synthon that awaits experimental exploitation.
Introduction
Living matter is for a great part governed by intermolecular recognition phenomena such as substrate/inhibitor/protein binding,1–4 signalling events5–8 and cell–cell interactions.9–11 Intramolecular phenomena such as the folding of proteins12–14 or DNA/RNA15–18 are governed by the same physical forces. The design and synthesis of molecules that can influence such processes are the basis of many inquiries in supramolecular chemistry,19–23 molecular biology24–27 and pharmacology.28–30 Underpinning the design process is knowing which molecular fragments will engage in favourable intermolecular interactions, i.e. knowing which supramolecular synthons one can use.31–33 Prominent among these are synthons that rely on hydrogen bonding or aromatic interactions, such as nucleosides, amino acids and sugars. Chemical modification of such natural synthons is common practice,34–37 while artificial supramolecular synthons that rely on other types of intermolecular interactions are rare. Halogen bonding is a noticeable exception,38 but in principle Chalcogen,39–41 Pnictogen,42–45 or Tetrel46–49 bonding interactions could similarly be exploited. That these interactions are not yet extensively used in (biochemical) research may in part be due to their novelty. Indeed, these interactions have only been studied in detail in the past decade or so.46,50–55 A major obstacle of their exploitation, however, is the synthetic accessibility of identified supramolecular synthons. In particular, the problem is how to incorporate a given synthon in a larger molecular framework. For example, the sulphur atom in SO2 and SO3 can participate in Chalcogen bonding interactions,56 but these entities lack a convenient chemical anchor point.We have recently highlighted that sp3 hybridized carbon – the most abundant tetrel atom in living matter – can be a supramolecular synthon.47,48 More specifically, the 1,1,2,2-tetracyanocyclopropane (TCCP) motif (Scheme 1) was identified as an electron poor bowl, apt to accommodate an electron rich guest.47 Two convenient (high yielding) synthetic routes towards this motif are shown in Scheme 1: reaction of a primary or secondary alkyl halide with tetracyanoethylene (top);57–60 and reaction of an aldehyde or ketone with malonitrile (bottom).61,62 In both instances, numerous variations of the R-bearing moieties are readily available and provide a convenient way to obtain a practically infinite amount of TCCP derivatives. Thus, TCCP provides a rather unique case of a synthetically versatile and accessible supramolecular synthon that awaits utilization by the molecular scientists.
 | ||
| Scheme 1 Synthetic routes to 1,1,2,2-tetracyanocyclopropane (TCCP) structures. The molecular electrostatic potential map of 3,3′-dimethyl-TCCP was computed at the DFT-B3LYP-6-31G* level of theory and the color code spans from −130 (red) to +150 (blue) kcal mol−1.63 | ||
Anticipating the experimental exploitation of TCCP, we here report on a comprehensive theoretical investigation of the binding interactions of a model for TCCP derivatives (where R1 = R2 = H) with three classes of compounds; commonly encountered small (neutral) molecules, common anions, and several aromatic systems including Nature's aromatic building blocks.
Results
For our enquiries we conducted computations based on density functional theory (DFT) at the BP86-D3/def2-TZVP level of theory and Table 1 summarized the results of the interaction of TCCP with common small molecules. For several of these molecule pairs, ab initio calculations at the MP2/def2-TZVP level of theory were also conducted (denoted ‘a’ in Table 1) to validate our use of the more economical DFT approach. The comparative results are given in the ESI† (Table S1) and are in excellent agreement: computed distances differ less than 3% and computed energies typically less than 10%. In all cases, the minimized complex was subjected to an ‘atoms in molecules’ (AIM) analysis in order to identify atoms engaging in bonding contacts.64 Graphical renderings of these analyses are depicted in Fig. S1 (ESI†), and Fig. 1 shows representative examples for some complexes with small neutral molecules.| Complex | Guest | ΔE (kcal mol−1) | D (Å) | ρ·100 (a.u.) |
