
High adsorption capacity of heavy metals on two-dimensional MXenes: an ab initio study with molecular dynamics simulation
Xun
Guo
 a,
Xitong
Zhang
ab,
Shijun
Zhao
a,
Qing
Huang
c and
Jianming
Xue
*ab
a,
Xitong
Zhang
ab,
Shijun
Zhao
a,
Qing
Huang
c and
Jianming
Xue
*ab
aState Key Laboratory of Nuclear Physics and Technology, Center for Applied Physics and Technology, School of Physics, Peking University, Beijing 100871, P. R. China. E-mail: jmxue@pku.edu.cn
bCAPT, HEDPS, and IFSA Collaborative Innovation Center of MoE College of Engineering Peking University, Beijing 100871, China
cNingbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo, Zhejiang 315201, China
First published on 12th November 2015
Abstract
Density functional theory (DFT) calculation is employed to study the adsorption properties of Pb and Cu on recently synthesized two-dimensional materials MXenes, including Ti3C2, V2C1 and Ti2C1. The influence of surface decoration with functional groups such as H, OH and F have also been investigated. Most of these studied MXenes exhibit excellent capability to adsorb Pb and Cu, especially the adsorption capacity of Pb on Ti2C1 is as high as 2560 mg g−1. Both the binding energies and the adsorption capacities are sensitive to the functional groups attached to the MXenes' surface. Ab initio molecular dynamics (ab-init MD) simulation confirms that Ti2C1 remains stable at room temperature after adsorbing Pb atoms. Our calculations imply that these newly emerging two-dimensional MXenes are promising candidates for wastewater treatment and ion separation.
1 Introduction
MXenes are a series of graphene-like two-dimensional (2D) materials, which are prepared by exfoliating the counterpart MAX phase in hydrofluoric acid (HF).1 MAX phases are a large family of layered ternary transition-metal carbides or nitrides with a chemical formula Mn+1AXn (n = 1, 2 or 3), where M is a transition metal, A is a group-A element (mostly IIIA or IVA group), and X is C or N.2 These MXenes exhibit unique electrical and chemical properties,2,3 they are suggested to be used in many practical applications, such as very recently Ti3C2 has been found to be an excellent anode material for Li ion batteries (LIBs).4–6Water pollution due to heavy metal ions, such as Pb and Cu, is a worldwide serious environmental problem due to the extreme toxicity of these ions even at very low concentrations.2,3 From the economical and efficiency point of views, adsorption is regarded as one of the most promising and widely used methods for the removal of these heavy ions.7 However, developing appropriate materials with excellent adsorption capacity is still the key challenge.
Though it seems that 2D materials are good candidates due to their large surface-to-volume ratio, most well-studied 2D materials, including MoS2, graphene and its oxide, have been proved to be not suitable for adsorbing heavy metal ions because of their weak van der Waals bonding.8 These newly emerging MXenes can be expected to overcome the disadvantage of weak adsorption, because MXenes are composed of transition metal elements such as titanium (Ti) and vanadium (V), which usually have high sorption affinity toward heavy metal ions.9
Recently, it has been reported that Ti3C2 has a high lead adsorption capacity of 140 mg g−1,8 and this primary result indicates that MXenes are good potential candidates for the removal of metal ions from wastewater. It should be noted that there are dozens of MXenes, so it is necessary to investigate other MXenes to seek other samples with better adsorbability. With the same MAX phases, MXenes can be divided into several groups according to their structure as M4X3, M3X2 and M2X1. Ti3C2 belonged to the M3X2 group, it is natural to expect that MXenes with M2X1 structure should have a higher adsorption capacity because M2X1 has a larger surface-to-volume ratio than its counterparts M3X2 and M4X3.
Currently, nearly all the MXenes are obtained by using the HF etching method, after which the surface of exfoliated MXenes was often decorated by functional groups, including H, OH and F.5,10 It has been reported that functionalized MXenes (f-MXene) have much lower Li capacity than their delaminated counterparts (bare MXene).5,11,12 For heavier ions like Pb and Cu, the influence of surface functional groups is still unclear,8 and this question is also needed to be clarified for future application of MXenes.
In our study, two kinds of the most studied M2X1 samples (V2C1 and Ti2C1) have been investigated, and Ti3C2 has also been considered in our calculation as a contrast. By using first-principles calculations based on density functional theory (DFT), we calculated the binding energies and adsorption capacities of toxic heavy metal elements Pb and Cu on Ti3C2, V2C1 and Ti2C1. The results indicate that the theoretical adsorption capacity of Pb on Ti2C1 can be as high as 2560 mg g−1 on bare MXenes, and 1280 mg g−1 on H-attached MXenes. These extremely high metal adsorption capacities suggest that MXenes hold great potential in the water treatment industry.
