
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical and physical processes in the retention of functional groups in plasma polymers studied by plasma phase mass spectroscopy†
Joonas
Ryssy
a,
Eloni
Prioste-Amaral
b,
Daniela F. N.
Assuncao
c,
Nicholas
Rogers
de,
Giles T. S.
Kirby
de,
Louise E.
Smith
ef and
Andrew
Michelmore
*ef
aSchool of Information Technology and Mathematical Sciences, University of South Australia, Mawson Lakes Campus, Mawson Lakes, SA 5095, Australia
bDepartment of Industrial Engineering, Universidade Federal de Sao Carlos, Sorocaba Campus, Highway John Leme dos Santos, Sao Paulo, 18052-780, Brazil
cDepartment of Materials Engineering, Centro Federal de Educacao Tecnologica de Minas Gerais, 5253 Amazonas Avenue, Nova Suíça, 30480-000, Belo Horizonte, Brazil
dFuture Industries Institute, University of South Australia, Mawson Lakes Campus, Mawson Lakes, SA 5095, Australia
eCooperative Research Centre for Cell Therapy Manufacturing, University of South Australia, Adelaide, South Australia 5000, Australia. E-mail: andrew.michelmore@unisa.edu.au
fSchool of Engineering, University of South Australia, Mawson Lakes Campus, Mawson Lakes, SA 5095, Australia
First published on 12th January 2016
Abstract
Surface engineering of functionalised polymer films is a rapidly expanding field of research with cross disciplinary implications and numerous applications. One method of generating functionalised polymer films is radio frequency induced plasma polymerisation which provides a substrate independent coating. However, there is currently limited understanding surrounding chemical interactions in the plasma phase and physical interactions at the plasma–surface interface, and their effect on functional group retention in the thin film. Here we investigate functionalised plasma polymer films generated from four precursors containing primary amines. Using XPS and fluorine tagging with 4-(trifluoromethyl)benzaldehyde, the primary amine content of plasma polymer films was measured as a function of applied power at constant precursor pressure. The results were then correlated with analysis of the plasma phase by mass spectrometry which showed loss of amine functionality for both neutral and ionic species. Surface interactions are also shown to decrease primary amine retention due to abstraction of hydrogen by high energy ion impacts. The stability of the plasma polymers in aqueous solution was also assessed and is shown to be precursor dependent. Increased understanding of the chemical and physical processes in the plasma phase and at the surface are therefore critical in designing improved plasma polymerisation processes.
Introduction
Surface engineering is growing as a field of research, and one that frequently crosses the disciplines of chemistry, physics and biology.1 Thin film coatings are added to surfaces as barrier coatings, or to impart specific properties such as hardness, roughness or wettability.2 Particularly in the area of micro and nanotechnology, the increase in surface to volume ratio as devices become ever smaller necessitates the ability to tailor surfaces with thin films (<10 nm) for a variety of applications.3Functionalised thin film surface coatings are used to improve biocompatibility,4 for biomolecule attachment,5 to fabricate anti-bacterial6 and non-fouling surfaces7 and as grafting platforms.8 Specific applications include drug delivery,9 biosensors,10 membranes for fuel cells,11 and surfaces for cell attachment, proliferation and delivery.12 In some cases a balance between adhesion and release must be achieved, requiring that chemical functionality be precisely controlled.13
Techniques for functionalizing surfaces include self-assembled monolayers (SAMs),14 direct chemical grafting,15 physisorption16 and UV-induced polymerisation.17 Factors limiting industrial uptake of these techniques include that they are wet chemical processes requiring subsequent drying stages, and the requirement for specific substrate chemistry (e.g. thiol grafting to gold substrates). Plasma polymerisation offers the ability to rationally design surface density of functional groups via a single step, substrate independent and solvent free process. While this technique was first reported in 196018 it was not until the 1980s when the ability to fabricate chemically functionalised surfaces was exploited.19 Through judicious choice of precursor and process parameters, surfaces can be fabricated with varying degree of functionality, but also degree of cross-linking in the plasma polymer bulk. The former determines their surface chemical properties such as surface potential and wettability,20 while the latter affects solubility and mechanical properties of the film.21
Recent studies have shown that understanding the physical processes of deposition from plasma can be used to simultaneously control functional group retention and cross-linking, enabling optimization of film properties. However, correlating process parameters (power input, gas flowrate etc.) with process outputs (film chemistry) is fraught as this misses the critical reactions that occur in the plasma phase, where energetic and reactive species are created which can deposit on surfaces.22 Therefore understanding the processes in the plasma phase23,24 and plasma–surface interactions25–27 is crucial. The plasma phase of organic plasmas quickly become very complex, with fragmentation and oligomerisation reactions occurring simultaneously.28 In principle, detailed understanding of these reactions should enable plasma polymer films to be tailored a priori. While this is not currently possible, detailed chemical and physical observation of the plasma is. Therefore, plasma phase mass spectrometry is a powerful, but under-utilized tool in the fabrication of functionalised plasma polymers.