|---|---|---|---|---|
| a Complex also computed at the MP2/def2-TZVP level of theory, as detailed in Table S1. b Also XH⋯NC(TCCP) hydrogen bonding present according to AIM analysis. c Alternate orientation also considered (respectively marked 2′/17′/18′/28′ in Fig. S1) but found to be less stable. d Another geometry where pyridine interacts with its π-cloud is less stable at −7.08 kcal mol−1 (see also complex 54 in Table 3). | ||||
| Control | ||||
| 1 | CH4a | −2.3 | 3.167a | 0.470 |
| O-donor atom(s) | ||||
| 2 | CO2a,c | −3.7 | 2.952 | 0.735 |
| 3 | H2Oa | −8.5 | 2.819 | 1.094 |
| 4 | (CH3)2Oa,b | −10.0 | 2.836 | 1.096 |
| 5 | 1,4-Dioxaneb | −11.4 | 2.824 | 1.129 |
| 6 | THFb | −11.2 | 2.778 | 1.223 |
| 7 | (CH3)2CO | −7.7 | 2.729 | 1.210 |
| 8 | EtOAcb | −10.8 | 2.752 | 1.190 |
| 9 | Urea | −11.1 | 2.687 | 1.310 |
| 10 | Lactameb | −13.4 | 2.759 | 1.220 |
| 11 | (CH3)2NC(O)Hb | −15.1 | 2.769 | 1.210 |
| 12 | CH3NO2 | −7.5 | 3.103 | 0.850 |
| 13 | H3POa | −10.3 | 2.727 | 1.231 |
| 14 | (CH3)3POa | −14.0 | 2.645 | 1.470 |
| 15 | H2SOa | −9.2 | 2.686 | 1.244 |
| 16 | (CH3)2SOa | −12.2 | 2.622 | 1.437 |
| Other-donor atoms | ||||
| 17 | COa,c | −3.2 | 3.302 | 0.599 |
| 18 | N2a,c | −2.6 | 3.168 | 0.604 |
| 19 | NH3a | −9.4 | 3.003 | 1.046 |
| 20 | N(CH3)3a,b | −11.2 | 3.177 | 0.831 |
| 21 | CH3CN | −7.3 | 2.962 | 0.963 |
| 22 | Pyridined | −9.5 | 3.010 | 1.025 |
| 23 | H2Sa | −4.5 | 3.400 | 0.654 |
| 24 | S(CH3)2a | −7.7 | 3.266 | 0.876 |
| 25 | PH3a | −4.9 | 3.567 | 0.665 |
| 26 | P(CH3)3a | −9.8 | 3.404 | 0.967 |
| 27 | CH2Cl2 | −4.9 | 3.968 | 0.580 |
| 28 | CCl4c | −4.2 | 3.686 | 0.558 |
 | ||
| Fig. 1 Molecular geometries of representative complexes of TCCP with small molecules, as computed at the BP86-D3/def2-TZVP level of theory (see also Table 1). The small red dots denote the bond-critical points according to an AIM analysis. | ||
The complexation energy with the control guest methane (−2.3 kcal mol−1) is very small and methane actually is not located in the electron poor binding pocket of TCCP (see Fig. S1, ESI†). All other guests do engage in tetrel bonding with the C2(CN)4 pocket, although in several structures additional hydrogen bonding with TCCP's N-atom(s) is also observed (i.e. in 4–6, 8, 10, 11, 20). These additional forces might explain the increased stability of these complexes over other, very similar ones. For example, the [H2O⋯TCCP] pair 3 has an energy of −8.45 kcal mol−1, solely due to O⋯C tetrel bonding interactions, while the additional hydrogen bonds with dimethyl ether (4), 1,4-dioxane (5) and THF (6) result in energies of about −11 kcal mol−1. The energies of other small molecules with O-donors (7–16) are very similar, between about −7 and −10 kcal mol−1. The strongest of these that do not have additional H-bonding according to AIM are trimethylphosphaneoxide 14 (−14.0 kcal mol−1) and dimethylsulfoxide 16 (−12.2 kcal mol−1). This is in line with the increased polarization of O in these molecules.
Other small molecules considered where an atom other than oxygen functions as electron donor (17–28) gave very similar energies, ranging between about −5 to about −10 kcal mol−1. Carbon monoxide (17) and dinitrogen (18) displayed the lowest predicted energies at about −3 kcal mol−1.
Interestingly, the series with H2O (3; −8.5 kcal mol−1, 2.82 Å) H2S (23; −4.5 kcal mol−1, 3.40 Å), H3N (19; −9.4 kcal mol−1, 3.00 Å) and H3P (25; −4.9 kcal mol−1, 3.57 Å) suggest that TCCP prefers ‘hard’ over ‘soft’ donor atoms, while the trend might also result from the longer distance required by the ‘soft’ second-row donors.