2 Method
All calculations were carried out based on DFT as implemented in the Vienna ab initio simulation package (VASP).13,14 The exchange correlation energy is described by the generalized gradient approximation (GGA) in the scheme proposed by Perdew–Burke–Ernzerhof (PBE).15 The optPBE method is used to account for the effect of van der Waals (vdW) interaction.16,17 Meanwhile, DFT+U correction was used during our calculations,18 with the consideration of 3d electrons in Ti, V and Cu. According to previous studies, the U value for pure transition metals (from Ti to Cu) should be in the range of 2–4 eV,19,20 so in our calculations the value of U was set to 3 eV.Each sample is modeled by a 3 × 3 supercell, including the bare MXene and f-MXene samples. Three kinds of functional groups, including F, OH and H, are considered in our calculation. Periodic boundary condition is applied and a large spacing of more than 20 Å between the MXene layer and its mirror image is used, which is far enough to avoid the interaction caused by periodic boundary. The total energy was converged to better than 10 meV for a plane wave cutoff of 500 eV and 15 × 15 × 1 Monkhorst–Pack k-point sampling for the Brillouin zone. For geometry relaxation we used the method of conjugate gradient energy minimization, and the convergence criterion for energy was 10−5 eV between two consecutive steps. The maximal force exerted on each atom is less than 0.01 eV Å−1 upon ionic relaxation.
The binding energy per atom for the adsorption of m adatoms (Pb or Cu) is defined as:
| Eb = (EMXene + m × EAtom − EMXene+Atom)/m | (1) |
We initialized the system with the adatom (Pb or Cu) right above the C atom, which is the most stable site for atom adsorption on MXenes. The distance between the adatom and the top M layer is 3 Å. The nudged elastic band (NEB) method was used to calculate the migration energy barrier for a single atom on the sample surface. In the calculation, ten intermediate images are created by interpolating between the initial and final states which are chosen as the local minima configurations obtained in the relaxation runs. Meanwhile ab-initio molecular dynamics (ab-init MD) simulation was also carried out to check the thermal stability of fully adsorbed MXene samples. Saturated configurations from the static calculation were used as the initial states, and a canonical NVT ensemble was simulated using the algorithm of Nose. The system was allowed to relax for 15 picoseconds in total with a time step of 3 femtoseconds, with the initial velocities set randomly according to the Maxwell–Boltzmann distribution. In these two cases, the k-points are 5 × 5 × 1 and the tangential force on each image is converged to 0.01 eV Å−1 in NEB calculation.
3 Result
In order to validate our approaches, we calculated the lattice parameters of pristine and decorated Ti3C2, V2C1 and Ti2C1, and the results are shown in Table 1. All the calculated values are in good agreement with previous results of pristine MXene: 3.071 Å (Ti3C2), 2.869 Å (V2C1) and 3.007 Å (Ti2C1).21 The decorated MXene sheets are constructed accordingly by saturating the surface M atoms with F, H and OH, respectively, based on the pristine MXene samples. All the functional groups are oriented above the topmost sites of C atoms on both sides of the MXene layers initially, then the new structure is fully optimized to locate the accurate position. After being decorated with functional groups (H, F or OH), the lattice parameters slightly decrease, a similar tendency has also been found in Andrey N. Enyashin and Alexander L. Ivanovskii's work,3 for the Ti3C2(OH)2 and Ti2C1(OH)2 double layer. A shorter lattice parameter means a stronger interaction, therefore the above results indicate that these MXenes become more stable after decorated by functional groups.| Ti3C2 | V2C1 | Ti2C1 | |
|---|---|---|---|
| a After adsorption, the lattice often becomes distorted and the lattice parameter is hard to meet the condition of a = b strictly, so we use the average lattice parameter (a + b)/2 instead. | |||
| Pristine | 3.145 | 2.957 | 3.104 |
| Decorated by F | 3.092 | 2.888 | 3.049 |
| Decorated by H | 3.083 | 2.850 | 3.025 |
| Decorated by OH | 3.131 | 2.872 | 3.036 |
| Single Pb adsorptiona | 3.152 | 2.954 | 3.075 |
| Single Cu adsorptiona | 3.146 | 2.957 | 3.073 |
| Single Pb adsorption with H decorationa | 3.087 | 2.856 | 3.033 |
| Single Cu adsorption with H decorationa | 3.083 | 2.850 | 3.026 |
| Fully Pb adsorptiona | 3.131 | 2.912 | 3.128 |
| Fully Cu adsorptiona | 3.135 | 2.906 | 3.104 |
| Fully Pb adsorption with H decorationa | 3.087 | 2.855 | 3.043 |
| Fully Cu adsorption with H decorationa | 3.069 | 2.813 | 3.032 |
Firstly, we calculate the binding energies of single adatoms on the bare and decorated samples and the results are plotted in Fig. 1. Clearly, the surface decoration remarkably decreases the binding energy for adsorbing Pb or Cu. The average binding energies of Pb and Cu on bare samples are all higher than 2.0 eV, and the adsorption of Pb on V2C1 has the highest binding energy of 4.5 eV.
