
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of conserved Cys residues in Brassica rapa auxin amidohydrolase: Cys139 is crucial for the enzyme activity and Cys320 regulates enzyme stability†
Ana
Smolko
a,
Filip
Šupljika
b,
Jelena
Martinčić
a,
Nina
Jajčanin-Jozić
b,
Marina
Grabar-Branilović
c,
Sanja
Tomić
c,
Jutta
Ludwig-Müller
d,
Ivo
Piantanida‡
*b and
Branka
Salopek-Sondi
*a
aDivision of Molecular Biology, Ruđer Bošković Institute, 10000 Zagreb, Croatia. E-mail: salopek@irb.hr; Fax: +385-1-4561-177; Tel: +385-1-4561-143
bDivision of Organic Chemistry and Biochemistry, Ruđer Bošković Institute, 10000 Zagreb, Croatia. E-mail: ivo.piantanida@irb.hr
cDivision of Physical Chemistry, Ruđer Bošković Institute, 10000 Zagreb, Croatia
dInstitute of Botany, Technische Universität Dresden, 01062 Dresden, Germany
First published on 26th February 2016
Abstract
Brassica rapa auxin amidohydrolase (BrILL2) participates in the homeostasis of the plant hormones auxins by hydrolyzing the amino acid conjugates of auxins, thereby releasing the free active form of hormones. Herein, the potential role of the two conserved Cys residues of BrILL2 (at sequence positions 139 and 320) has been investigated by using interdisciplinary approaches and methods of molecular biology, biochemistry, biophysics and molecular modelling. The obtained results show that both Cys residues participate in the regulation of enzyme activity. Cys320 located in the satellite domain of the enzyme is mainly responsible for protein stability and regulation of enzyme activity through polymer formation, as has been revealed by enzyme kinetics and differential scanning calorimetry analysis of the BrILL2 wild type and mutants C320S and C139S. Cys139 positioned in the active site of the catalytic domain is involved in the coordination of one Mn2+ ion of the bimetal center and is crucial for the enzymatic activity. Although the point mutation Cys139 to Ser causes the loss of enzyme activity, it does not affect the metal binding to the BrILL2 enzyme, as has been shown by isothermal titration calorimetry, circular dichroism spectropolarimetry and differential scanning calorimetry data. MD simulations (200 ns) revealed a different active site architecture of the BrILL2C139S mutant in comparison to the wild type enzyme. Additional possible reasons for the inactivity of the BrILL2C139S mutant have been discussed based on MD simulations and MM-PBSA free energy calculations of BrILL2 enzyme complexes (wt and C139S mutant) with IPA-Ala as a substrate.
1. Introduction
Auxin amidohydrolases belong to the metallopeptidase family M20, subfamily M20D (MEROPS: the Peptidase Database, http://merops.sanger.ac.uk/),1 and are related to the amidohydrolase superfamily. The enzymes of the amidohydrolase superfamily catalyze the hydrolysis of a wide range of substrates containing amide or ester functional groups at carbon and phosphorus centers.2 Reactions catalyzed by enzymes in this superfamily include deamination, dephosphorylation, decarboxylation, dechlorination, and isomerization, and are important in amino acid and nucleotide metabolisms as well as biodegradation of agricultural and industrial compounds. Many enzymes in the M20 peptidase family are Zn2+-dependent, but the auxin amidohydrolases prefer Mn2+ as a metal cofactor. They participate in the regulation of the activity of the plant hormones auxins.3,4 Auxins are well known as crucial plant hormones involved in many aspects of growth and development, such as cell division, cell elongation, root and shoot growth, flower and fruit development, and tropisms. High concentrations of free auxins, of which the most common is indole-3-acetic acid (IAA), are toxic for plants, so only about 5% of the total concentration of auxin molecules in plants is in the free (active) form, while the rest is stored in inactive forms, mostly as amino acid and sugar conjugates.3 Different plant species have distinct profiles of auxin conjugates; monocots store preferably ester conjugates, whereas dicots synthesize mostly amide conjugates.5Auxin amidohydrolases specifically hydrolyze the amide bond of amino acid conjugated auxins, releasing free active compounds. Such hydrolases (named ILR, IAR and ILL) were first cloned from Arabidopsis thaliana,6–8 and the purified recombinant proteins showed activity towards the conjugates of the most common auxin IAA, with a variety of amino acids. Homologous amidohydrolases from Triticum aestivum, Medicago truncatula, and Brassica rapa were found to be more active towards amino acid conjugates of long-chained auxins: indole-3-butyric acid (IBA-Ala) and indole-3-propionic acid (IPA-Ala) in comparison to IAA-Ala.9–11
Although these proteins have been known for quite some time, their structure–function relationship is still not clear. The first X-ray structure of a plant enzyme (ILL2 from Arabidopsis thaliana) has been reported as an apoenzyme,12,13 still missing details about the substrate binding site and the amino acid residues important for substrate specificity. Based on the modelling results, a potential substrate binding cleft has been proposed for the Arabidopsis enzyme (AtILL2),13 as well as for the Brassica rapa enzyme (BrILL2),11 in which several substrate binding modes for the preferred long-chained auxin conjugates were additionally predicted. Details of the active site of a similar enzyme, (S)-ureidoglycolate amidohydrolase, have recently been reported.14 Structurally, auxin amidohydrolases are characterized by two perpendicular domains with the larger catalytic domain bearing a binuclear metal center, and the smaller ‘‘satellite’’ domain, usually functioning as a polymerization site (Fig. 1). The larger domain contains the active site and consists of β-strands with α-helical bundles on both sides. The smaller satellite domain adopts an α/β-sandwich topology. In other M20 peptidases, the satellite domain serves as a dimerization interface. However, auxin amidohydrolases function as monomers and need a reducing agent, such as dithiothreitol (DTT) for their activity in the enzyme assay in vitro.11
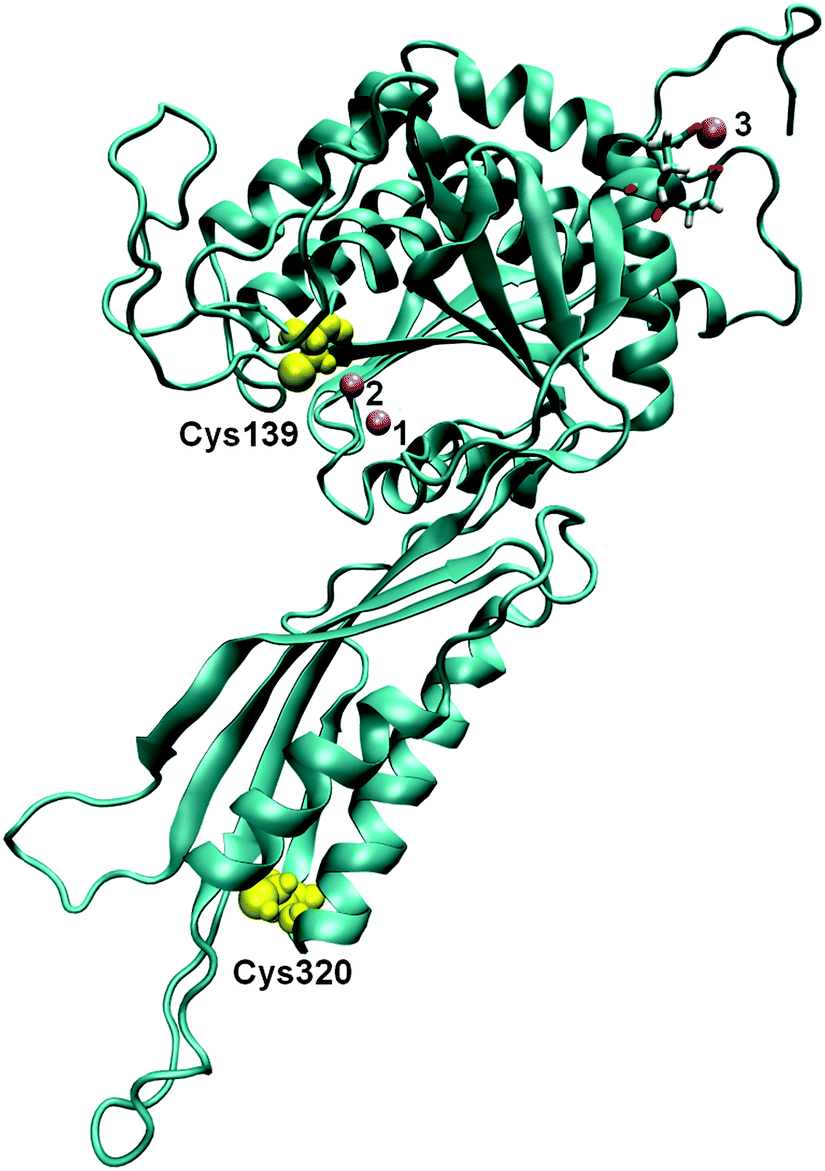 | ||
| Fig. 1 The 3D structure of BrILL2 obtained by comparative modelling. Three Mn2+ ions are presented as violet spheres (numbered 1, 2 and 3). Two of them (1 and 2) are placed in the active site within the protein according to their position in the structure of an amidohydrolase, SACOL0085, from methicillin-resistant Staphylococcus aureus (pdb: 4EWT). The position of the third one was determined by molecular modelling. The positions of two cysteine residues are shown as yellow spheres: Cys139 near the active site within the catalytic domain and Cys320 within the smaller satellite domain. | ||
Based on the sequence homology, auxin amidohydrolases contain two highly conserved cysteine residues.11 It has been proposed previously that one of the two conserved Cys residues in the B. rapa ILL2 enzyme, Cys139, is a part of the active site and coordinates the metal co-factor.11
Based on our previous finding that the activity of BrILL2 depends on a reducing agent, and since molecular modelling indicated the importance of Cys, we undertook this study to examine the potential role of the conserved Cys residues in this metalloenzyme. For that purpose, Cys mutants (C139S and C320S as well as the double mutant C139,320S) were generated and comparative biochemical characterization of the mutant and the wild type enzymes was performed, including the determination of enzyme kinetics with the substrate IPA-Ala. Furthermore, our research involved biophysical (HR-ICP-MS, ITC, CDS, and DSC) and theoretical (MD simulations) studies in order to examine metal binding to the enzyme, and the importance of Cys139 in the coordination of the metal cofactor. Finally, the role of Cys residues in the enzyme activity regulation will also be discussed.