All anionic guests appears to sit comfortably within the electron poor bowl shape of TCCP, and are held in place solely by one or multiple Tetrel bonding interactions, as evidenced by the AIM analyses (see Fig. 2 for representative examples, but also Fig. S1, ESI†). The interacting energies are summarized in Table 2, and range typically from −19.4 kcal mol−1 for SCN− (43) to −32.8 kcal mol−1 for acetate (32). The two complexes with the largest energies (37 and 38) concern dianions (SO42− = −68.9 kcal mol−1; SO32− = −88.1 kcal mol−1). The latter (38) might well have some covalent character; the C–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N angles (143°) deviate significantly from the ∼170–180° observed in the monoanionic complexes. The C–CN angles in 37 are about 164°, indicating that there is much less covalent character.
N angles (143°) deviate significantly from the ∼170–180° observed in the monoanionic complexes. The C–CN angles in 37 are about 164°, indicating that there is much less covalent character.
 | ||
| Fig. 2 Molecular geometries of representative complexes of TCCP with anions, as computed at the BP86-D3/def2-TZVP level of theory (see also Table 2). The small red dots denote the bond-critical points according to an AIM analysis. | ||
| Complex | Guest | ΔE (kcal mol−1) | D (Å) | ρ·100 (a.u.) |
|---|---|---|---|---|
| Anions with O-donor atoms and hydride | ||||
| 29 | HO− | −57.7 | 2.286 | 2.850 |
| 30 | HCO2− | −31.8 | 2.720 | 1.890 |
| 31 | CH3CO2− | −32.8 | 2.700 | 1.940 |
| 32 | HOCO2− | −29.5 | 2.720 | 1.860 |
| 33 | ClO4− | −18.6 | 2.810 | 1.410 |
| 34 | NO3− | −26.0 | 2.748 | 1.690 |
| 35 | H2PO4− | −29.2 | 2.737 | 1.890 |
| 36 | HSO4− | −24.1 | 2.764 | 1.740 |
| 37 | SO42− | −65.9 | 2.482 | 2.810 |
| 38 | SO32− | −88.1 | 2.520 | 7.490 |
| 39 | H− | −43.4 | 2.409 | 1.680 |
| Anions with N-donor atoms | ||||
| 40 | N3− | −26.3 | 2.558 | 1.910 |
| 41 | OCN− | −25.6 | 2.831 | 2.120 |
| 42 | CN− | −22.6 | 2.696 | 1.670 |
| 43 | SCN− | −19.4 | 2.681 | 1.670 |
| Anions with halogen donor atoms | ||||
| 44 | F− | −52.1 | 2.311 | 2.800 |
| 45 | Cl− | −30.4 | 2.907 | 1.650 |
| 46 | Br− | −24.8 | 3.104 | 1.410 |
| 47 | I− | −19.6 | 3.369 | 1.160 |
| 48 | BF4− | −17.2 | 2.755 | 1.350 |
| 49 | PF6− | −14.8 | 2.857 | 1.200 |
From the series with hydroxide 29 (−57.7 kcal mol−1) > formate 30 ≈ acetate 31 ≈ hydrogen carbonate 32 (about −30 kcal mol−1) > perchlorate 33 (−18.6 kcal mol−1) it seems evident that the interacting energies decrease when the negative charge becomes more spread out over a larger anion (although the hydride result (39) breaks this trend, likely due to the short distance). The anions where a N-donor atom formally bears the negative charge (40–43) bind weaker (about −20–25 kcal mol−1) than the small anions with O-donor atoms (e.g. formate 30 with −31.8 kcal mol−1). In the series with halogen donor atoms (44–49) there is a clear trend with energies ranging from −52.1 kcal mol−1 for F− (44) to −14.8 kcal mol−1 for PF6− (49).
In general the interaction energies reported in Tables 1 and 2 are in good agreement with the MEP values of the guest molecules on their negative regions. For instance in the neutral O/N Lewis bases the MEP values vary from −58 kcal mol−1 [for (CH3)3PO] to −12 kcal mol−1 (for N2). Moreover, for the mono-anionic guests, the MEP values vary from −216 kcal mol−1 (F−) to −125 kcal mol−1 (PF6−), in line with the interaction energies observed for their corresponding complexes. The SO32− dianionic guest exhibits the most negative MEP value (−247 kcal mol−1) and the largest interaction energy (see Table 2).