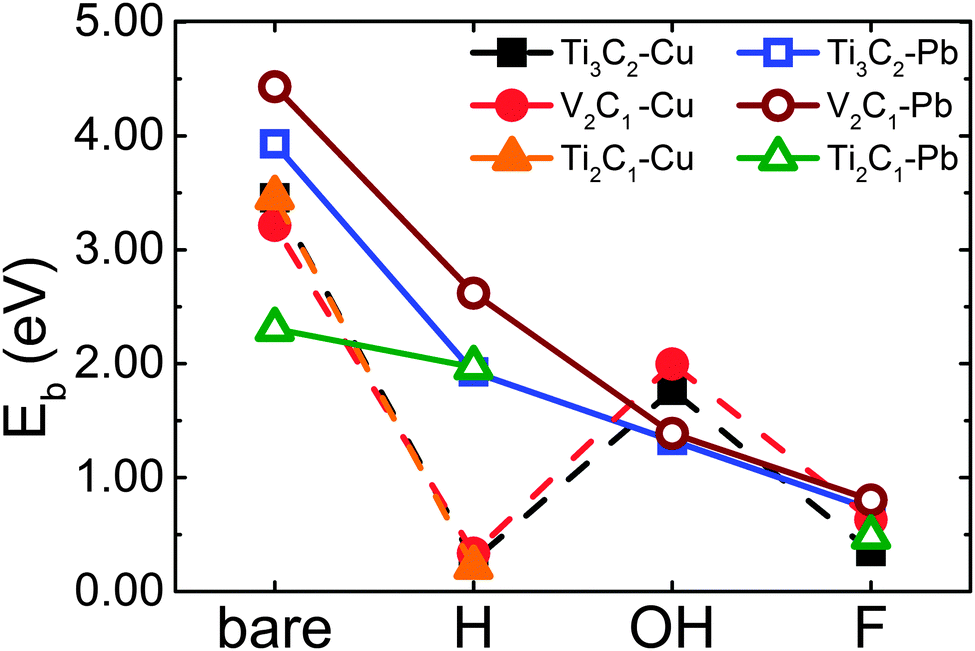 | ||
| Fig. 1 Binding energy Eb of single Cu (solid lines) and Pb (dotted lines) atoms changes with different surface functional groups on Ti3C2, V2C1 and Ti2C1. | ||
After surface decoration, the binding energy of Pb on the f-MXene sample decreases dramatically from ∼2.5 to ∼0.5 eV in the order: bare > H > OH > F. As to Cu, the changing tendency does not follow that of Pb adsorption. In particular the binding energies of Cu on all the H-attached samples are less than 0.3353 eV, which means H-decorated MXenes are not good candidates for adsorbing Cu. Therefore, considering that the binding energy of Pb on H-attached samples is higher than 2 eV, it could be expected that in acid environments, MXenes can only adsorb Pb atoms while leaving Cu atoms in wastewater.
In contrast, in cases of OH decoration, the binding energies for Cu can reach up to 1.765 eV (on Ti3C2(OH)2) and 1.996 eV (on V2C1(OH)2), and are higher than the binding energies of Pb (1.325 eV on Ti3C2(OH)2 and 1.388 eV on V2C1(OH)2). According to Halsey and George's theory, the binding energy difference can lead to preferential adsorption.22 This means OH-attached samples prefer to adsorb Cu rather than Pb in an alkaline environment, in which –OH is rich and the solid surface is usually OH-attached.
For F-decoration, the binding energies of Pb and Cu on each sample are all very limited, and are in the range of 0.3526–0.8040 eV. The system even cannot locate the stable Cu adsorption site for Ti2C1F2 since the interaction is too weak to reach a stable adsorbing configuration. Therefore, the F decorated surface is not suitable for heavy metal ion adsorption.