2. Results and discussion
2.1. Importance of the conserved Cys residues for enzyme activity and stability
The auxin amidohydrolase BrILL2 contains two highly conserved cysteine residues (Cys139 and Cys320) (Fig. 1). Many proteins and enzymes in the cell contain cysteine residues which have a sulfhydryl group in their side chain and with the labile proton which makes it a chemical hot spot for a wide variety of biochemical interactions. Such a thiol group provides sites for the formation of coordinate bonds with metal ions, sites for electrophilic reactions, and an easily reversible site for oxidation/reduction chemistry. The importance of Cys residues for the BrILL2 enzyme activity was firstly noticed by pre-treatments of the BrILL2 wild type (wt) enzyme with iodo-acetamide (I-acetamide). The enzyme irreversibly lost activity due to alkylation by I-acetamide. Based on the proposed model of the active site, Cys139 is suggested as a residue of the binding pocket and potentially participates in metal binding. To prove this assumption, we generated mutants in which Cys residues were replaced by Ser amino acids, and investigated the role of Cys residues in the regulation of enzyme activity.The results of CD spectropolarimetry of the mutant enzymes (ESI,† Fig. S1) confirmed that none of the mutations changed the secondary structure of the protein, thus excluding structure perturbations as possible reasons for changes in the enzyme activity. Based on our specific enzyme activity assay, the BrILL2C139S mutant was inactive, while the BrILL2C320S mutant appeared to be approximately 38% less active in comparison to BrILL2wt (Table 1). Furthermore, enzyme kinetics experiments with the substrate IPA-Ala were performed with the active mutant C320S and wt enzymes. As can be seen the Km value has not been changed for the C320S mutant, while the Kcat was 1.6-fold lower for the C320S mutant in comparison to the wt. It seems that the Cys320 residue influenced the enzyme activity regardless of its distance from the active site. So what could be the role of a distant Cys residue in the regulation of BrILL2 activity? As we already mentioned, auxin amidohydrolases are active in the monomeric form. In the absence of a reducing agent, BrILL2 is prone to form dimers and even higher oligomers, which are not active in an in vitro enzyme assay. To investigate whether Cys residues have a role in the oligomer formation, we have applied gradient SDS-PAGE and Western blot analysis using anti-His-tag antibodies and compared the potential of the BrILL2wt and the mutants C139S, C320S and C139,320S for polymerization under reducing vs. non-reducing conditions (Fig. 2). As can be seen, the wild type BrILL2 forms dimers and higher oligomers under non-reducing conditions. Similarly, both C139S and C320S are capable of forming dimers to a lesser extent than BrILL2wt, whereas this capacity is completely absent in the double C139,320S mutant. To learn more about the enzyme stability under reducing and non-reducing conditions, differential scanning calorimetry (DSC) measurements were done (Table 2) in parallel with Western blot analysis (Fig. 2).
| Protein | Specific activity (nmol mg−1 min−1) | V max (μM min−1) | K m (mM) | K cat (min−1) | K cat/Km (mM−1 min−1) |
|---|---|---|---|---|---|
| Wt | 2261 ± 320 | 0.0316 ± 0.005 | 0.61 ± 0.3 | 157 ± 25 | 258.4 |
| C139S | nd | nd | nd | nd | nd |
| C320S | 1143 ± 18 | 0.0195 ± 0.002 | 0.59 ± 0.18 | 97 ± 11 | 163.7 |
| C139,320S | nd | nd | nd | nd | nd |
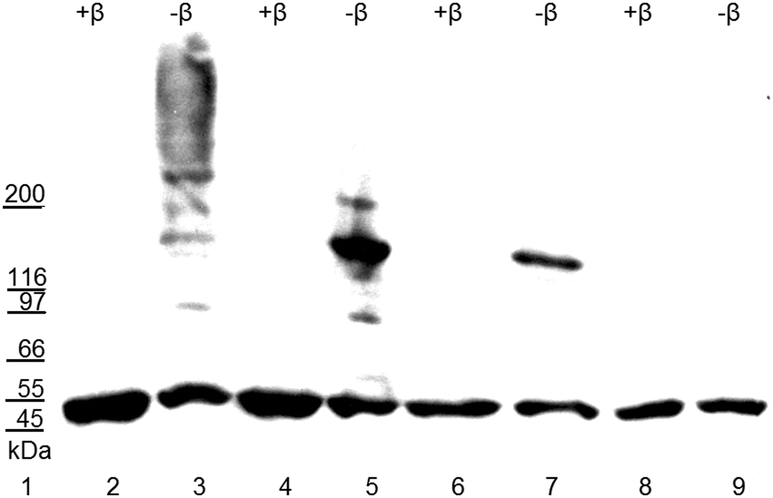 | ||
| Fig. 2 Electro-blot of the purified recombinant BrILL2 enzymes (wt and mutants) resolved by SDS-PAGE in 4–20% gradient gel under non-reducing conditions (−β), and reducing conditions by adding β-mercaptoethanol (+β). Proteins were blotted onto a PVDF membrane and visualized by chemiluminescence with the use of anti-His-tag antibodies. Lane: 1, molecular weight markers; 2–3, wt BrILL2; 4–5, mutant C139S; 6–7, mutant C320S; 8–9, double mutant C139,320S. 4 μg (lanes 6–9) or 5 μg (lanes 2–5) of proteins was loaded onto gel lanes. | ||
| BrILL2 | T max/°C | ΔrH/kJ mol−1 | ΔrS/J K−1 mol−1 |
|---|---|---|---|
| wt −β | 56.4 | 60.6 | 183.7 |
| wt +β | 54.4 | 89.6 | 273.5 |
| C139S −β | 56.8 | 93.8 | 284.3 |
| C139S +β | 55.2 | nd | nd |
| C320S −β | 53.4 | 59.6 | 182.4 |
| C320S +β | 52.2 | 16.8 | 51.6 |
DSC measures the excess heat capacity of a solution (Cp) as a function of temperature. When a protein changes its thermodynamic state (e.g., unfolds), a heat capacity change (ΔrCp) is observed.15 This change is due to the fact that the heat required to increase the temperature of a solution of unfolded proteins is greater than that required for a solution of folded proteins. Tmax is an indicator of thermostability and generally, the higher the Tmax, the more thermodynamically stable the protein. Integration of the Cpversus T curve yields the transition enthalpy (ΔrH) and the shift in the baseline yields the ΔrCp. The value ΔrH, calculated from the area under the transition peak, is correlated with the content of the ordered secondary structure within a protein.16
Denaturation of the BrILL2 enzyme is thermodynamically irreversible (ESI,† Fig. S11–S13), so all the observed thermodynamic data were obtained from the first scan. The denaturation process is also slightly kinetically controlled (ESI,† Fig. S11) so the analysis of the thermograms on the basis of equilibrium thermodynamics is ruled out. As can be seen in Table 2, the Tmax for the non-reduced wt enzyme was 56.4 °C. The point mutation Cys139 to Ser did not influence the Tmax of the enzyme, while the mutation Cys320 to Ser caused a decrease of the Tmax by approximately 3 °C. This suggests that the mutant C320S is less stable in the non-reduced form, compared to the wt and C139S mutant. Studies on similar systems are rather scarce in the literature, and, so far, there are no available data on the representatives of the M20 peptidase family. We compared thermodynamic data of several peptidases with the herein obtained data for BrILL2. Carboxypeptidase B is a globular enzyme, but it is of a somewhat smaller size (procarboxypeptidase B: 45.5 kDa and carboxypeptidase B: 34.5 kDa) in comparison to BrILL2 (49.8 kDa). DSC analysis revealed similar irreversible thermal denaturation for carboxypeptidase B (Tmax = 55–70 °C), but larger ΔrH values (500–800 kJ mol−1) in comparison to BrILL2 (60–90 kJ mol−1).17 Furthermore, procarboxypeptidase A and carboxypeptidase A from porcine pancreas (46.4 kDa and 34.8 kDa, respectively) revealed also irreversible thermal denaturation in a significantly larger temperature span (Tmax = 40–70 °C), and a ΔrH value of approximately 600 kJ mol−1.18 Leucine aminopeptidase revealed a Tmax almost identical to the herein studied BrILL2 (57 ± 0.5 °C) and a three-fold higher ΔrH value of 80 ± 0.1 kcal mol−1 (335 ± 0.4 kJ mol−1).19 Thus, the compared enzymes, having similar general functions, and approximately comparable sizes are characterised by similar thermodynamic parameters of denaturation.