As it appears from the data collected in Table 3, small isolated π-systems like ethene (50) and ethyne (51) bind to TCCP with about −5 kcal mol−1. Small conjugated systems such as benzene (53) bind even stronger (about −7 kcal mol−1), while larger condensed hydrocarbons (55–60) such as pyrene (58) bind stronger still (about −10 kcal mol−1). As is apparent from the AIM analyses shown in Fig. 3, all these complexes are held together mainly by tetrel bonding interactions (in some cases perhaps stabilized by weak CN⋯HC polar contacts).
| Complex | Guest | ΔE (kcal mol−1) | D (Å) | ρ·100 (a.u.) |
|---|---|---|---|---|
| a Another geometry where pyridine interacts with its N-atom is more stable at −9.5 kcal mol−1 (see also complex 22Table 1). b No interaction with the π-system. | ||||
| Simple isolated π-systems (control) | ||||
| 50 | Ethene | −5.6 | 3.458 | 0.531 |
| 51 | Ethyne | −4.3 | 3.431 | 0.528 |
| Simple extended π-systems | ||||
| 52 | Cyclobutadiene | −6.9 | 3.502 | 0.660 |
| 53 | Benzene | −8.5 | 3.601 | 0.649 |
| 54 | Pyridinea | −7.1 | 3.697 | 0.629 |
| Larger extended π-systems | ||||
| 55 | Naphtalene | −8.5 | 3.800 | 0.222 |
| 56 | Antracene | −10.1 | 3.727 | 0.585 |
| 57 | Phenanthrene | −11.4 | 3.600 | 0.527 |
| 58 | Pyrene | −11.9 | 3.581 | 0.710 |
| 59 | Triphenylene | −11.8 | 3.665 | 0.657 |
| 60 | Coronene | −12.6 | 3.698 | 0.692 |
| Fullerenes | ||||
| 61 | C60 | −7.7 | 3.762 | 0.672 |
| 62 | CNT (8,0) | −11.6 | 3.545 | 0.723 |
| 63 | CNT (10,0) | −12.3 | 3.540 | 0.710 |
| 64 | CNT (12,0) | −12.7 | 3.549 | 0.686 |
| Nature's aromatic building bocks | ||||
| 65 | Model of Tyr | −8.1 | 3.650 | 0.671 |
| 66 | Model of Trp | −11.7 | 3.627 | 0.693 |
| 67 | Model of Hisb | −11.6 | 2.928 | 1.159 |
| 68 | Model of His+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
−17.6 | 1.841a | 3.500 |
| 69 | Adenine | −11.2 | 3.710 | 0.643 |
| 70 | Guanine | −11.3 | 3.228 | 0.598 |
| 71 | Thymineb | −11.5 | 2.767 | 1.217 |
| 72 | Cytosineb | −14.4 | 2.925 | 1.432 |
| 73 | Uracilb | −10.6 | 2.787 | 1.160 |
 | ||
| Fig. 3 Molecular geometries of representative complexes of TCCP with π-systems, as computed at the BP86-D3/def2-TZVP level of theory (see also Table 3). The small red dots denote the bond-critical points according to an AIM analysis. | ||
It is interesting to note that the binding energy peaks at coronene (60; −12.6 kcal mol−1), which can be seen as a model for graphene. Likewise, the binding energies calculated with several fullerenes (61–64) are substantial and strongest for a model of carbon nanotube (12,0) at −12.6 kcal mol−1 (64).
Also noteworthy is the positioning of TCCP over pyrene in 58 and triphenylene in 59; apparently TCCP prefers the periphery over the center. It is known what Li+ also preferentially binds to a peripheral ring in large condensed hydrocarbons.65 However, in 60 the TCCP sits perfectly above the center of the coronene.
Encouraged by the energies computed with small molecules and aromatic systems, we expected that Nature's aromatic building blocks could bind to TCCP as well. The computational verifications of this expectation are listed in Table 3 as complexes 65–73 and Fig. 4 shows the molecular structure and AIM analysis of several representative examples. Models of tyrosine 65 (−8.1 kcal mol−1) and tryptophan 66 (−11.7 kcal mol−1) interact much like condensed hydrocarbons, binding to TCCP with their π-electrons. Histidine 67 (−11.6 kcal mol−1) seems to prefer binding to TCCP with its N-atom. When protonated, histidine moves away from TCCP's electron poor binding pocket and instead establishes a strong hydrogen bond with one of the N-atoms in TCCP. The binding energies computed with the nucleobases (69–73) are very similar at about −11 kcal mol−1. Adenine (69) and guanine (70) bind with their π-surfaces, while the thymine (71), cytosine (72) and uracil (73) interact with their lone-pair electrons on O and/or N and additional hydrogen bonding.