To explain the irregular drop in the binding energy of Cu on H-decorated MXene samples, the Bader Charge Analysis code23 was used to compare the charge transfer characteristics of Cu and Pb on different surfaces, and the results are shown in Table 2. In most cases, the adsorption of heavy metal ions on MXene samples is mainly chemical since the amount of charge transfer is large enough. Anyhow, for the fully hydrogenated samples, the charge transfer between Cu and H-decorated surfaces is close to zero, resulting in very small binding energy as shown in Fig. 1. What is more, the amount of charge transfer of Pb on a H-decorated surface is in the sequence of V2C1H2 > Ti3C2H2 > Ti2C1H2, and the binding energies illustrated in Fig. 1 also have the same order. Briefly speaking, the difference in binding energies is mainly attributed to the amount of charge transfer for f-MXene samples. And according to the present result, this theory can explain most of the phenomena in our calculation.
| MXene | Charge transfer for Pb (e−) | Charge transfer for Cu (e−) |
|---|---|---|
| T13C2 | +0.40 | +0.63 |
| V2C1 | +0.32 | +0.48 |
| Ti2C1 | +0.52 | +0.66 |
| Ti3C2H2 | −0.38 | −0.045 |
| V2C1H2 | −0.54 | −0.081 |
| Ti2C1H2 | −0.34 | −0.028 |
But why there is such a large difference between Cu and Pb adsorption on H-decorated samples is still a difficult problem, and cannot be simply attributed to the amount of charge transfer. To understand it we calculate the difference density of electron (DDOE) for these samples as shown in Fig. 2. It can be seen from it that there is a completely different charge transfer mechanism for Pb and Cu adsorption. When Pb adsorbs onto Ti3C2, the electron obtained by Pb is mainly provided by the top Ti layer, because the distribution of electron density of deeper atom layers do not change much after adsorption (compared with the near atoms in the same layer). But for Cu, the inner C atom layer also participates in the interaction, which makes the Coulomb interaction more complicated: the change in the polarity of the inner C atom would provide an extra repulsive force (as in Fig. 2(a2)), or the change in the polarity of the upper layer atom (as in Fig. 2(b2)) diminishes the interaction.
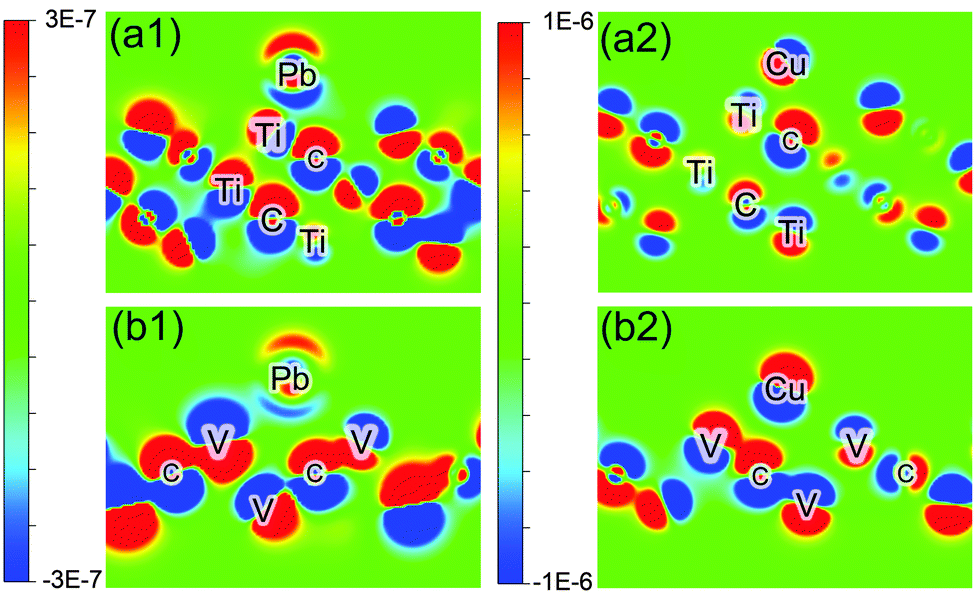 | ||
| Fig. 2 Difference electron density (DDOE) plot (in e− Å−3) of Pb adsorbed onto bare Ti3C2 (a1), V2C1 (b1), and Cu onto bare Ti3C2 (a2), V2C1 (b2). | ||
It should also be noticed that in Table 2, Pb transfers less electrons than Cu on bare surfaces, and they could abnormally gain electrons from the host. Obviously it reveals that the surfaces are far from saturated and this large amount of unpaired electrons cannot actually exist in a real situation. But further analysis of this phenomenon is still meaningful because it could help us understand the mechanism of heavy metal ion adsorption.
To explore the adsorption capacity of MXenes, we increase the number of adatoms from 1, 2, 4, and 8 until up to 16 for a 3 × 3 monolayer MXene, and bare and H-decorated MXenes were selected as examples. Following the previous notation,24 we employ the notations Ax[Mn+1Cn]1−x and Ax[Mn+1CnH2]1−x to quantitatively account for the concentration of Pb and Cu (A represents Pb or Cu, n = 1 or 2). All adatoms are added symmetrically at each side of the most stable site (the top of C atoms) in every step, until the adsorption is saturated. The binding energies as a function of Pb/Cu concentration x are summarized in Fig. 3. It indicates that the binding energies of adatoms vary along with their content, and the values are mainly beyond 2 eV, which means that Pb and Cu atoms bind strongly with the MXene layer. This result favors the adsorption process of these samples significantly because the adsorption can spontaneously occur if there is no kinetic transport resistance.