Amine functionalised surfaces are useful for electrostatic/covalent attachment of biomolecules, and have been demonstrated to improve cell adhesion.29 However, in general, primary amine group density increases film solubility in aqueous solution as they act as sites for water to hydrogen bond. The current study aims to elucidate the processes of functional group loss in plasma polymerisation for four commonly used amine containing precursors. Elemental and functional group surface analysis by XPS are correlated with plasma phase mass spectrometry. Analysis reveals the physical and chemical processes in the plasma phase and at the surface which lead to loss of amine functionality.
Methods
Reagents
Precursors 1,2-ethylenediamine (EDA), allylamine (AA), heptylamine (HA) and 1,3-diaminopropane (DAP) were purchased from Sigma Aldrich (>97%) and used as received. Structures of the precursors are given in Fig. 1. 4-(Trifluoromethyl) benzaldehyde (TFBA) was also purchased from Sigma Aldrich and was stored under an Argon atmosphere to avoid reaction with nitrogen in the air. Glass coverslips were purchased from ProSciTech and were cleaned prior to use by rinsing in ethanol before drying under a stream of dry nitrogen. | ||
| Fig. 1 Chemical structures of precursors used. Primary amine functional groups are highlighted in red. | ||
Plasma reactor and diagnostics
A parallel plate reactor was used throughout, as previously characterized.30 The reactor consisted of a grounded 30 cm diameter steel cylinder with a height of 25 cm. The electrode was an internal 28 cm diameter plate located approximately 1 cm below the top of the reactor. RF power at 13.56 MHz was supplied to the top electrode via a Coaxial Power Systems (UK) generator (RFG050) with a matching network (AMN150). The chamber was evacuated using a rotary pump to a base pressure of below 1 × 10−3 mbar. Precursor vapours were introduced to the chamber via a needle valve (Chell, UK) after several freeze/thaw cycles of the precursor to remove dissolved air. The initial pressure in the chamber was maintained at 1 × 10−2 mbar throughout. For deposition experiments, the RF power was typically applied for 20 min, resulting in plasma polymer films of around 20 nm thickness. Substrates used were 13 mm coverslips, which were placed near the center of the grounded bottom plate. After deposition of plasma polymers, the substrates were stored in sealed containers prior to subsequent analysis.A Hiden Analytical quadrupole mass spectrometer (EQP 1000) was used to measure the neutral and positively charged species in the plasma phase. The mass spectrometer was differentially pumped using a turbo molecular pump to a base pressure below 5 × 10−6 mbar via a 300 μm orifice. For residual gas analysis of the neutral mass spectrum (RGA), the influent gas was ionized using an electron impact source at 20 μA and 70 eV. For positive ions, the instrument was tuned to the protonated precursor (M + H+) and the ion energy distribution recorded. The instrument was then tuned to the peak intensity ion energy, and the mass spectrum acquired. Spectra were corrected for instrument transmission by assuming the peak intensity was proportional to m−1 as advised by the manufacturer.
X-ray photoelectron spectroscopy (XPS)
A SPECS SAGE XPS system was used to obtain XP spectra with an Mg Kα radiation source operating at 10 kV and 20 mA. The system included a Phoibos 150 hemispherical analyzer, with an MCD-9 detector. Survey spectra were recorded between 0–1000 eV at a pass energy of 100 eV with energy steps of 0.5 eV to determine the elements present on the surface of the plasma polymer films. High resolution spectra were then recorded for selected peaks using 0.1 eV energy steps at a pass energy of 20 eV. All spectra were corrected for charging effects by setting the aliphatic carbon peak to 285 eV.31 Processing and component fitting of the spectra were performed using CasaXPS (Neal Fairley, UK).Fluorine derivatization of primary amines
TFBA was used to label and quantify the primary amine concentration on the surface of the plasma polymer films using the Quantitative Elemental Analysis (QEA) method.32 Covalent coupling of TFBA to primary amines allows the surface concentration of primary amines to be determined from XPS data using the eqn (1). | (1) |
Results
XPS surface analysis
Fig. 2 shows the XP spectra of DAP plasma polymer produced at 1 × 10−2 mbar and 4 W applied RF power, which is representative of other plasma polymers produced under different conditions and with other precursors. The spectra shows characteristic peaks for C1s (285 eV) and N1s (399 eV) as expected for amine functionalised plasma polymers, and a small O1s (532 eV) peak due to either residual oxygen in the chamber during plasma, or uptake of oxygen from the atmosphere post-plasma. The C1s peak consists mainly of a C–C/H peak at 285, but also exhibits asymmetry due to the incorporation of C–NHx bonds which manifests in a shoulder at around 286 eV. Uptake of oxygen post-plasma contribute C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–CO groups which manifest at 287.9 eV and 288.1 eV respectively.33 Also shown are the spectra of the same plasma polymer surface after derivatization with TFBA. Successful coupling between primary amines and the TFBA is evident by the appearance of an F1s peak (690 eV) in the survey spectrum, and a small peak in the C1s at around 292.5 eV characteristic of C–F3 groups.