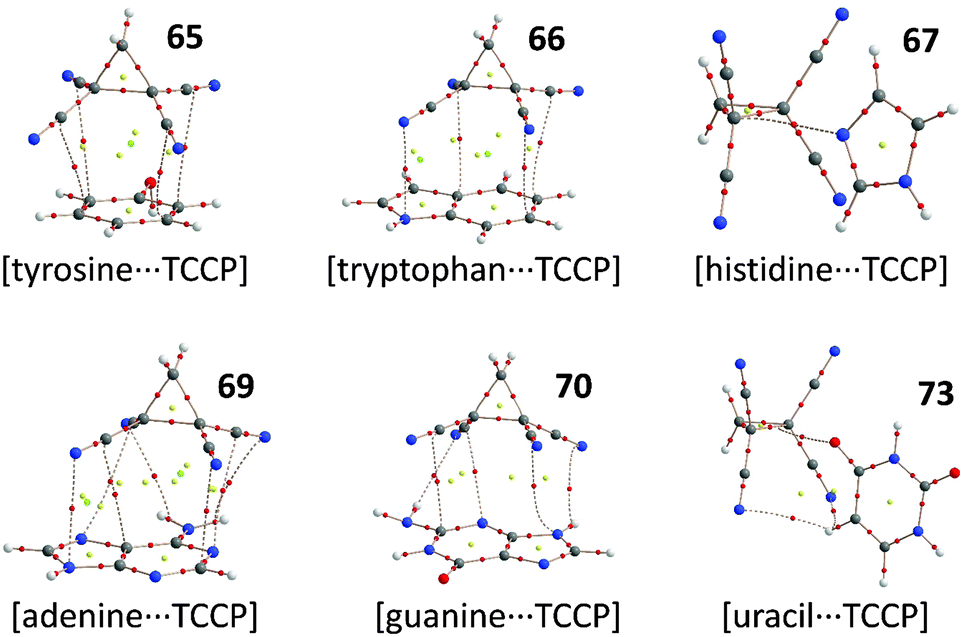 | ||
| Fig. 4 Molecular geometries of complexes of TCCP with some of Nature's aromatic building blocks, as computed at the BP86-D3/def2-TZVP level of theory (see also Table 3). The small red dots denote the bond-critical points according to an AIM analysis. | ||
Next, we wondered how a host molecule with several appropriately-spaced TCCP units would interact with some size-complementary electron rich guests. To this end we conjured one bipodal and two tripodal claw-like hosts (Fig. S2, ESI†) in which the linking unit assures an appropriate space in between TCCP-moieties and also allows for the correct angles so that the C2(CN)4 ‘binding pockets’ can face each other. We computed interacting energies with a selection of guests (see Table 4). The molecular geometries of selected complexes are shown in Fig. 5 (the whole series is shown in Fig. S3, ESI†). AIM analyses were also performed and revealed tetrel bonding in all cases (not shown due to congested graphics).
| Complex | Guest | ΔEa (kcal mol−1) | D (Å) |
|---|---|---|---|
| a Energies relative to unbound hosts in its energy minimal conformer (as estimated by a Monte Carlo MMFF simulation prior to the DFT minimum energy calculation). For geometries see Fig. S2 (compounds 88–90). | |||
| Bipodal host | |||
| 74 | H2O | −5.6 | 3.226 |
| 75 | H2S | −5.7 | 3.466 |
| 76 | Benzene | −12.4 | 3.622 |
| 77 | Pyridine | −9.6 | 3.634 |
| 78 | F− | −67.1 | 2.489 |
| 79 | Cl− | −41.3 | 3.005 |
| 80 | Br− | −34.1 | 3.177 |
| 81 | I− | −27.0 | 3.411 |
| Tripodal hosts | |||
| 82 | BF4− | −27.5 | 3.403 |
| 83 | ClO4− | −31.5 | 3.403 |
| 84 | NO3− | −41.1 | 2.718 |
| 85 | BF4− | −23.4 | 3.381 |
| 86 | ClO4− | −22.2 | 2.811 |
| 87 | NO3 | −22.9 | 2.649 |
 | ||
| Fig. 5 Molecular geometries of complexes of bipodal (top) and tripodal (bottom) TCCP-hosts with some electron rich guests, as computed at the BP86-D3/def2-TZVP level of theory (see also Table 4). | ||
The bipodal host interacts with some neutral and ‘flat’ molecules with about −5 to −10 kcal mol−1 (74–77); while the interaction of the spherical halide anions is much larger varying between −30 and −70 kcal mol−1 (78–81). The two tripodal hosts seem to complement the tetrahedral anions BF4− (82, 85), ClO4− (83, 86) as well as the trigonal planar NO3− (84, 87) with interacting energies of about −20 to −40 kcal mol−1.