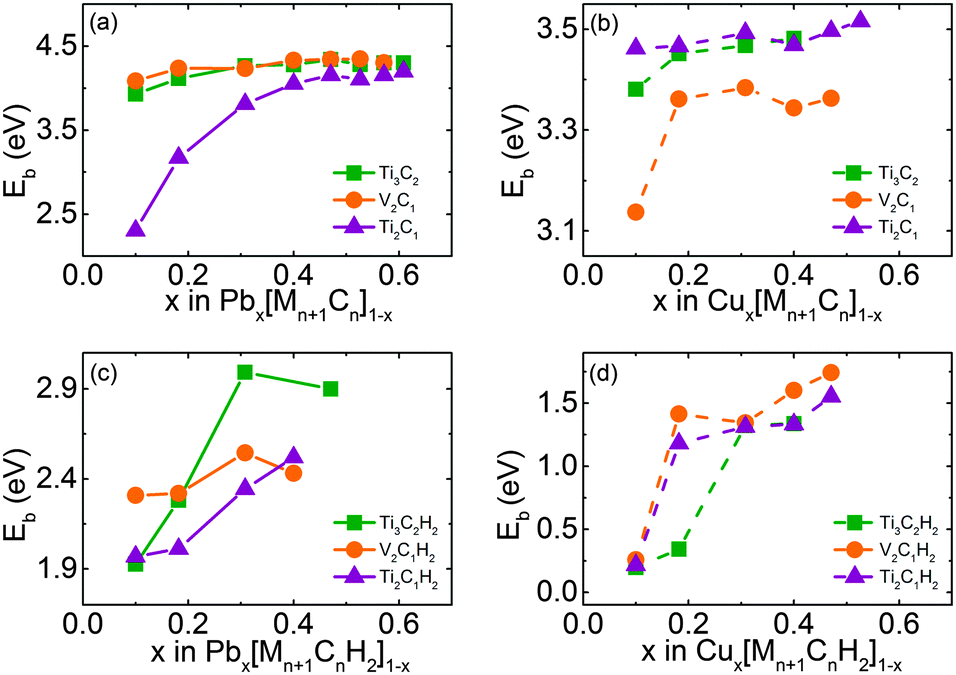 | ||
| Fig. 3 Dependence of the binding energies of Pb and Cu on their concentration x on Mn+1Cn and Mn+1CnH2 monolayer: Pb (a) and Cu (b) on bare samples, compared with Pb (c) and Cu (d) on H-functionalized samples. M represents Ti or V, and n = 1 or 2. | ||
For Pb on bare Ti3C2, the adsorption is saturated when the x value increases to 0.6087 (corresponding to 1920 mg g−1). As to bare V2C1 and Ti2C1, the maximal x value is only 0.5714. Though their x value is a little lower than that of bare Ti3C2, their adsorption capacities are much higher, can reach up to 2430 and 2560 mg g−1, respectively, resulting from the lower mass density. After being decorated with H, the x values of Pb adsorption on all three samples decline to the same value of 0.4000 (corresponding to 820, 1210 and 1280 mg g−1 respectively). In other words, the mass capacities decrease more than 50% after surface decoration with H for all samples. But it should be noticed that these theoretical capacities are nearly ten times (about four times for H-decorated samples) higher than other nano-materials such as graphene oxide, whose maximal experimental capacity is no more than 200 mg g−1.25–27 And up to now, the experimental value of Pb adsorption capacity is only 140 mg g−1, it could be expected that this value will be greatly increased in the future.
The results regarding Cu adsorption are shown in Fig. 3(b) and (d). The x values of Cu on bare Ti3C2, V2C1 and Ti2C1 are 0.4000, 0.4706 and 0.5263, corresponding to 250, 500 and 660 mg g−1 respectively. Obviously, the adsorption capacity of Cu atoms is much lower compared to that of Pb for bare MXenes. Apart from the mass difference between Pb and Cu, the adsorption sites occupied by Cu atoms are much less (2–6 sites) than Pb on bare samples. What is interesting is that decoration with H does not change the adsorption capacity of Cu, the maximal x values are still maintained at 0.4000 and 0.4706 for Ti3C2 and V2C1, and for Ti2C1, the x value only declines from 0.5263 to 0.4076, indicating that adsorbability of Cu is not sensitive to H-decoration. On the other hand, H-decoration greatly enlarges the difference between the binding energies of Pb and Cu adsorption. As a result, the preferential adsorbability of Pb compared to Cu will also be greatly enhanced.