O and N–CO groups which manifest at 287.9 eV and 288.1 eV respectively.33 Also shown are the spectra of the same plasma polymer surface after derivatization with TFBA. Successful coupling between primary amines and the TFBA is evident by the appearance of an F1s peak (690 eV) in the survey spectrum, and a small peak in the C1s at around 292.5 eV characteristic of C–F3 groups.
 | ||
| Fig. 2 Survey (left) and C1s (right) XP spectra of DAP plasma polymer deposited at 4 W, before (bottom) and after (top) TFBA derivatization. | ||
Nitrogen to carbon ratio
The nitrogen to carbon ratio (N/C) for plasma polymer films was determined by XPS and the results are presented in Fig. 3a. EDA showed the highest N/C at around 0.38, followed by DAP (0.3), AA (0.18) and HA (0.08). These values are slightly lower but similar to previous reports despite variations in operating pressure, reactor geometry etc., as shown in Table 1. The N/C was remarkably constant with power for each precursor. The trend follows the stoichiometric N/C of each precursor, (1, 0.67, 0.33, 0.14 respectively) but with approximately 40–60% of the nitrogen content being lost during the deposition process, again in agreement with previous studies.33,34 This is presumably due to fragmentation of the precursor in the plasma by electron impacts, and smaller fragments, such as ˙NH2, being readily pumped out of the chamber. | ||
Fig. 3 (a) Nitrogen to carbon ratio for AA ( ), EDA ( ), EDA ( ), DAP ( ), DAP ( ) and HA ( ) and HA ( ) as a function of RF power. (b) Primary amine group surface concentration for AA, EDA, DAP and HA as a function of RF power. Initial pressure was 1 × 10−2 mbar. ) as a function of RF power. (b) Primary amine group surface concentration for AA, EDA, DAP and HA as a function of RF power. Initial pressure was 1 × 10−2 mbar. | ||
Primary amine retention
The primary amine surface concentration, calculated from TFBA derivatization, is presented in Fig. 3b. In general, the primary amine concentration decreases with RF power for each precursor. Interestingly, the N/C data presented in Fig. 3a does not predict the primary amine concentration. EDA exhibited the highest N/C of around 0.38, but the primary amine concentration was much lower than DAP, particularly at low power. The N/C for AA was around 2.5 times higher than HA, but the primary amine concentrations were very similar, particularly at high RF power.Neutral mass spectrometry
Fig. 4 shows the neutral mass spectrometry of the four precursors without the plasma being ignited. The AA spectrum shows a dominant peak at 56 m/z which corresponds to the precursor (M − H)+ indicating the AA molecule is relatively stable under electron impacts. This may be due to the resonant double bond for allylic compounds, which for AA may exist as C1C2 or C2C3 and therefore strengthens the bonds in the carbon backbone of the molecule. Peaks are also evident at 30 m/z (CH2–NH2), corresponding to cleavage of the precursor at the C–C bond as previously observed,25 and 41 (CH2–CHCH2) and 17 m/z (NH3) corresponding to cleavage of the C–N bond. The peak at 28 m/z is assigned to either CH2CH2+ or loss of two hydrogens from the 30 m/z peak.
 | ||
| Fig. 4 Electron impact spectra of AA, EDA, DAP and HA at 1 × 10−2 mbar without plasma being ignited (left) and with plasma at 10 W (right). | ||
For EDA, DAP and HA, the precursor peaks at 60, 74 and 114 m/z are relatively small indicating that these precursors are less stable than AA when subjected to high energy electron impacts. This has previously been explained for EDA as being due to weakening of the C–C bond because of the proximity of two electronegative NH2 groups.37 A similar explanation can be postulated for DAP as the precursor is only one carbon longer than EDA, so weakening of the C–C bonds would be less pronounced but still offers a plausible explanation. For both EDA and DAP, a major peak is seen at 30 m/z, the same CH2–NH2 peak seen for AA. The major peak for DAP is observed at 57 m/z, corresponding to loss of NH2 from the precursor.