These energies are generally larger compared to the analogous interaction with a single TCCP unit (Tables 2 and 3). For example, 78 (−67.1 kcal mol−1) is about 30% more stable than 44 (−52.08 kcal mol−1) and 82 (−27.5 kcal mol−1) is about 60% more stable than 48 (−17.17 kcal mol−1). That the stabilization is not strictly additive is likely a result of some repulsive interactions in the complex (e.g. CN⋯NC), some strain on the conformation of the host (e.g. the Ar–CC–CH2 units in 82 and 84 are not perfectly linear), and/or the decreased electronegativity of the guest upon binding to one TCCP moiety.
Discussion and conclusions
From the above results it is clear that TCCP derivatives can accommodate a plethora of guest molecules that bear lone-pair electrons, π-electrons and/or a negative charge. The main mode of interaction with these electron rich entities is tetrel bonding with TCCP's electron deficient C2(CN)4 bowl. Hydrogen bonding with the cyano N-atoms may further stabilize the complex (e.g. complex 6 with THF).The binding energies of about −10 kcal mol−1 observed with various neutral guest molecules are comparable in strength to strong hydrogen bonding involving charge-neutral H-bonding pairs.66 The values of about −15 to −30 kcal mol−1 – typically observed with various anions – is truly remarkable because they are comparable in strength to very strong (ionic) hydrogen bonding.66 The exceptionally large enthalpies computed for H− (−43.4 kcal mol−1) HO− (−57.7 kcal mol−1) and F− (−52.1 kcal mol−1) even far exceed the common benchmark for strong hydrogen bonding (about −35 kcal mol−1).66
The large energies of formation computed between TCCP and (models of) fullerenes (about −10 kcal mol−1) was somewhat expected, as TCCP's bowl-like shape and electron positive core are complementary to the concave shape and electron rich surface of fullerenes. This complementarity hints towards the potential of TCCP derivatives to act as facial amphiphiles to help mobilize these carbon isomorphs in solution.67–70 Other charge-neutral supramolecular approaches for binding fullerenes indeed seem far less apt. For example, typical binding energies of hydrogen-π and halogen-π interactions are estimated at about 1–5 kcal mol−1,71,72 while not being shape-complementary to fullerenes at all.
Perhaps the most important result is the difference in geometric preferences of TCCP binding to (models of) amino acids and nucleobases. This implies that TCCP derivatives might selectively nest themselves in proteins and DNA/RNA-type molecules. In this context it is worth mentioning that TCCP derivatives are expected to be poorly hydrated in aqueous solution (no strong H-bond donors) and thus also interact with biomolecules by virtue of the hydrophobic effect. The potential of TCCP derivatives to bind strongly and selectively to biomolecules implies that TCCP might be engineered to influence the functioning of biomachineries, which in turn might have pharmacological implications. Additionally, the bipodal and tripodal TCCP hosts illustrate that strategically placed TCCP-units may greatly enhance the affinity for a guest molecule, just like multiple H-bond donors within a protein can result in high affinity binding to a ligand.
In summary we highlighted that TCCP is an accessible supramolecular synthon that acts as an ‘electron sponge’, mainly by virtue of tetrel bonding interactions. Its unique bowl-like shape, electron deficient core, and (presumed) hydrophobic character make TCCP-derivatives a promising new addition to the (bio)chemists toolbox (e.g. the PDB is void of TCCP-like ligands). As a result, following this theoretical exploration we anticipate that experimental exploitation of this unit will soon unveil its functional potential.
Acknowledgements
A. B. and A. F. thank DGICYT of Spain (projects CTQ2014-57393-C2-1-P and CONSOLIDER INGENIO CSD2010–00065, FEDER funds) for funding.References
- S. Jones and J. M. Thornton, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13 CrossRef CAS.
- L. Lo Conte, C. Chothia and J. Janin, J. Mol. Biol., 1999, 285, 2177 CrossRef CAS PubMed.
- D. D. Boehr, R. Nussinov and P. E. Wright, Nat. Chem. Biol., 2009, 5, 789 CrossRef CAS PubMed.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357 CrossRef CAS PubMed.
- B. Baker, P. Zambryski, B. Staskawicz and S. P. DineshKumar, Science, 1997, 276, 726 CrossRef CAS PubMed.