With the exception of mass difference, another possible reason for the low adsorption capacity of Cu on these MXene samples is the repulsive interaction between the inner carbon atom and Cu. As described above, Cu would gain electrons from the inner C atom layer: once an adsorption site was occupied by a Cu atom, the binding energy of near sites would be strongly influenced, which leads to the reduction of Cu adsorption capacity. So for the adsorption of multiatomic layer 2D materials, the influence of the inner layer is significant.
Volume expansion caused by atom adsorption is also one of the most concerned characteristics for materials used in wastewater treatment. Table 1 illustrates that the maximal lattice change at the highest Pb adsorption condition is only 0.45%, 1.5% and 0.77% for Ti3C2, V2C1 and Ti2C1, respectively. These values are low enough since the expansion rate of fully lithiated Ti3C2 is about 0.5%,28 and we believe that materials used in LIBs need more strict requirement of the expansion rate than water treatment materials. As to the H-decorated samples, the lattice parameters of Ti3C2H2, V2C1H2 and Ti2C1H2 only increase by factors of 0.13%, 0.18% and 0.60%, respectively, after complete adsorption of Pb. All these MXenes and f-MXenes are robust against heavy metal ion insertion and do not suffer from structural expansion, which is very important in practical applications.
4 Discussion
It should be noticed that these binding energies do not include the zero-point energy and entropic contributions. The relative stability of these adsorption configurations can be changed by these corrections, but the general trends are unlikely to be affected, as previously pointed out.28 The above calculations assume the samples to be fixed at 0 K, to test the stability of these Pb-saturated samples at room temperature (about 300 K), we have carried out a 15 ps ab-init MD simulation. Ti2C1 and Ti2C1H2 are chosen as representatives, because their binding energies of Pb are the lowest among all samples. The simulation uses a canonical NVT ensemble and the temperature is set to 300 K. Four snapshots of the simulation at 0, 5, 10 and 15 ps are shown in Fig. 4.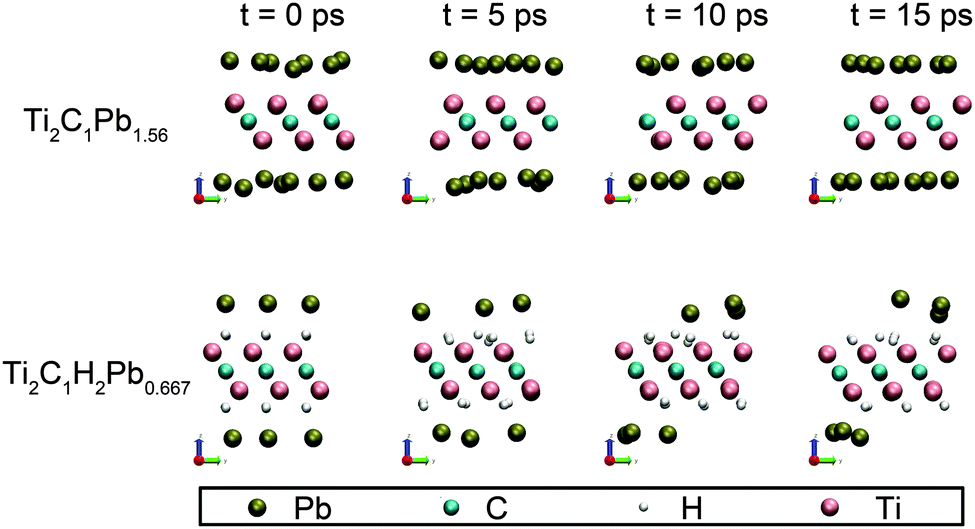 | ||
| Fig. 4 Snapshot of fully Pb adsorbed Ti2C1 and Ti2C1H2 that endure 15 ps ab-init MD simulation at 300 K. | ||
Ab-init MD simulation shows that all the adatoms remain stable on the surface of Ti2C1 and Ti2C1H2 at 300 K within 15 ps, indicating that Pb-adsorbed Ti2C1 and Ti2C1H2 samples are stable at room temperature. On the other hand, we observed that Pb atoms jump frequently on the surface from one adsorption site to the adjacent sites. To understand this behavior, the migration energy barrier was calculated using the NEB method and the results are shown in Fig. 5. The migration energy barriers of Pb on the three bare MXenes are as low as 0.0442, 0.0607 and 0.0238 eV, respectively. H-decoration slightly decreases the Pb migration energy for V2C1 (0.00580 eV) and Ti2C1 (0.0120 eV), but for Ti3C2, the migration energy increases to 0.0678 eV. All in all, these migration barrier energies are very low, so that the adatoms could migrate frequently on the surfaces of all these samples.