All enzymes showed lower Tmax values and consequently certain protein destabilization upon adding a reducing agent. The ΔrH value for the C320S mutant is significantly lower than that measured for the wt and C139S mutant, in both, polymeric and monomeric forms. These results suggest that both Cys residues contribute to the regulation of enzyme activity through the formation of non-active polymers by disulfide bonds. However, the Cys320 residue seems to be more important in protein stabilization, while Cys139 is directly responsible for the enzyme activity. We attempted to purify BrILL2 in the presence of a reducing agent, i.e. in the monomeric form. However, the purified protein was inactive. This confirmed that BrILL2 is highly unstable in the monomeric form. Werner et al.20 reported that the monomeric form of allantoate amidohydrolase is the active form of the enzyme, but is less stable than polymeric forms which is in agreement with our finding. Based on our experimental results, we propose a possible enzyme activity regulation in vivo by dissociation of polymers in the presence of natural reductants. Our hypothesis is that auxin amidohydrolases are active in the monomeric form, but, as such a form is highly unstable, the enzymes have a tendency to stabilize by forming polymers in vivo. To prove the effectiveness of the potential natural reducing agents on the activation of the BrILL2 enzyme, we examined the influence of various naturally occurring reducing agents such as reduced glutathione (GSH), ascorbic acid (AA), and the amino acid Cys on enzyme activity (Table 3). GSH, AA and Cys were as equally potent in the activation of BrILL2 as DTT and β-mercaptoethanol which are routinely used in the enzyme assay. These results again implicated a lower activity of the C320S mutant in comparison to the wt. Both enzymes were reversibly inactivated in the absence of any reducing agent, while both of them lost activity irreversibly upon alkylation with I-acetamide (Table 3). The activity of auxin amidohydrolases could be regulated on demand through redox-switches in the plant cell which include natural reducing agents such as GSH, AA and Cys. Good examples in the plant kingdom are seed proteins that undergo redox changes during development and germination.21 Upon synthesis, proteins become oxidized to a more stable disulfide (S–S) state during maturation and drying. Upon germination, the proteins are converted back to the reduced state to facilitate mobilization. The importance of highly conserved Cys residues in the stabilization of protein and redox regulation of activity has also been demonstrated in one type of protein tyrosine phosphatases (PTPs) (Src homology 2 domain containing PTPs (SHPs)).22 Furthermore, glycine decarboxylase (P-protein) was also reported as an enzyme regulated by cellular redox homeostasis.23 The formation of homodimers by disulfide bonds is subject to regulation by the redox status of the cell and restricts access of the substrate to the active site. Under conditions favoring catalysis, the disulfide bond is broken enabling the opening of an unlocked position of the active site.23
| BrILL2wt | BrILL2C320S | BrILL2C139S | |
|---|---|---|---|
| No thiol agent | nd | nd | nd |
| DTT (1 mM) | 1.87 ± 0.19 | 1.64 ± 0.67 | nd |
| β-MeOH (1 mM) | 1.55 ± 0.70 | 1.62 ± 1.1 | nd |
| GSH (1 mM) | 1.13 ± 0.41 | 0.70 ± 0.03 | nd |
| AA (1 mM) | 0.99 ± 0.66 | 0.16 ± 0.07 | nd |
| Cys (1 mM) | 1.24 ± 0.85 | 1.24 ± 0.51 | nd |
| I-Acetamide | nd | nd | nd |
2.2. Role of the conserved Cys139 residue in metal coordination
Metalloproteins and metalloenzymes that contain binuclear active sites are prevalent in nature.24 A structurally similar (S)-ureidoglycolate amidohydrolase has been reported to contain two Mn2+ ions in the active site.14 In order to experimentally confirm the number of Mn2+ ions bound to the BrILL2 active site, and to investigate whether Cys139 participates in the Mn2+ binding, high resolution inductively coupled plasma mass spectrometry (HR-ICP-MS) and isothermal titration calorimetry (ITC) analyses of BrILL2wt and the C139S mutant were performed.
HR-ICP-MS of the wt and C139S mutant were done in samples following purification (as described in Methods), in samples which were chelated by EDTA and samples which were saturated by Mn2+ after chelation by EDTA. The results showed that the purified BrILL2wt enzyme was slightly saturated with Mn2+ during protein production (Mn2+/enzyme ratio: 0.114). The Mn2+/apoenzyme ratio was 0.068, while the Mn2+/enzyme ratio upon saturation with Mn2+ was 5.4 which was much higher than that expected based on the previous modelling results (ESI,† Table S1).
To confirm these findings, ITC measurements were done by using a multiple injection method. Fig. 3 shows a representative ITC titration profile of BrILL2wt with the MnCl2·4H2O (the ITC titration profile of the C139S mutant is shown in Fig. S2, ESI†). The reverse titration profile of MnCl2·4H2O with BrILL2wt is shown in Fig. S4, ESI†. The blank titration (titration of MnCl2·4H2O into the buffer solution) (ESI,† Fig. S3) was subtracted from each ITC titration (titration of BrILL2wt with MnCl2·4H2O). Each of the peaks in the figures corresponds to a single injection. The areas under these peaks were determined by integration to yield the associated injection heats, which were plotted against the respective molar ratios. The data points reflect the experimental injection heats, while the solid lines reflect calculated fits of data. Fitting procedures were performed using a model with one set of sites. The equilibrium constant, binding stoichiometry, reaction enthalpy change, entropy contribution, and Gibbs energy change obtained from the calorimetric data are summarized in Table 4. It is noteworthy that the results obtained from titration of BrILL2wt with MnCl2 (Table 4; N = 5.6 ± 0.3; log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ka = 5.5 ± 0.1; ΔrG = −31.6 ± 0.8 kJ mol−1) and reverse titration (Fig. S4 (ESI†), N = 5.6 ± 0.4; logKa = 5.4 ± 0.2; ΔrG = −30.7 kJ mol−1) are almost identical. The results show no significant differences in titration profiles of binding MnCl2 to BrILL2wt (Fig. 3) and the C139S mutant (ESI,† Fig. S2). In general, the binding of MnCl2 to BrILL2wt and the BrILL2C139S mutant is favored by small negative enthalpies and large positive entropy changes. For example, the binding of Mn2+ to the TroR enzyme was also reported as an entropy-driven process.25 The fitting of the experimental data revealed the binding stoichiometry of Mn2+/enzyme (wt or C139S) in the range of 5–6, which is in agreement with the HR-ICP-MS results. The enzymes studied here had a His-tag (hexa histidine-tag) for purification reasons, and the His-tag is well known to bind not only Ni2+ cations, but other cations as well.26,27 Therefore, we also performed ITC experiments of a single His-tag with MnCl2, which resulted in surprisingly high binding constants and a Mn2+/His-tag stoichiometry equal to 2:1 (Table 4). The results obtained suggest that in MnCl2–enzyme titrations two Mn2+ ions are located in the active site of the enzyme, two are bound to the His-tag, and the rest (1 or 2) are electrostatically bound to the non-specific sites along the enzyme. Further modelling analysis confirmed this hypothesis (see Section 2.2.3).