- S. Akira, K. Takeda and T. Kaisho, Nat. Immunol., 2001, 2, 675 CrossRef CAS PubMed.
- W. Strober, P. J. Murray, A. Kitani and T. Watanabe, Nat. Rev. Immunol., 2006, 6, 9 CrossRef CAS PubMed.
- E. Meylan, J. Tschopp and M. Karin, Nature, 2006, 442, 39 CrossRef CAS PubMed.
- E. Ruoslahti and M. D. Pierschbacher, Science, 1987, 238, 491 CAS.
- C. Kasper, H. Rasmussen, J. S. Kastrup, S. Ikemizu, E. Y. Jones, V. Berezin, E. Bock and I. K. Larsen, Nat. Struct. Biol., 2000, 7, 389 CrossRef CAS PubMed.
- E. H. Chen and E. N. Olson, Science, 2005, 308, 369 CrossRef CAS PubMed.
- M. J. Gething and J. Sambrook, Nature, 1992, 355, 33 CrossRef CAS PubMed.
- A. Sali, E. Shakhnovich and M. Karplus, Nature, 1994, 369, 248 CrossRef CAS PubMed.
- C. M. Dobson, Nature, 2003, 426, 884 CrossRef CAS PubMed.
- A. H. J. Wang, G. J. Quigley, F. J. Kolpak, J. L. Crawford, J. H. van Boom, G. van der Marel and A. Rich, Nature, 1979, 282, 680 CrossRef CAS PubMed.
- B. N. Conner, T. Takano, S. Tanaka, K. Itakura and R. E. Dickerson, Nature, 1982, 295, 294 CrossRef CAS PubMed.
- A. H. J. Wang, S. Fujii, J. H. van Boom, G. A. van der Marel, S. A. A. van Boeckel and A. Rich, Nature, 1982, 299, 601 CrossRef CAS PubMed.
- M. C. Nagan, S. S. Kerimo, K. Musier-Forsyth and C. J. Cramer, J. Am. Chem. Soc., 1999, 121, 7310 CrossRef CAS.
- J. M. Lehn, Angew. Chem., Int. Ed., 1990, 29, 1304 CrossRef.
- J. F. Stoddart, Chem. Soc. Rev., 1979, 8, 85 RSC.
- A. P. Davis and R. S. Wareham, Angew. Chem., Int. Ed., 1999, 38, 2978 CrossRef.
- J. H. van Maarseveen, J. N. H. Reek and J. W. Back, Angew. Chem., Int. Ed., 2006, 45, 1841 CrossRef CAS PubMed.
- T. J. Mooibroek, J. M. Casas-Solvas, R. L. Harniman, C. M. Renney, T. S. Carter, M. P. Crump and A. P. Davis, Nat. Chem., 2015 DOI:10.1038/nchem.239.
- J. W. Trauger, E. E. Baird and P. B. Dervan, Nature, 1996, 382, 559 CrossRef CAS PubMed.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, S. Qiu, Y. S. Ding, W. Gao, J. Stuckey, K. Krajewski, P. P. Roller, Y. Tomita, D. A. Parrish, J. R. Deschamps and S. M. Wang, J. Am. Chem. Soc., 2005, 127, 10130 CrossRef CAS PubMed.
- C. C. Lee, J. A. MacKay, J. M. J. Frechet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517 CrossRef CAS PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185 CrossRef CAS PubMed.
- I. D. Kuntz, Science, 1992, 257, 1078 CAS.
- S. L. Schreiber, Science, 2000, 287, 1964 CrossRef CAS PubMed.
- M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46 CrossRef PubMed.
- V. R. Thalladi, B. S. Goud, V. J. Hoy, F. H. Allen, J. A. K. Howard and G. R. Desiraju, Chem. Commun., 1996, 401 RSC.
- A. Nangia and G. R. Desiraju, Top. Curr. Chem., 1998, 198, 57 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342 CrossRef CAS PubMed.
- SPR – Carbohydrate Chemistry, RSC Publishing, Cambridge (UK), 1968-present Search PubMed.
- Amino Acids, Peptides and Proteins in Organic Chemistry, ed. A. B. Hughes, Wiley-VCH, Weinheim, 2009, vol. 2 Search PubMed.
- Recent Advances in Nucleosides_Chemistry and Chemotherapy, ed. C. K. Chu, Elsevier Science, Amsterdam (NL), 2002 Search PubMed.