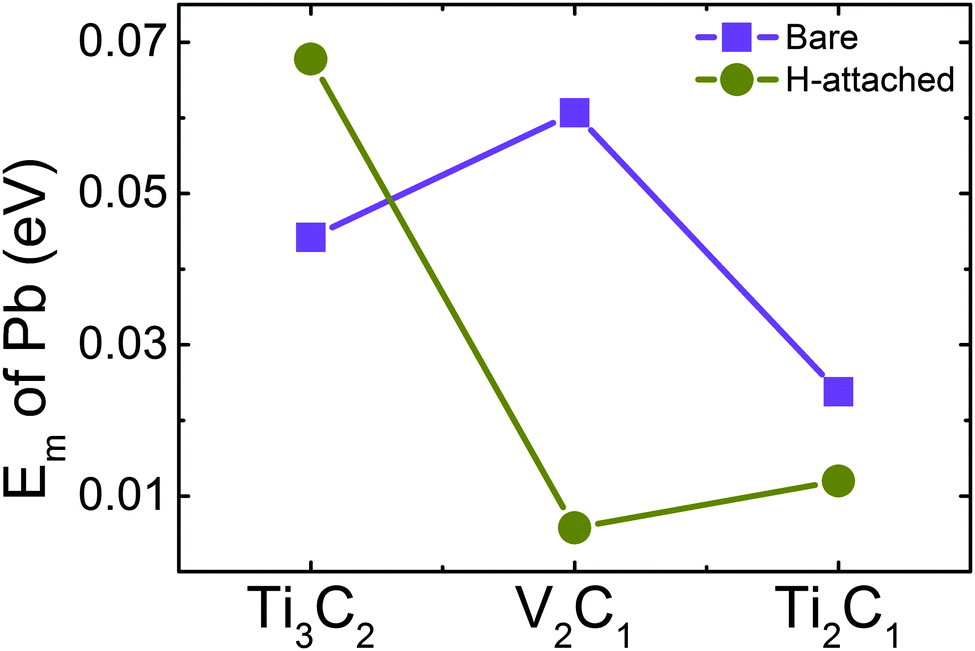 | ||
| Fig. 5 Comparison of migration energy barrier Em of a single Pb atom on bare and H-decorated samples. | ||
Considering that the binding energies of Pb on these samples vary between 2 and 4 eV, while the migration energy barriers are only within 0.07 eV, Pb atoms prefer to migrate on the surface from site to site rather than jumping off the sample surface due to thermal disturbance. These low migration energy barriers of Pb are quite close to the Li migration energy (about 0.02–0.05 eV) on MXenes,28 which illustrates that migration energy on the MXene surface is not sensitive to the type of adatom. It has been reported that the surface decoration has a significant influence on the diffusion of Li atoms on Ti3C2, and the migration energy would be increased after surface decoration.12,28 Our result exhibits the similar trend for Ti3C2, but more interesting thing is that the migration energy barrier of V2C1 and Ti2C1 becomes lower after H-decoration. Meanwhile, the migration energy of Ti2C1 is significantly lower than V2C1 and Ti3C2. Although the low migration energy barrier can decrease the equilibration time, it should also be noticed that for most samples, the difference between energy barriers is small enough, so we do not take it as an important factor to evaluate their properties.
Another important factor to take into account is the volume density after adsorption. As previously mentioned, fully Pb-adsorbed V2C1 and Ti2C1 have the same x value, their capacity difference is mainly determined by the discrepancy of mass density. But V2C1 has a shorter lattice parameter than Ti2C1, indicating that its volume density of adsorbed Pb is (about 15%) higher than Ti2C1. Our calculation also shows that fully Pb-adsorbed V2C1 has a lower expansion rate than Ti2C1, which means that the adsorption structure of V2C1 is more stable than Ti2C1. So from an application perspective, V2C1 is more practical and promising.
In summary, our results show that MXenes possess extremely high Pb adsorption capacities. No matter whether on the bare or H-attached MXenes, the theoretical lead adsorption capacities are much higher than other 2D materials. And we also confirm that Ti2C1 and V2C1 have a higher lead capacity than Ti3C2. In addition, apart from the MXene structure, the metal element M also has an influence on the adsorption performance, for instance, Ti2C1 has a slightly higher Pb adsorption capacity than V2C1, but its average binding energy (about 4.1 eV per atom) is a little smaller than that of V2C1 (about 4.3 eV per atom). In practical applications, MXenes with M2X1 structure is preferred, and of course, if it has enough high binding energy. Even with the same structure like M2X1, it is still possible to optimize the adsorption performance of heavy metal atoms by choosing a suitable M element.