Ka = 5.5 ± 0.1; ΔrG = −31.6 ± 0.8 kJ mol−1) and reverse titration (Fig. S4 (ESI†), N = 5.6 ± 0.4; logKa = 5.4 ± 0.2; ΔrG = −30.7 kJ mol−1) are almost identical. The results show no significant differences in titration profiles of binding MnCl2 to BrILL2wt (Fig. 3) and the C139S mutant (ESI,† Fig. S2). In general, the binding of MnCl2 to BrILL2wt and the BrILL2C139S mutant is favored by small negative enthalpies and large positive entropy changes. For example, the binding of Mn2+ to the TroR enzyme was also reported as an entropy-driven process.25 The fitting of the experimental data revealed the binding stoichiometry of Mn2+/enzyme (wt or C139S) in the range of 5–6, which is in agreement with the HR-ICP-MS results. The enzymes studied here had a His-tag (hexa histidine-tag) for purification reasons, and the His-tag is well known to bind not only Ni2+ cations, but other cations as well.26,27 Therefore, we also performed ITC experiments of a single His-tag with MnCl2, which resulted in surprisingly high binding constants and a Mn2+/His-tag stoichiometry equal to 2:1 (Table 4). The results obtained suggest that in MnCl2–enzyme titrations two Mn2+ ions are located in the active site of the enzyme, two are bound to the His-tag, and the rest (1 or 2) are electrostatically bound to the non-specific sites along the enzyme. Further modelling analysis confirmed this hypothesis (see Section 2.2.3).
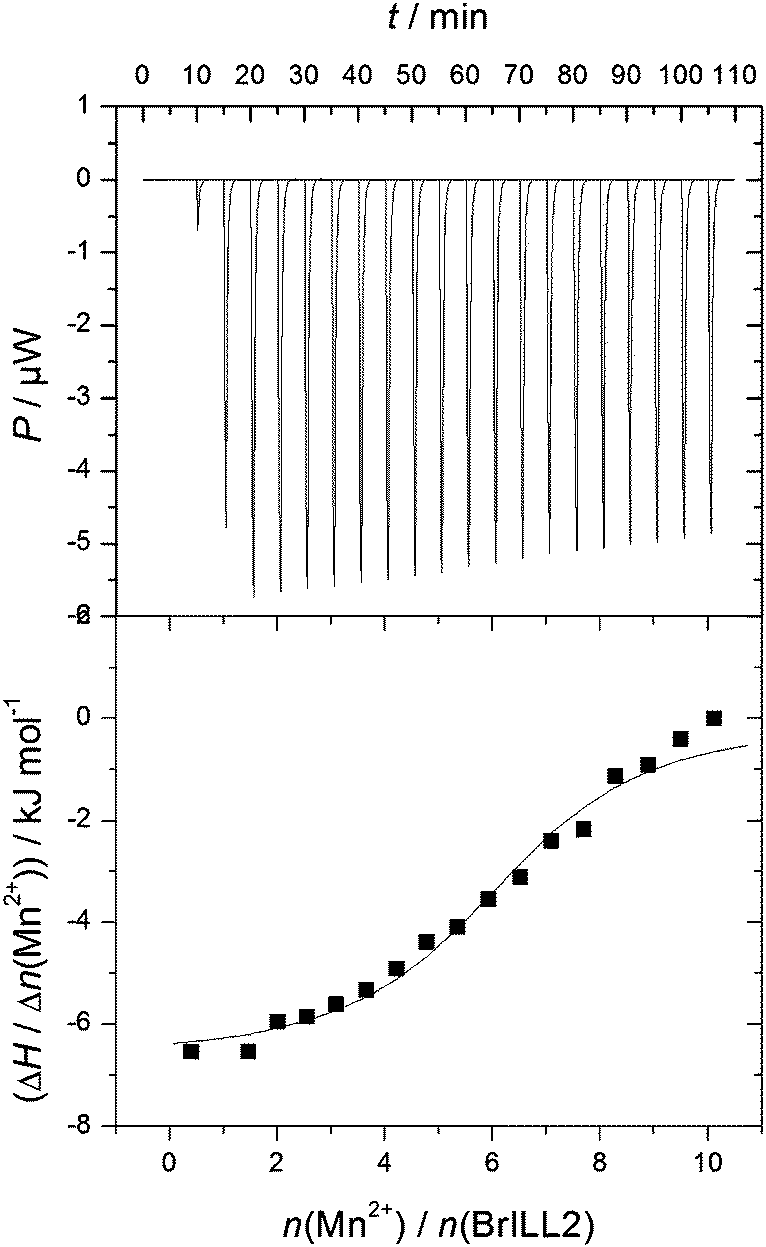 | ||
| Fig. 3 Representative ITC titration profile of the BrILL2wt enzyme (5 × 10−6 mol dm−3) with MnCl2·4H2O (2.5 × 10−4 mol dm−3). | ||
| Enzyme | N | logKa |
ΔrH/kJ mol−1 | ΔrS/J K−1 mol−1 | TΔrS/kJ mol−1 | ΔrG/kJ mol−1 |
|---|---|---|---|---|---|---|
| BrILL2wt | 5.6 ± 0.3 | 5.5 ± 0.1 | −6.2 ± 0.6 | 85.1 ± 3.5 | 26.3 | −31.1 ± 0.8 |
| BrILL2C139S | 5.5 ± 0.2 | 5.6 ± 0.1 | −8.6 ± 0.4 | 78.2 | 23.3 | −31.9 |
| His-tag | 2.0 | 4.6 ± 0.1 | 1.5 ± 0.1 | 92.2 ± 0.8 | 27.5 | −26.1 ± 0.3 |
| Enzyme | T max/°C | ΔrH/kJ mol−1 | ΔrS/J K−1 mol−1 |
|---|---|---|---|
| BrILL2wt | 56.5 ± 0.1 | 54.6 ± 6.0 | 165.7 ± 18.1 |
| Mn–BrILL2wt complex | 56.2 ± 0.2 | 73.9 ± 19.4 | 224.5 ± 58.9 |
| BrILL2C139S | 56.8 | 93.8 ± 2.1 | 284.3 ± 6.5 |
| Mn–BrILL2C139S complex | 57.2 ± 0.3 | 46.7 ± 2.6 | 141.3 ± 7.8 |
MD simulations of the ligand free enzyme: the protein structure and the metal binding site. 200 ns long MD simulations of the native BrILL2 and the BrILL2C139S mutant, with serine in two different protonation states, Cys139Ser and Cys139Ser−, with and without Mn2+ ions bound into the assumed active site were performed. The results obtained by the MD simulations are in agreement with the experimental DSC and CD data which suggested that neither the presence of the manganese ions nor the Cys139 to Ser mutation affects the protein 3D structure. The presence of the metal ions has no influence on the overall protein structure during the MD simulations, and no significant differences in the structure and flexibility/stability of the wt protein and the mutant were traced over 200 ns of MD simulations in solvent at room temperature (Fig. 4, and ESI,† Fig. S5). Similarly, the active site architecture, especially the surrounding of the metal ion at position 2, i.e. the more buried ion (Mn2+ at position 1 is closer to the active site entrance than Mn2+ at position 2, and so is more solvated) was not affected by the Cys139 to Ser mutation (ESI,† Fig. S6). However, while in the mutant with the deprotonated serine 139 (C139S−) the position of manganese ions in the active site does not differ significantly from their position in the wild type enzyme and their average distance is about 4 Å during all 200 ns MD simulations; in the variant with neutral serine, C139S, the metal ions moved away from each other during the simulations and after 200 ns they were about 7 Å apart from each other (ESI,† Fig. S7 and Table S2).