- Chemical Synthetic Biology, ed. P. L. Luisi and C. Chiarabelli, John Wiley & Sons, Chichester (UK), 2011 Search PubMed.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386 CrossRef CAS PubMed.
- M. Iwaoka, S. Takemoto and S. Tomoda, J. Am. Chem. Soc., 2002, 124, 10613 CrossRef CAS PubMed.
- P. Sanz, O. Mó and M. Yáñez, Phys. Chem. Chem. Phys., 2003, 5, 2942 RSC.
- D. B. Werz, R. Gleiter and F. Rominger, J. Am. Chem. Soc., 2002, 124, 10638 CrossRef CAS PubMed.
- S. Zahn, R. Frank, E. Hey-Hawkins and B. Kirchner, Chem. – Eur. J., 2011, 17, 6034 CrossRef CAS PubMed.
- P. Kilian, A. M. Z. Slawin and J. D. Woollins, Chem. – Eur. J., 2003, 9, 215 CrossRef CAS PubMed.
- S. Scheiner, J. Chem. Phys., 2011, 134, 094315 CrossRef PubMed.
- J. E. Del Bene, I. Alkorta, G. Sanchez-Sanz and J. Elguero, J. Phys. Chem. A, 2012, 116, 9205 CrossRef CAS PubMed.
- A. Bauza, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317 CrossRef CAS PubMed.
- A. Bauza, T. J. Mooibroek and A. Frontera, Chem. – Eur. J., 2014, 20, 10245 CrossRef CAS PubMed.
- A. Bauza, T. J. Mooibroek and A. Frontera, Chem. Commun., 2014, 50, 12626 RSC.
- S. J. Grabowski, Phys. Chem. Chem. Phys., 2014, 16, 1824 RSC.
- P. Politzer, K. E. Riley, F. A. Bulat and J. S. Murray, Comput. Theor. Chem., 2012, 998, 2 CrossRef CAS.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178 RSC.
- J. S. Murray, P. Lane, T. Clark and P. Politzer, J. Mol. Model., 2007, 13, 1033 CrossRef CAS PubMed.
- J. S. Murray, P. Lane and P. Politzer, Int. J. Quantum Chem., 2007, 107, 2286 CrossRef CAS.
- J. S. Murray, P. Lane and P. Politzer, J. Mol. Model., 2009, 15, 723 CrossRef CAS PubMed.
- J. S. Murray, K. E. Riley, P. Politzer and T. Clark, Aust. J. Chem., 2010, 63, 1598 CrossRef CAS.
- L. M. Azofra, I. Alkorta and S. Scheiner, Theor. Chem. Acc., 2014, 133, 1586 CrossRef.
- K. Kayukova and N. Nikolaev, Russ. J. Org. Chem., 2004, 40, 1382 CrossRef.
- O. E. Nasakin, P. M. Lukin and A. V. Sadovoi, Russ. J. Org. Chem., 1993, 29, 1598 Search PubMed.
- E. Sheverdov, S. Nasakin and K. Chernushkin, Russ. J. Org. Chem., 2000, 70, 1251 Search PubMed.
- J.-Y. Lee and H. K. Hall, J. Org. Chem., 1990, 55, 4963 CrossRef CAS.
- M. N. Elinson, A. N. Vereshchagin, N. O. Stepanov, A. I. Ilovaisky, A. Y. Vorontsov and G. I. Nikishin, Tetrahedron, 2009, 65, 6057 CrossRef CAS.
- N. Noroozi Pesyan, M. A. Kimia, M. Jalilzadeh and E. Sahin, J. Chin. Chem. Soc., 2013, 60, 35 CrossRef.
- J. S. Murray and P. Politzer, Comput. Mol. Biosci., 2011, 1, 153 CrossRef CAS.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9 CrossRef CAS.
- J. F. Gal, P. C. Maria, M. Decouzon, O. Mó, M. Yáñez and J. L. M. Abboud, J. Am. Chem. Soc., 2003, 125, 10394 CrossRef CAS PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229 CrossRef CAS PubMed.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS PubMed.
- D. A. Britz and A. N. Khlobystov, Chem. Soc. Rev., 2006, 35, 637 RSC.
- D. M. Guldi, F. Zerbetto, V. Georgakilas and M. Prato, Acc. Chem. Res., 2005, 38, 38 CrossRef CAS PubMed.
- A. Forni, S. Pieraccini, S. Rendine and M. Sironi, J. Comput. Chem., 2014, 35, 386 CrossRef CAS PubMed.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp06350g |
| This journal is © the Owner Societies 2016 |