5 Conclusions
First-principles calculations based on density functional theory have been carried out to investigate the adsorption of Pb and Cu atoms on MXenes to explore its potential applications. Our results show that surface decoration can significantly influence the binding energies and adsorption capacities of these samples. The theoretical capacity is quite promising compared with other commercial materials, no matter whether on bare or H-decorated samples. Meanwhile, we find that the capacities and binding energies of Cu are significantly lower than those of Pb, because Cu has a stronger interaction with the inner layer atoms, which we believe could lead to preferential adsorption behavior. Lastly, the fully Pb-adsorbed MXenes are quite stable at room temperature, since we cannot see any dissociation for all saturated MXenes in 15 ps, and the volume changes are all very small. Our findings show that the high Pb capacity, excellent thermal stability, potential capacity controllability and preferential adsorbability make MXenes, especially V2C1, good candidates as cleaning or ion separating materials in the situation of heavy metal pollution.Acknowledgements
This work was financially supported by NSAF (Grant No. U1230111), NSFC (Grant No. 91226202 and 91426304) and the China Postdoctoral Science Foundation (Grant No. 2014M550561).References
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed.
- A. N. Enyashin and A. L. Ivanovskii, J. Phys. Chem. C, 2013, 117(26), 13637–13643 CAS.
- Y. Xie, M. Naguib, V. N. Mochalin, M. W. Barsoum, Y. Gogotsi, X. Yu, K. W. Nam, X. Q. Yang, A. I. Kolesnikov and P. R. Kent, J. Am. Chem. Soc., 2014, 136, 6385–6394 CrossRef CAS PubMed.
- M. Naguib, J. Halim, J. Lu, K. M. Cook, L. Hultman, Y. Gogotsi and M. W. Barsoum, J. Am. Chem. Soc., 2013, 135, 15966–15969 CrossRef CAS PubMed.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed.
- Q. Zhang, Q. Du, M. Hua, T. Jiao, F. Gao and B. Pan, Environ. Sci. Technol., 2013, 47, 6536–6544 CAS.
- Q. Peng, J. Guo, Q. Zhang, J. Xiang, B. Liu, A. Zhou, R. Liu and Y. Tian, J. Am. Chem. Soc., 2014, 136, 4113–4116 CrossRef CAS PubMed.
- T.-Y. Ma, X.-J. Zhang, G.-S. Shao, J.-L. Cao and Z.-Y. Yuan, J. Phys. Chem. C, 2008, 112, 3090–3096 CAS.
- O. Mashtalir, M. Naguib, B. Dyatkin, Y. Gogotsi and M. W. Barsoum, Mater. Chem. Phys., 2013, 139, 147–152 CrossRef CAS.
- O. Mashtalir, M. Naguib, V. N. Mochalin, Y. Dall'Agnese, M. Heon, M. W. Barsoum and Y. Gogotsi, Nat. Commun., 2013, 4, 1716 CrossRef PubMed.
- Q. Tang, Z. Zhou and P. Shen, J. Am. Chem. Soc., 2012, 134, 16909–16916 CrossRef CAS PubMed.
- G. Kresse and J. Furthmĺźller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmĺźller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. Klimes, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed.
- J. Klimes, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- A. I. Liechtenstein and J. Zaanen, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, R5467–R5470 CrossRef CAS.
- F. Aryasetiawan, K. Karlsson, O. Jepsen and U. Schönberger, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 125106 CrossRef.
- B. Cox, M. Coulthard and P. Lloyd, J. Phys. F: Met. Phys., 1974, 4, 807 CrossRef CAS PubMed.
- M. Kurtoglu, M. Naguib, Y. Gogotsi and M. W. Barsoum, MRS Commun., 2012, 2, 133–137 CrossRef CAS.
- G. Halsey, J. Chem. Phys., 1948, 16, 931 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- G. A. Tritsaris, E. Kaxiras, S. Meng and E. Wang, Nano Lett., 2013, 13, 2258–2263 CrossRef CAS PubMed.
- Y.-H. Li, S. Wang, J. Wei, X. Zhang, C. Xu, Z. Luan, D. Wu and B. Wei, Chem. Phys. Lett., 2002, 357, 263–266 CrossRef CAS.
- X. Deng, L. Lu, H. Li and F. Luo, J. Hazard. Mater., 2010, 183, 923–930 CrossRef CAS PubMed.
- M. R. Lasheen, I. Y. El-Sherif, D. Y. Sabry, S. T. El-Wakeel and M. F. El-Shahat, Desalin. Water Treat., 2013, 1–10 Search PubMed.
- S. Zhao, W. Kang and J. Xue, J. Phys. Chem. C, 2014, 118, 14983–14990 CAS.
| This journal is © the Owner Societies 2016 |