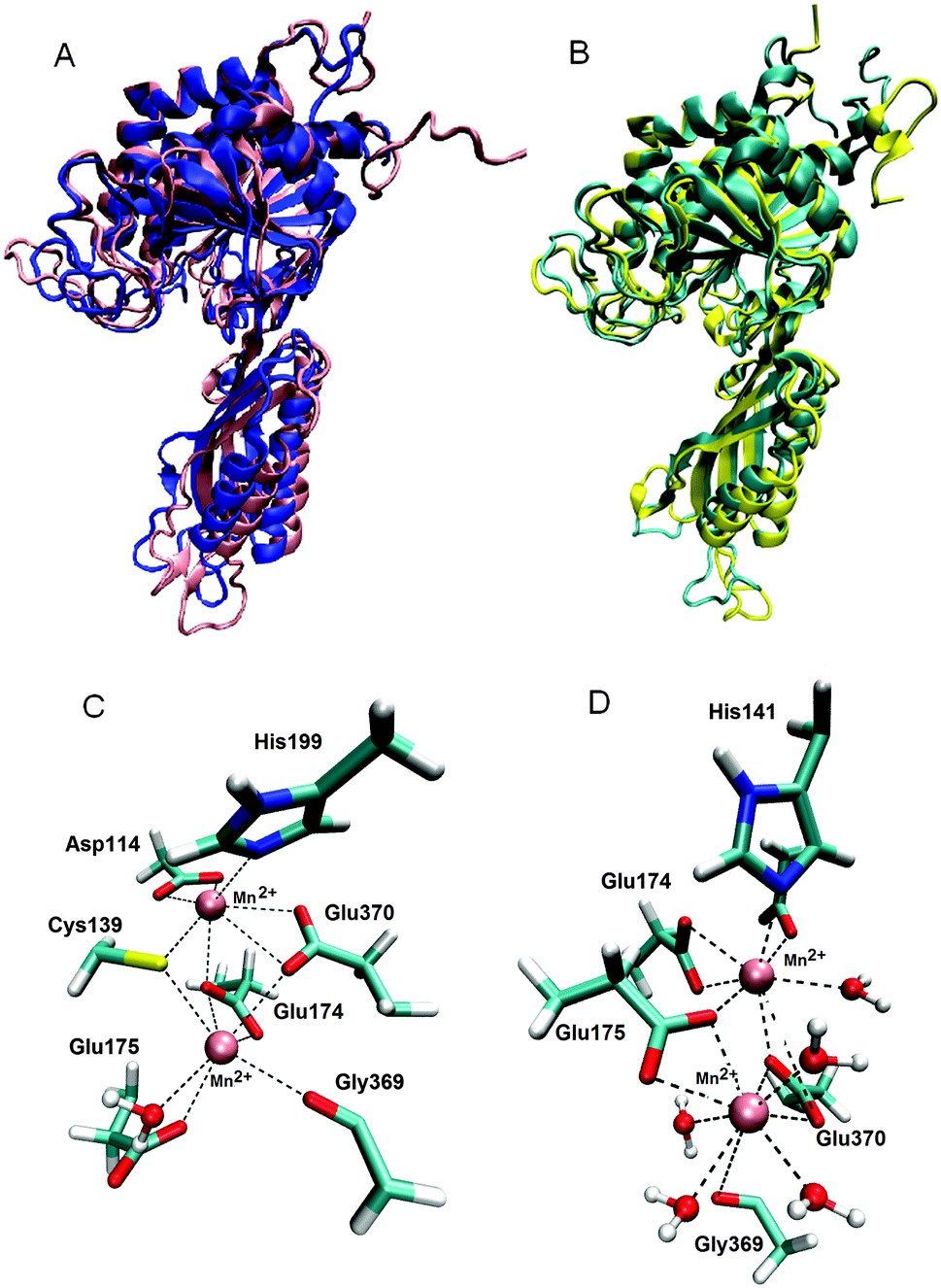 | ||
| Fig. 4 (A) Alignment of the BrILL2wt protein structures obtained after MD simulations in the presence (pink) and absence of Mn2+ ions (blue). (B) Alignment of the mutated (C139S−) structures obtained after MD simulations in the presence (yellow) and absence of Mn2+ ions (cyan). (C) Mn2+ surrounded by the wt protein. All residues (side chains) and water molecules within 3 Å of metal ions are displayed. The lower Mn2+ ion (Mn2+ 1), the metal ion which is closer to the active site entrance, is surrounded (coordinated) by Cys139, Glu174, Glu175, Gly369, Glu370 and one water molecule. The upper Mn2+ ion (Mn2+ 2) is coordinated by Asp114, Cys139, Glu174, His199 and Glu370. (D) Mn2+ surrounded by the mutated protein C139S−. The lower Mn2+ (Mn2+ 1) is coordinated by Glu175, Gly369, Glu370 and four water molecules. The upper Mn2+ ion (Mn2+ 2) is coordinated by Asp114, His141, Glu174, Glu175, Glu370 and two water molecules, see ESI,† Table S3. | ||
The other possible metal ion binding sites were investigated by placing four ions at the minimal energy positions at the protein surface and performing 22 ns long MD simulations. During the simulations only one of the ions from the protein surface, the ion at position 3 near Glu357 (average distance 2.63 Å) and Glu358 (average distance 2.53 Å) (Fig. 1) remained anchored to the protein. The two Mn2+ ions in the enzyme active site together with one Mn2+ ion at the protein surface nicely correlate with the experimentally determined number of Mn2+ ions (3.4) that bind to the BrILL2 enzyme without a His-tag, the wild type and the C139S mutant.
MD simulations of the BrILL2–IPA complexes. Since the metal cations are constituents of the auxin amidohydrolase active site, their significantly different coordination in the C139S variant is a direct explanation of catalytic inactivity of this mutant. In order to elucidate possible reasons for the loss of the enzymatic activity of the C139S mutant we performed MD simulations of BrILL2 complexed with IPA-Ala, for which experiments have shown that it is the most favored BrILL2 substrate.11 For each system, the solvated complexes with the wt protein and the C139S mutant, 200 ns of simulations was accomplished.
Within the wt protein we identified two slightly different ligand binding modes, BM1 and BM2 (ESI†, Fig. S8 and S9). In the mutated complexes the orientation of the substrate differs from the orientations determined in the wt protein (ESI,† Fig. S8 and S9). In all complexes, IPA-Ala coordinates either one or both of the Mn2+ ions during the simulations (Fig. 5 and ESI,† Table S3, Fig. S8 and S9).
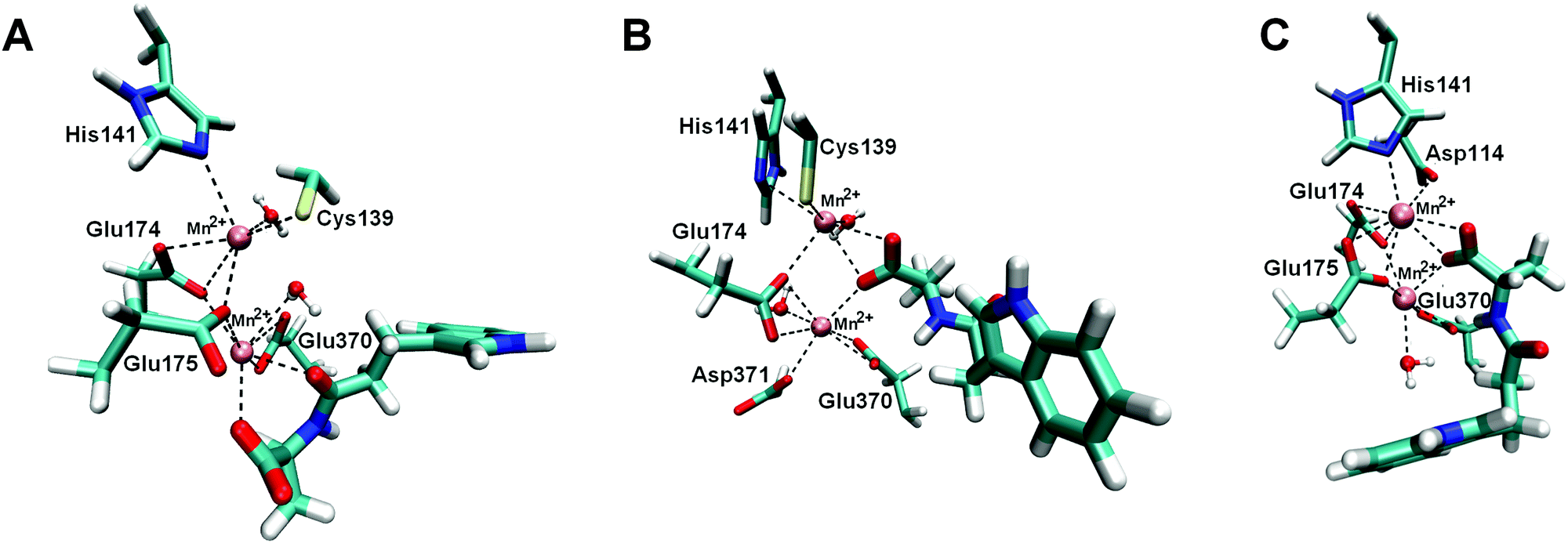 | ||
| Fig. 5 Binding sites of BrILL2–IPA-Ala complexes obtained during MD simulations. Two manganese ions are shown as violet spheres. IPA-Ala is shown in a thicker sticks representation. For simplicity only side chains of amino acids are shown. (A) Complex BrILL2wt–IPA-Ala, binding mode 1 (BM1). The distance between IPA-Ala and Mn2+ is approximately 2.5 Å (shown as dashed lines). (B) Complex BrILL2wt–IPA-Ala, binding mode 2 (BM2). The distance between IPA-Ala and Mn2+ is approximately 2.6 Å (shown as dashed lines). (C) Complex BrILL2C139S−–IPA-Ala. Distance between IPA-Ala and Mn2+ is approximately 2.6 Å (shown as dashed lines). For details of coordination see the ESI,† Table S3. | ||
According to the MM_PBSA calculations performed at the trajectories generated during the last 8 ns (of the total 200 ns of MD simulations in water) the complexes between the wt protein and IPA-Ala are more stable than the mutated C139S−–IPA-Ala (ESI,† Table S4).