For HA, the precursor only has one terminal NH2 group with a 7 carbon chain backbone, so weakening of the C–C bonds by electronegative groups is not predicted to be as important. However, the increased molecular weight gives a much higher cross-sectional area for electron impacts, resulting in increased fragmentation of the precursor. Nevertheless, the peak at 30 m/z is the dominant peak, with minor peaks at 17, 44 and 57 m/z.
With the plasma ignited at 10 W, the neutral mass spectra look remarkably similar for all four precursors as shown in Fig. 4. The major peak for all precursors occurs at 27 or 28 m/z, with smaller peaks at 17, 30, 41 and 57 m/z. The peaks at 27, 28 m/z probably originate from CH2–NH2, (30 m/z) with subsequent loss of hydrogen atoms with further electron impacts, either in the plasma phase, or in the ionizing step in the mass spectrometer. This would result in either a diradical (˙HC–NH˙),38 or an unsaturated fragment where the two radicals combine to form a CN bond (HCNH). Similarly, the series of peaks at 39, 41, 42 and 43 m/z may be due to loss of hydrogen from CH2–CH2–NH2. Therefore, the chemistry of the neutral species in the plasma phase appears to be quite constant for all four precursors. Scheme 1 shows examples of some reactions which occur in the plasma phase for DAP.
 | ||
| Scheme 1 Plasma phase reactions which may occur for DAP, highlighting loss and retention of primary amine groups. | ||
The neutral mass spectra for DAP were normalized, and then normalized distributions subtracted from the electron impact spectra with no plasma. These data are shown in Fig. 5, which indicates the change in mass distribution at different powers. Peaks assigned to species containing primary amines (30, 44, 57 m/z) decrease in relative intensity as power is increased. These species are fragmented by loss of hydrogen to 27, 28, 40, 42, 56 m/z etc. which may include hydrogen being lost from –NH2 to form –NH˙ or –˙N˙, all of which increase in intensity with power. Alternatively, hydrogen may be lost from the CHx groups. The analysis in Fig. 5 shows that increasing the applied plasma power increases the likelihood of primary amines being fragmented in the plasma phase to secondary and tertiary amine radicals. Similar analysis for AA, EDA and HA shows similar trends but with even greater fragmentation of the primary amine peaks as power is increased for EDA (see ESI,† Fig. S1).
 | ||
| Fig. 5 Change in neutral species distribution with applied power for DAP compared to electron impact with no plasma. | ||
Positive ion mass spectrometry
The positive ion mass spectra are presented in Fig. 6. These data are normalized by the precursor molecular weight such that primary precursor and oligomeric species can be compared. At 10 W, the dominant peak for all precursors was the protonated precursor (M + H)+, which is formed by proton transfer between the precursor M, and H3O+.39 Smaller fragment peaks are observed similar to the neutral mass spectra. For example, for DAP, the protonated precursor peak is seen at 75 m/z (see Scheme 1), with smaller fragment peaks observed at 44, 30 and 17 m/z. These small fragments are particularly high in intensity for AA and HA.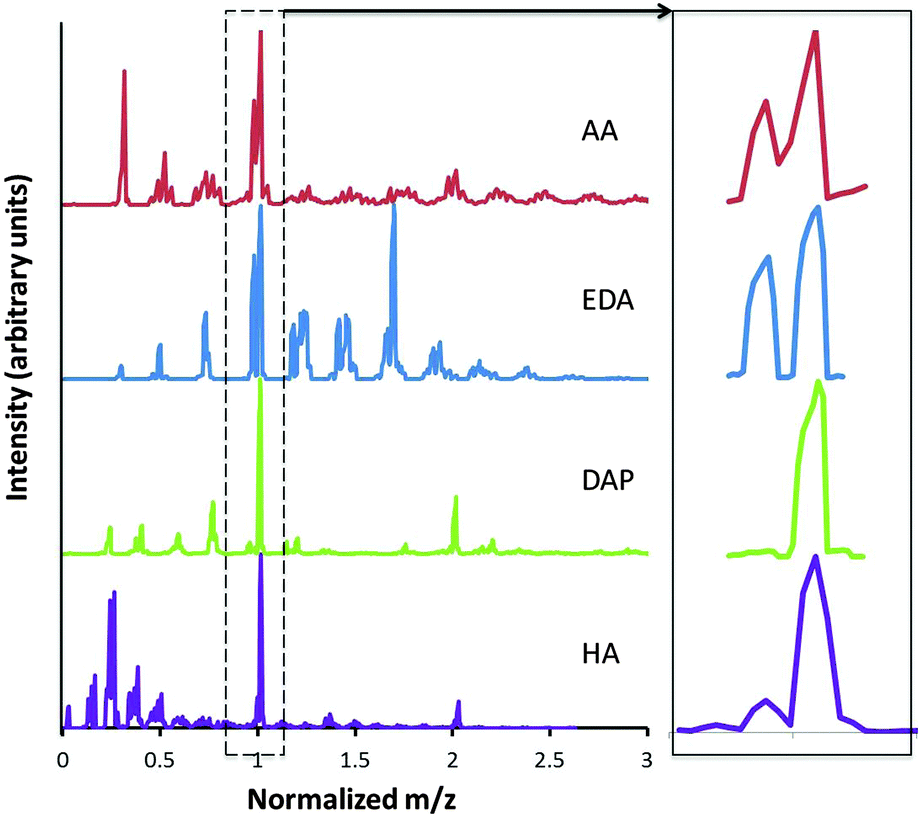 | ||
| Fig. 6 Positive ion spectra of AA, EDA, DAP and HA plasmas at 10 W (left) and (M − H)+ and (M + H)+ peaks (right). Masses have been normalized by the precursor mass for comparison of intact precursor and oligomer ions. | ||
Unlike the neutral mass spectra though, large, positively charged oligomeric species are observed in the plasma phase following eqn (1).40
| R(NH2)jH+ + R(NH2)j → [R(NH2)j]2H+ | (2) |