In the most stable BrILL2–IPA-Ala binding mode (BM1), both the substrate carbonyl and carboxyl groups coordinate the Mn2+ ion closer to the active site entrance, position 1 (ESI,† Table S5). The indole ring is stabilized by the hydrophobic interactions with Arg203, Leu217, Ile366 and Phe385 (weak face to edge type of stacking interaction) (ESI,† Fig. S9). The substrate NH group of the peptide bond is stabilized by the hydrogen bond to the Gly369 carbonyl, while the methyl group is in the neighbourhood of the hydrophobic Leu177 side chain. The water molecules in the active site are frequently replaced by the water molecules from the bulk; however, for a few of them the exchange ratio is lower than for the others pointing to their better stabilization (ESI,† Table S6). One of such resistant water molecules was found close to the peptide carbonyl of IPA-Ala. This water molecule could be activated either by the carboxyl group of Glu175 or by the carboxyl group of the substrate itself.
In the BM2, IPA-Ala coordinates both Mn2+ ions with its carboxyl group. The indole ring is stabilized by hydrophobic interactions with Arg203 and Leu217. Besides, the carbonyl oxygen of Leu217 occasionally interacts with the indole NH group during the simulations (ESI,† Fig. S9 and Table S5). Thr201 also participates in the indole ring stabilization. The hydrophobic part of the propyl group is stabilized by the side chain of Leu217, and its methyl group with the Ala397 methyl. The NH group of the peptide bond is stabilized by the Glu370 carboxyl; besides this carboxyl group forms a strong H-bond with a water molecule close to the substrate carbonyl. It might be that Glu370 activates this water molecule which then performs the nucleophilic attack to the carbonyl carbon. In the next step, Glu370 donates the H from the activated water to the substrate NH group which is additionally stabilized with the Gly369 oxygen.
In the C139S− mutant, IPA-Ala coordinates both Mn2+ ions with its carboxyl group (ESI,† Fig. S9 and Table S5). The indole ring is stabilized by the hydrophobic interactions with Phe447 and Ile366, while Gly369 carbonyl interacts with the indole hydrogen H1. Despite significant fluctuations, the Arg203 side chain constantly approaches the substrate during MD simulation (ESI,† Fig. S10). In the final structure, it interacts electrostatically with the substrate carbonyl group and hinders nucleophilic attack of water molecules required for the enzymatic reaction.
3. Experimental
3.1. Materials
Commercial, analytical grade chemicals and solvents were used if not stated otherwise.The plasmid pTrcHis2-Topo containing the previously cloned cDNA corresponding to the BrILL2 protein (called simply pBrILL2)28 was used for the expression of the wt enzyme and as a template for generating the mutants.
The bacterial strain E. coli XL-10 Gold (Stratagene, La Jolla, CA, USA) was used for plasmid propagation, and E. coli BL21-Codon-Plus (DE3)-RIL+ (Stratagene, La Jolla, CA, USA) was used for the synthesis of the BrILL2wt protein and the mutants BrILL2C139S, BrILL2C320S, and BrILL2C239,320S. The substrate IPA-Ala was synthesized as previously published.9
3.2. Site-directed mutagenesis and sequencing
Site-directed mutagenesis was performed on the recombinant plasmid pBrILL2. Point mutations of the BrILL2 gene C139S and C320S were carried out using the QuickChange II XL Site-Directed Mutagenesis kit (Stratagene, LaLoya, CA, USA) by using the corresponding pair of primers custom synthesized by Invitrogen (USA). Primers for C139S mutation:Forward: 5′-GGTAAAATGCACGCTTCTGGACACGACCG-3′ and Reverse: 5′-CCGTCGTGTCCAGAAGCGTGCATTTTACC-3′; and for C320S mutation:
Forward: 5′-CGGATGCATTGGACCTCTGAACTGTTGC-3′ and
Reverse: 5′-GCAACAGTTCAGAGGTCCAATGCATCCG-3′.
The double mutant C139,320S was generated by using the mutant plasmid pBrILL2C139S already obtained and the corresponding pair of primers for introducing the C320S mutation.
The DNA sequences of the final constructs were analyzed using an automated sequence analyser ‘‘ABI PRISM_3100-Avant Genetic Analyzer’’ (Applied Biosystem, USA), using a ABI PRISM BigDye Terminator v3.1 Ready Reaction Cycle Sequencing Kit, and commercial T7 forward and T7 reverse primers.
3.3. Protein synthesis and purification
The recombinant proteins of BrILL2, wt and mutants were produced by heterologous expression in the E. coli strain BL21(DE3)RIL+ as previously described.11 For the protein purification and dialysis, as well as subsequent analysis, all buffers were prepared in high purity Milli-Q water obtained from a Millipore purification system (resistivity less than 18 MΩ cm). To isolate recombinant proteins, the bacterial pellet was resuspended in buffer (50 mM phosphate or 50 mM Tris buffer, 300 mM NaCl, 10 mM imidazole, 50 μg mL−1 PMSF, 1 mg mL−1 lysozyme, 0.1% Triton X-100, pH 8.0) and then 10 mL per g of pellet, following incubation on ice for 30 min, was frozen in liquid nitrogen and thawed. The freeze–thaw cycle was repeated two times. The resulting lysate was supplemented with 10 μg DNAse I, (Boehringer Mannheim GmbH), incubated at room temperature (at approximately 23 °C) for 20 minutes and centrifuged (10000g, 40 minutes, 4 °C). proteins were purified by affinity chromatography on a Ni–NTA agarose column according to the manufacturer's instructions (Qiagen). The proteins recovered were quantified according to Bradford's method and subsequently analyzed by SDS-PAGE (12.5% acrylamide).29 Fractions of high purity were pooled and dialyzed overnight at 4 °C against 50 mM Tris, 300 mM NaCl (pH 8.0) to remove the excess of imidazole. Further buffer exchange was performed as described below for the ICP-MS and calorimetric analysis. The dialyzed protein was shortly stored at 4 °C for further studies.
3.4. Circular dichroism spectropolarimetry (CDS)
To investigate the secondary structure of the enzyme mutants obtained in comparison to the wt, CD spectra of recombinant proteins were recorded on a Jasco J-815 CD spectropolarimeter with automatic temperature control using a quartz cuvette of 0.1 mm path length. Enzyme samples were prepared for CD measurements by exchange into 50 mM phosphate buffer, pH 8.0. The protein concentrations used were from 0.31 to 0.39 mg mL−1. The CD spectra were collected in the 190–300 nm range, and analyzed for the protein secondary structure as previously described.303.5. Western blotting
For the analysis of protein polymerization capacity, Western blotting of wt and mutant proteins were performed. The purified proteins, unreduced and reduced by β-mercaptoethanol, were separated by SDS-PAGE in a 4–20% (w/v) polyacrylamide gel. Proteins were then transferred to a polyvinylidene difluoride (PVDF, Millipore, USA) membrane. Following blocking with 5% (w/v) non-fat dry milk (Carl Roth, Germany), blots were incubated in the penta-His antibody (Qiagen), using an antibody/buffer ratio of 1:2000. Horse radish peroxidase (HRP)-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology) was employed as the second antibody in a 1:10000 ratio to the buffer, and chemiluminescence detection was performed using luminol as a substrate and p-coumaric acid as an enhancer according to the method reported by Mruk and Cheng.31
3.6. Enzyme assay
Enzyme reactions were run in 100 mM Tris-HCl pH 8.0 in the presence of 0.2 mM MnCl2 and 1 mM DTT at 37 °C using 0.5 mM IPA-Ala as a substrate IPA-Ala. The effect of the additional reducing agents (β-mercaptoethanol, reduced glutathione, cysteine, and ascorbic acid) was also examined at a concentration of 1 mM. To examine the effect of alkylation of Cys residues on enzyme activity, proteins were also incubated with I-acetamide according to Lundblad's method32 prior to activity measurements. Enzyme kinetics were determined for BrILL2, wt and C320S, by calculating initial reaction rates (v0) in the concentration range of the substrate (IPA-Ala) 0.1–2.0 mM. The progress of the cleavage reaction was monitored by HPLC according to the method reported by Savić et al.113.7. High resolution inductively coupled plasma mass spectrometry (HR-ICP-MS)
For the ICP-MS analysis of the BrILL2 protein, the protein was prepared in three forms: (1) enzyme immediately after purification by affinity chromatography, with a buffer exchanged 3 times into 50 mM Tris-HCl, pH 8.0; (2) an apoenzyme obtained by a 3 hour dialysis in 10 mM EDTA, 50 mM Tris, 100 mM NaCl, pH 8.0, which was afterwards dialyzed as in (1); and (3) a holoenzyme prepared by firstly chelating the non-specifically bound metal ions with a 3 hour dialysis in 10 mM EDTA, 50 mM Tris, 100 mM NaCl, pH 8.0, following 3 times exchange into 50 mM Tris, 100 mM NaCl, pH 8.0, and then saturated by 1 mM MnCl2 in the same dialysis buffer. Excess of MnCl2 was removed by further exchanging the buffer 3 times into 50 mM Tris-HCl, pH 8.0. All containers used in ICP-MS were previously cleaned by soaking for several days in 10% HNO3 solution and rinsed with Milli-Q water before use. The measurements of 55Mn were performed in the medium resolution mode on a high-resolution inductively coupled plasma mass spectrometer (HR ICPMS Element 2, Thermo Finnigan, Germany) equipped with an autosampler ESI-a SC-2 DX FAST (Elemental Scientific, USA). The typical instrumental conditions and measurement parameters used throughout the work were reported previously.33 External calibration was performed using standard solutions prepared from multielement stock standard solutions for trace elements (Analitika, Czech Republic) in which a single element standard solution of U (Aldrich, USA) was added.3.8. Isothermal titration calorimetry (ITC)
To study the metalation of BrILL2 with Mn2+ ions by ITC and DSC, the protein solution obtained as in Section 3.3 was further dialyzed to remove any unbound metal ions for 3 h with 10 mM EDTA, 50 mM Tris-HCl, 100 mM NaCl, pH 8.0 and the removal of EDTA was achieved by exchanging the dialysis buffer 3 times into 50 mM Tris-HCl, pH 8.0 and 3 times into 50 mM cacodylate, pH 7.4. In order to examine metal binding to the wt and C139S mutant, isothermal titration calorimetry (ITC) experiments were performed on a MicroCal VP-ITC microcalorimeter (MicroCal, Inc., Northampton, MA, USA). Origin 7.0 software, supplied by the manufacturer was used for data analysis. The reference cell was filled with ultrapure water. In the experiments, one aliquot of 2 μL and 28 aliquots of 10 μL of the compound MnCl2 (c = 2.5 × 10−4 mol dm−3) were injected from a rotating syringe (307 rpm) into the isothermal cell, equilibrated at 25.0 °C, containing 1.4406 mL of enzyme (c = 5 × 10−6 mol dm−3).The spacing between each injection was in the range 240–300 s. The initial delay before the first injection was 600 s in all experiments. All solutions used in ITC experiments were degassed prior to use under vacuum (0.64 bar, 10 min) to eliminate air bubbles.