Similar to the comparative analysis of the neutral species in Fig. 5, Fig. 7 shows the change in distribution of the positive species for DAP with applied power relative to 2 W plasma. In the region below M + H+ at 75 m/z, the relative amounts of small fragments increased with power, as expected due to increased electron density in the plasma and greater likelihood of fragmentation due to electron impacts. The relative amount of dimer (2M + H)+ increased with power. This can be explained by an increase in the amount of reactive species in the plasma phase as the plasma density increases. This increases the likelihood of ionized precursor molecules colliding and forming dimers. Of particular interest is the decrease in the relative amounts of species between M + H+ and 2M + H+. The peaks at 131, 117, 103 and 89 m/z correspond to 2M + H+ after the loss of NH2, and then subsequent CH2 groups.
 | ||
| Fig. 7 Change in positive ion distribution with applied power for DAP compared to 2 W plasma. | ||
Stability
Plasma polymer films at each condition were prepared such that their thicknesses were ∼20 nm. To assess the stability of these films, they were placed in Milli-Q water for 1 hour without agitation, then rinsed briefly before being dried under dry nitrogen. The samples were then analysed by XPS. Fig. 8 shows the silicon signal as detected by XPS as a function of initial primary amine content. The presence of silicon in the spectra indicates the dissolution of the film, as the depth of analysis for XPS is of the order of 5–10 nm.41 The results show that HA and AA are very stable, with only minor amounts of silicon present when the primary amine content was high (i.e. at low power). EDA showed a significant increase in the silicon signal, particularly at high primary amine concentrations. This is consistent with previous measurements using the current reactor, although at a higher pressure,25 which showed that EDA films were unstable except at high power. Indeed, from the data it is likely that the EDA films at low power were almost completely dissolved. Interestingly, DAP also exhibited silicon peaks at high primary amine content, but the EDA and DAP plots do not overlap; the dissolution of DAP films occurs at much higher primary amine concentrations that EDA. DAP plasma polymers with 3% primary amine content were still stable, while EDA plasma polymers with the same primary amine content were almost completely dissolved. | ||
Fig. 8 Stability of AA ( ), EDA ( ), EDA ( ), DAP ( ), DAP ( ) and HA ( ) and HA ( ) plasma polymers as shown by change in nitrogen content (top) and appearance of the substrate silicon peak (bottom) after washing in Milli-Q water for 1 hour. ) plasma polymers as shown by change in nitrogen content (top) and appearance of the substrate silicon peak (bottom) after washing in Milli-Q water for 1 hour. | ||
Also shown is the change in nitrogen concentration after contact with water. HA and AA decreased in nitrogen only slightly, with DAP showing a slightly higher decrease. Plasma polymers at lower power decreased in nitrogen more than higher power. This is consistent with the primary amine content, as it has been shown that primary amines provide sites for water to hydrogen bond and thus dissolve short attached hydrocarbon chains.42,43 EDA showed the largest decrease in nitrogen content, particularly at low power, despite the fact that DAP had a higher primary amine content (see Fig. 3). The lower than expected decrease in nitrogen at 2 W and 4 W for EDA is probably due to the plasma polymer being almost completely dissolved, leaving a very thin layer in intimate contact with the substrate. It has been shown previously that the chemistry of amines44 and other plasma polymers45 close to substrates is different to that in the bulk of the film.