Microcalorimetric experiment directly gave three parameters: reaction enthalpy change (ΔrH), binding constant (Ka) and stoichiometry (N). The value of ΔrG was calculated from the binding constant (ΔrG = −RTlnK) and the reaction entropy change was calculated from the binding enthalpy and Gibbs energy (ΔrS = (ΔrH − ΔrG)/T).
3.9. Differential scanning calorimetry (DSC)
Differential scanning calorimetry (DSC) was used to investigate the melting temperature of the BrILL2wt enzyme and the mutants using the instrument Nano DSC from TA Instruments. The DSC scans were obtained against a blank Na-cacodylate buffer (pH = 8.0) from 10 °C to 100 °C at heating rates of 1 °C min−1 and 2 °C min−1. The corresponding baseline (buffer–buffer) scans were subtracted from the unfolding/folding scans prior to their normalization and analysis. In stability experiments under non-reducing and reducing conditions, the protein concentrations were in the range 0.2–0.5 mg mL−1. Monomeric forms of proteins were measured upon addition of 5 mM β-mercaptoethanol. To examine the metal binding effect, the DSC scans were measured for proteins: BrILL2wt enzyme (0.25–0.5 mg mL−1), Mn–BrILL2wt complex (0.2–0.4 mg mL−1), mutant BrILL2C139S (0.25 mg mL−1) and Mn–BrILL2C139S complex (0.20 mg mL−1).3.10. Molecular modelling methods
The initial systems for our computational study were derived from the previously constructed and studied models.11 The 3D protein structure was obtained by homology modelling using the X-ray structure of the IAA-amino acid hydrolase from Arabidopsis thaliana, AtILL2 (pdbs: 1XMB and 2Q43)12,13 as a template, and the initial position of metal ions was based on the position of the manganese ions in the recently determined structure of amidohydrolase, SACOL0085, from methicillin-resistant Staphylococcus aureus (pdb: 4EWT).34 Both of the mentioned hydrolases belong to the M20 family. The amino acid sequence identity between AtILL2 and BrILL2 is 76%, while the identity between SACOL0085 and BrILL2 is 34%. The structure of S. aureus SACOL0085 amidohydrolase possesses two manganese ions in the active binding site, so we used it for creating the active site of BrILL2. We performed molecular modelling studies of the ligand free enzyme, wild type and the C139S mutants, in the presence and absence of manganese ions. The wild type protein (the van der Waals parameters are shown in the ESI,† Table S7) and two C139S mutants with different protonation states of the serine side chain (neutral and deprotonated) were simulated for 200 ns each. The mutants were built from the equilibrated wild type structure by replacing Cys139 with Ser, either neutral (C139S), MUT1 or deprotonated (C139S−), MUT2. Since the parameters for the deprotonated Ser were not available, we built them using the AMBER1435 module antechamber combined with semiempirical AM1 calculations. In order to find out how manganese ions influence the protein structure, we performed molecular dynamic simulations (200 ns) of the wild type (WT) and mutated C139S enzymes with and without the Mn2+ ions in the enzymatic binding site. Furthermore, the other possible stable metal ion sites at the protein surface were searched. For this purpose, the four Mn2+ ions were placed in the minimal energy positions at the protein surface and their behaviour during 22 ns-long MD simulations was investigated.In order to find out possible reasons for inactivity of the C139S mutant, the BrILL2–IPA-Ala complex was studied as well by molecular modelling. For this purpose, two initial orientations of IPA-Ala in the active site of the wild type enzyme were simulated for 200 ns each and the binding free energies were calculated. The complex with the mutated C139S enzyme was constructed using the energetically more favourable ligand binding mode and the complex was also simulated for 200 ns. The initial orientation of the substrate was based on the previously studied and reported models of the complex.11
The parameters for the ligand IPA-Ala were derived using antechamber and parmchk modules of AMBER14.35
Parameterization was accomplished within the general AMBER force field GAFF, AMBER ff12SB36 (ligand free enzyme) and ff14SB37 (complexes). Each of the built complexes and the ligand free enzyme variants was placed in an octahedral box filled with the TIP3P type water molecules.38 A water buffer of 11 Å was used. Na+ ions were used for the system neutralization. The solvated complexes were geometry optimized using the steepest descent and conjugate gradient methods, 2500 steps of each. After optimization the systems were equilibrated for 0.6 ns in three steps. During each of the 0.2 ns long steps the temperature was increased by 100 K. The equilibrated systems were subjected to productive molecular dynamic (MD) simulations at constant temperature and pressure (300 K, 1 atm). During the first 2 ns of MD simulations the time step was 1 fs and after that 2 fs and the temperature was kept constant by using Langevin dynamics with a collision frequency of 1 ps−1. Simulations were performed using periodic boundary conditions (PBCs) by the AMBER14 program pmemd. The long range electrostatic interactions were calculated using the particle mesh Ewald (PME) method and a cut-off distance of 11 Å for the pairwise interactions in the direct space. The binding free energies were calculated using the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA)39 approach as implemented in AMBER12.40,41
4. Conclusions
Based on the experimental and molecular modelling research, we confirmed that two conserved Cys residues (Cys139 and Cys320) of Brassica rapa auxin amidohydrolase BrILL2 play important roles in the regulation of enzyme activity and enzyme stabilization. The Cys320 residue is mainly responsible for protein stabilization through participation in the formation of non-active polymer forms. DSC measurements resulted in Tmax values which are the indicators of protein thermostability. Accordingly, the monomeric, active enzyme is less stable than the polymeric, non-active forms. Furthermore, the Cys320 to Ser point mutation significantly destabilized the BrILL2 enzyme. Based on the mutagenesis experiments it was shown that Cys139 is crucial for the enzyme activity. However ITC, CDS and DSC data showed that Cys139 to Ser mutation did not affect Mn2+ ion binding to the enzyme, suggesting that Cys139 is not crucial for metal binding. The theoretical results revealed that Cys139 is a part of the bimetal active site and participates in the coordination of the metal co-factor Mn2+. While the position of manganese ions in the active site of BrILL2 (wild type) remained stable (their average distance is about 4 Å) during 200 ns of MD simulations, the metal ions in the BrILL2C139S mutant moved away from each other during the simulations, and after 200 ns they were separated by about 7 Å. According to the molecular modelling results, we assume that the preferred substrate IPA-Ala binds into the BrILL2 active site either in the orientation identified as BM1 or BM2. In each of these orientations, there is a water molecule close to the amide bond carbon appropriate to actively participate in the amide bond hydrolysis, i.e. to be activated and to perform the nucleophilic attack to carbonyl carbon. In the BrILL2wt–IPA-Ala_BM1 complex the water molecule could be activated either by the carboxyl group of Glu151 or by the carboxyl group of the substrate itself, while in the BrILL2wt–IPA-Ala_BM2 Glu370 might activate the water molecule. In the C139S mutant (neutral Ser), the active site architecture differs from that in the wild type complexes and this is probably the main reason for its inactivity. However, if we assume that Ser at position 139 is deprotonated, the active site architecture is similar to that in the active site, and the reason for its inactivity is not so obvious. The possible explanations for the inactivity of the S139S− variant are (a) lower binding affinity for the preferred IPA-Ala substrate in comparison to the wild type enzyme and (b) lack of water molecules appropriate for the nucleophilic attack.Abbreviations
| CDS | Circular dichroism spectropolarimetry |
| DSC | Differential scanning calorimetry |
| EDTA | Ethylenediaminetetraacetic acid |
| HR-ICP-MS | High resolution inductively coupled plasma mass spectrometry |
| IAA | Indole-3-acetic acid |
| IAA-Ala | Conjugate of indole-3-acetic acid (IAA) with amino acid alanine (Ala) |
| IBA | Indole-3-butyric acid |
| IBA-Ala | Conjugate of indole-3-butyric acid (IBA) with amino acid alanine (Ala) |
| IPA | Indole-3-propionic acid |
| IPA-Ala | Conjugate of indole-3-propionic acid (IPA) with amino acid alanine (Ala) |
| IPTG | Isopropyl-β-D-1-thiogalactopyranoside |
| ITC | Isothermal titration calorimetry |
| PMSF | Phenylmethylsulfonyl fluoride |
| wt | Wild type |
Acknowledgements
We kindly thank Dr sc. Marija Matković for help with CDS measurements and Niko Bačić for assistance with HR-ICP-MS analysis. We are also grateful to Dr sc. Marija Abramić for critical reading of the manuscript. This work has been supported by the FP7-REGPOT-2012-2013-1 Grant Agreement no. 316289 and by the Alexander van Humboldt Institutional partnership between Ruđer Bošković Institute and the Technische Universität Dresden.References
- N. D. Rawlings, M. Waller, A. J. Barrett and A. Bateman, Nucleic Acids Res., 2014, 42, D503–D509 CrossRef CAS PubMed.