Discussion
Retention of functionality in plasma polymerisation is dependent on two processes; fragmentation/oligomerisation in the plasma phase due to electron impacts, and deposition and sputtering processes at the surface.Plasma phase
Electrons heated by the applied electric fields in the plasma typically have a Druyvesteyn distribution of temperatures.46 Average electron temperatures of 3 eV are typical, but a small proportion may have temperatures greater than 10 eV. Electrons impacting molecules with energies of 3–5 eV can result in fragmentation due to dissociation reactions, giving rise to radical species. C–H and N–H bonds are relatively strong (typically ∼100–110 kcal mol−1) while C–C and C–N bonds are slightly weaker (∼85–90 kcal mol−1).37 Thus, in the case of a single electron impact with an organic molecule containing amine groups, the most likely bonds to be broken would be the C–C and C–N bonds. This is shown in Fig. 4 for the RGA with no plasma data which gives rise to ˙NH2, ˙CH2–NH2, and ˙CH2–CH2–NH2 species. As the applied RF power is increased, the plasma and electron density increases which increases the probability of molecules undergoing multiple electron impacts. Therefore it is more likely that C–H and N–H bonds may be broken as shown in Fig. 5 where the relative intensity of primary amine containing groups are decreased. Thus retention of primary amine groups in radical species is high at low applied RF power, but increasing the RF power leads to increased probability of primary amines losing hydrogen and being converted into secondary and tertiary amines.Higher energy electron impacts (>10 eV) result in ionization of neutrals. For all precursors, at low power the dominant ionic species is the protonated monomer, retaining the primary amine functionality. However loss of hydrogen also occurs, which may be due to release of excess energy from the ionizing electron impact, or secondary electron impacts. For the M + H+ ion, loss of hydrogen occurs most readily for EDA and AA, while precursor ions of HA and DAP are more stable. While the chemistry of the neutral species are dominated by fragmentation reactions, ionic oligomeric species are formed in the plasma phase, vastly increasing the complexity of the ion chemistry. There is competition between oligomerisation and fragmentation reactions, as shown by the formation of the 2M + H+ ion, but then fragmentation of this oligomer to smaller species, for example 2M − NH2+. EDA in particular shows fragmentation of dimer oligomers.
Therefore, from the plasma phase analysis, DAP appears to favour structures which retain primary amine functionality, while in contrast EDA exhibits noticeably more loss of hydrogen which can contribute to loss of primary amines.
Surface interactions
In the initial stages of deposition, the surface is activated by high energy ion impacts which cause scission of the bonds at the surface of the substrate. The radical sites which result can then enable plasma phase species to deposit, forming a covalent bond between the depositing molecule and the surface (see Scheme 2). After this initial stage, plasma polymers grow via three main mechanisms; direct ionic deposition, radical termination and radical propagation.47 Radical propagation can only occur via carbon–carbon double bonds, which are not present in EDA, DAP and HA. Therefore EDA, DAP and HA deposition must be dominated by ionic deposition and radical termination reactions. AA contains an allylic bond, which in traditional polymerisation have been shown to polymerize slowly and have increased probability of terminating.48 It has been shown that plasma deposition of allylic precursors is dominated by ionic processes.49 It is possible for species containing double bonds to be created in the plasma through formation of diradicals, however the fraction of such species would be extremely low. Thus, radical propagation reactions can largely be discounted as a growth mechanism for the precursors studied here. | ||
| Scheme 2 Surface interactions of high energy ions, creating radical sites, enabling radical grafting. | ||
Due to the sheath voltages which are created by the plasma, at low pressure ions are accelerated to surfaces and arrive at energies of a few tens of eV and this increases with power due to an increase in electron temperature.50 These ion impacts can result in ion deposition (see below) or creation of radical sites. The resulting radical sites are then available for radical termination reactions with plasma phase radical species. Unlike ions, radicals are not accelerated to the surface due to sheath voltages, and so arrive at the surface by diffusion at approximately ambient temperature. Radicals then utilize chemical free energy gained in the plasma phase, rather than kinetic energy to drive deposition. Thus if radical processes were the only mechanism by which plasma polymers could grow, the process of radical termination reactions result in retention of functionality, and the limiting factor is retention in the plasma phase.
However, ions are also important in plasma polymerisation. While radical species utilize chemical free energy to adsorb to the plasma polymer surface, ions must utilize kinetic energy to overcome any energetic barrier to deposition. Ionic adsorption has been shown to be important in the deposition of AA.51 This is in part due to the fact that AA is a stable precursor, as it exhibits low fragmentation into radical species in the plasma phase compared to other precursors studied here, as shown in Fig. 4. The sticking probability of ionic species is dependent on the energy with which the ions impact the surface. Increasing the ion energy increases the sticking probability of the ion,52 but also increasing ion energy above ∼15 eV increases the rate of ablation and damage to the existing surface.53 Plasma polymers are typically deficient in hydrogen due to abstraction of hydrogen26,54 and it is likely that some of the lost hydrogen will be from surface amine groups. Thus, retention of ionic functional amine groups in the plasma phase may not lead to retention in the deposited thin film, as they may be fragmented/rearranged after subsequent ion impacts on the surface. In fact, ions which do not deposit on the surface may still reduce functional group retention by abstracting hydrogen from radicals and ions already deposited. The ion energy for the plasmas used in this study varied from ∼10 eV at 2 W up to ∼28 eV at 20 W. Thus, at low power, while the sticking probability of the ions is expected to be low, loss of functionality due to surface bombardment should be minimized. However, at higher power we can expect a high degree of fragmentation/rearrangement on the surface, and thus loss of primary amine groups.
Stability
Stability in aqueous media of amine functionalised plasma polymers is determined by primary amine density and the degree of cross-linking of the plasma polymer. Cross-linking increases the average molecular weight of the polymer chain attached to each amine group25 which decreases solubility. In addition to losing amine functionality, ion bombardment increases the cross-link density for plasma polymers.55 AA and HA plasma polymers are low in primary amines (maximum ∼2%) and thus are quite insoluble in aqueous solution, with only a small loss of nitrogen from the surface. In contrast, EDA and DAP plasma polymers exhibit solubility at high primary amine concentrations, but their behaviours are quite different. EDA plasma polymers with only ∼1% primary amine concentration exhibit some solubility, and at 2% are almost completely soluble. DAP plasma polymers on the other hand are stable up to ∼3% primary amine. The difference in solubility between EDA and DAP plasma polymers with the same primary amine density may be related to the relative densities of secondary amines (which are still soluble, but less so than primary amines) or an increase in cross-linking of the plasma polymers. Hegemann et al. describe the effect of momentum transfer from high energy ions to the surface in the densification of plasma polymers.56 For ions of different masses which traverse the plasma sheath, while the kinetic energy delivered to the surface is constant, the momentum transfer is proportional to m1/2. The average ion masses for EDA and DAP at 10 W are 83.9 and 96.0 amu respectively. Thus, with larger ions for DAP, the momentum transfer to the growing DAP plasma polymer would be higher, resulting in a higher degree of cross linking.Conclusions
Plasma phase mass spectrometry is a powerful tool for measuring reactions in the plasma phase, and the species which impact surfaces in plasma polymerisation. For the four precursors studied here, nitrogen to carbon ratio is not a good predictor of surface functionality. This is due to the ease with which some precursors (EDA in particular) fragment with electron impacts in the plasma losing primary amine groups, while others (e.g. DAP) favour structures which retain primary amines.While plasma phase reactions are important, plasma–surface interactions can also be the cause of loss of functionality due to sputtering, and certainly affect the degree of cross-linking in the surface. These factors combined determine the functional group density and solubility of aminated plasma polymers, and thus their practicality. Increased understanding of the processes in the plasma phase and at the surface are therefore critical in intelligently designing improved plasma polymerisation processes.
References
- P. Roach, T. Parker, N. Gadegaard and M. R. Alexander, Surf. Sci. Rep., 2010, 65, 145–173 CrossRef CAS.
- M. Ohring, The Materials Science of Thin Films, Academic Press, Hoboken, New Jersey, 1992 Search PubMed.
- S. Prakash, M. B. Karacor and S. Banerjee, Surf. Sci. Rep., 2009, 64, 233–254 CrossRef CAS.
- F. Variola, F. Vetrone, L. Richert, P. Jedrzejowski, J.-H. Yi, S. Zalzal, S. Clair, A. Sarkissian, D. F. Perepichka, J. D. Wuest, F. Rosei and A. Nanci, Small, 2009, 5, 996–1006 CrossRef CAS PubMed.
- K. S. Siow, L. Britcher, S. Kumar and H. J. Griesser, Plasma Processes Polym., 2006, 3, 392 CrossRef CAS.
- T. Michl, B. R. Coad, M. Doran, M. Osiecki, M. H. Kafshgari, N. H. Voelcker, A. Husler, K. Vasilev and H. J. Griesser, Chem. Commun., 2015, 51, 7058–7060 RSC.