- C. M. Seibert and F. M. Raushel, J. Biochem., 2005, 44, 6383–6391 CrossRef CAS PubMed.
- J. Ludwig-Müller, J. Exp. Bot., 2011, 62, 1757–1773 CrossRef PubMed.
- C. S. Westfall, A. M. Muehler and J. M. Jez, J. Biol. Chem., 2013, 288, 19304–19311 CrossRef CAS PubMed.
- A. Bajguz and A. Piotrowska, Phytochemicals, 2009, 20, 957–969 CrossRef PubMed.
- B. Bartel and G. R. Fink, Science, 1995, 268, 1745–1748 CAS.
- R. T. Davies, D. H. Goetz, J. Lasswell, M. N. Anderson and B. Bartel, Plant Cell, 1999, 11, 365–367 CrossRef CAS.
- S. LeClere, R. Tellez, R. A. Rampey, P. T. Seiichi, S. P. T. Matsuda and B. Bartel, J. Biol. Chem., 2002, 277, 20446–20452 CrossRef CAS PubMed.
- J. J. Campanella, A. F. Olajide, V. Magnus and J. Ludwig-Müller, Plant Physiol., 2004, 135, 2230–2240 CrossRef CAS PubMed.
- J. J. Campanella, S. M. Smith, D. Leibu, S. Wexler and J. Ludwig-Müller, J. Plant Growth Regul., 2008, 27, 26–38 CrossRef CAS.
- B. Savić, S. Tomić, V. Magnus, K. Gruden, K. Barle, R. Grenković, J. Ludwig-Müller and B. Salopek-Sondi, Plant Cell Physiol., 2009, 50, 1587–1599 CrossRef PubMed.
- E. J. Levin, D. A. Kondrashov, G. E. Wesenberg and G. N. Phillips, Structure, 2007, 15, 1040–1052 CrossRef CAS PubMed.
- E. Bitto, C. A. Bingman, L. Bittova, N. L. Houston, R. S. Boston, B. G. Fox and G. N. Phillips Jr., Proteins, 2009, 74, 61–71 CrossRef CAS PubMed.
- I. Shin, K. Han and S. Rhee, J. Mol. Biol, 2014, 426, 3028–3040 CrossRef CAS PubMed.
- G. Bruylants, J. Wouters and C. Michaux, Curr. Med. Chem., 2005, 12, 2011–2020 CrossRef CAS PubMed.
- P. Privalov, Microcalorimetry of Macromolecules: The Physical Basis of Biological Structures, Wiley, 1st edn, 2012 Search PubMed.
- F. Conejero-Lara, J. M. Sanchez-Ruiz, P. L. Mateo, F. J. Burgos, J. Vendrell and F. X. Aviles, Eur. J. Biochem., 1991, 200, 663–670 CrossRef CAS PubMed.
- J. M. Sanchez-Ruiz, j. L. Lopez-Lacomba, P. L. Mateo, M. Vilanova, M. A. Serra and F. X. Aviles, Eur. J. Biochem., 1988, 176, 225–230 CrossRef CAS PubMed.
- A. V. Hernandez-Moreno, F. Villasenor, E. Medina-Rivero, N. O. Perez, L. F. Flores-Ortiz, G. Saab-Rincon and G. Luna-Barcenas, Int. J. Biol. Macromol., 2014, 64, 306–312 CrossRef CAS PubMed.
- A. K. Werner, I. A. Sparkes, T. Romeis and C.-P. Witte, Plant Physiol., 2008, 146, 418–430 CrossRef CAS PubMed.
- B. B. Buchanan and Y. Balmer, Annu. Rev. Plant Biol., 2005, 56, 187–220 CrossRef CAS PubMed.
- C.-Y. Chen, D. Willard and J. Rudolph, J. Biochem., 2009, 48, 1399–1409 CrossRef CAS PubMed.
- D. Hasse, E. Andersson, G. Carlsson, A. Masloboy, M. Hagemann, H. Bauwe and I. Andersson, J. Biol. Chem., 2013, 288, 35333–35345 CrossRef CAS PubMed.
- M. Jarenmark, H. Carlsson and E. Nordlander, C. R. Chimie., 2007, 10, 433–462 CrossRef CAS.
- Y. Liu, L. Wei, W. Yaozhui, J. Yindi and T. Xiangshi, Metallomics, 2013, 5, 1448–1457 RSC.
- T. H. Evers, M. A. M. Appelhof, E. W. Meijer and M. Merkx, Protein Eng., Des. Sel., 2008, 21, 529–536 CrossRef CAS PubMed.
- M. Rowinska-Zyrek, D. Witkowska, S. Potocki, M. Remellib and H. Kozlowski, New J. Chem., 2013, 37, 58–70 RSC.
- A. Schuller and J. Ludwig-Müller, New Phytol., 2006, 171, 1–13 CrossRef PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- N. Jajčanin-Jozić, S. Deller, T. Pavkov, P. Macheroux and M. Abramić, Biochimie, 2010, 92, 89–96 CrossRef PubMed.
- D. D. Mruk and C. Y. Cheng, Spermatog., 2011, 1, 121–122 CrossRef PubMed.
- R. Lundblad, Chemical Reagents for Protein Modification, CRC Press, 3rd edn, 2004, pp. 352 Search PubMed.
- Ž. Fiket, V. Roje, N. Mikac and G. Kniewald, Croat. Chem. Acta, 2007, 80, 91–100 Search PubMed.
- T. S. Girish, B. Vivek, M. Colaco, S. Misquith and B. Gopal, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2013, 69, 103–108 CrossRef CAS PubMed.
- D. A. Case, J. T. Berryman, R. M. Betz, D. S. Cerutti, T. E. Cheatham, III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, T. Luchko, R. Luo, B. Madej, K. M. Merz, G. Monard, P. Needham, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, R. Salomon-Ferrer, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, D. M. York and P. A. Kollman, AMBER, 2015, University of California, San Francisco Search PubMed.
- Å. A. Skjevik, B. D. Madej, R. C. Walker and K. Teigen, J. Phys. Chem. B, 2012, 116, 11124–11136 CrossRef PubMed.
- C. J. Dickson, B. D. Madej, Å. A. Skjevik, R. M. Betz, K. Teigen, I. R. Gould and R. C. Walker, J. Chem. Theory Comput., 2014, 10, 865–879 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- M. R. Lee, Y. Duan and P. A. Kollman, Proteins, 2000, 39, 309–316 CrossRef CAS.
- D. A. Case, T. A. Darden, T. E. Cheatham, III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, 2012, University of California, San Francisco Search PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: CDS Fig. S1, ITC Fig. S2–S4 and MD simulations Fig. S5–S10 of BrILL2 wt and mutants, DSC Fig. S11–S13 and Tables S1–S7. See DOI: 10.1039/c5cp06301a |
| ‡ Corresponding author for biophysical part of research. |
| This journal is © the Owner Societies 2016 |