- S. Bouaidat, C. Berendsen, P. Thomas, S. G. Petersen, A. Wolff and J. Jonsmann, Lab Chip, 2004, 4, 632–637 RSC.
- B. R. Coad, K. E. Styan and L. Meagher, ACS Appl. Mater. Interfaces, 2014, 6, 7782–7789 CAS.
- K. Vasilev, N. Poulter, P. Martinek and H. J. Griesser, ACS Appl. Mater. Interfaces, 2011, 3, 4831–4836 CAS.
- F. S. H. Krismastuti, H. Bayat, N. H. Voelcker and H. Schönherr, Anal. Chem., 2015, 87, 3856–3863 CrossRef CAS PubMed.
- Z. Jiang and Z. Jiang, J. Membr. Sci., 2014, 456, 85–106 CrossRef CAS.
- N. G. Walker, A. Mistry, L. E. Smith, P. C. Eves, S. Forster, G. Tsaknakis, S. Watt and S. MacNeil, Tissue Eng., Part C, 2012, 18, 143–1552 CrossRef CAS PubMed.
- G. Kirby, L. Vandenpoel, J. Pinxteren, R. Short and A. Michelmore, Cytotherapy, 2015, 17, S14 CrossRef.
- A. Hucknall, S. Rangarajan and A. Chilkoti, Adv. Mater., 2009, 21, 2441–2446 CrossRef CAS.
- B. Dong, H. Jiang, S. Manolache, A. C. Lee Wong and F. S. Denes, Langmuir, 2007, 23, 7306–7313 CrossRef CAS PubMed.
- C. D. Heyes, J. Groll, M. Moller and G. U. Nienhaus, Mol. BioSyst., 2007, 3, 419–430 RSC.
- P. Wang, K. L. Tan, E. T. Kang and K. G. Neoh, J. Mater. Chem., 2001, 11, 2951–2957 RSC.
- J. Goodman, J. Polym. Sci., 1960, 44, 551–552 CrossRef CAS.
- H. K. Yasuda, Plasma Polymerisation, Academic Press, London, U.K., 1985 Search PubMed.
- M. Miyama, Y. Yang, T. Yasuda, T. Okuno and H. K. Yasuda, Langmuir, 1997, 13, 5494–5503 CrossRef CAS.
- A. Michelmore, D. A. Steele, D. E. Robinson, J. D. Whittle and R. D. Short, Soft Matter, 2013, 9, 6167–6175 RSC.
- B. Chapman, Glow Discharge Processes, John Wiley and Sons, Chichester UK, 1980 Search PubMed.
- S. Ligot, M. Guillaume, P. Gerbaux, D. Thiry, F. Renaux, J. Cornil, P. Dubois and R. Snyders, J. Phys. Chem. B, 2014, 118, 4201–4211 CrossRef CAS PubMed.
- L. Denis, D. Cossement, T. Godfroid, F. Renaux, C. Bittencourt, R. Snyders and M. Hecq, Plasma Processes Polym., 2009, 6, 199–208 CrossRef CAS.
- C. Daunton, L. E. Smith, J. D. Whittle, R. D. Short, D. A. Steele and A. Michelmore, Plasma Processes Polym., 2015, 12, 817–826 CrossRef CAS.
- S. Peter, K. Graupner, D. Grambole and F. Richter, J. Appl. Phys., 2007, 102, 0533304 CrossRef.
- J. Trieschmann and D. Hegemann, J. Phys. D: Appl. Phys., 2011, 44, 475201 CrossRef.
- L. O'Toole, A. J. Beck, A. P. Ameen, F. R. Jones and R. D. Short, J. Chem. Soc., Faraday Trans., 1995, 91, 3907–3912 RSC.
- P. Hamerli, T. Weigel, T. Groth and D. Paul, Biomaterials, 2003, 24, 3989–3999 CrossRef CAS PubMed.
- A. Michelmore, J. D. Whittle, R. D. Short, R. W. Boswell and C. Charles, Plasma Processes Polym., 2014, 11, 833–841 CrossRef CAS.
- G. Beamson and D. Briggs, High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database, John Wiley and Sons, Chichester, U.K., 1992 Search PubMed.
- J. C. Ruiz, S. Taheri, A. Michelmore, D. E. Robinson, R. D. Short, K. Vasilev and R. Forch, Plasma Processes Polym., 2014, 11, 888–896 CrossRef CAS.
- T. R. Gengenbach and H. J. Griesser, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2191–2206 CrossRef CAS.
- C.-G. Golander, M. W. Rutland, D. L. Cho, A. Johansson, H. Ringblom, S. Jonsson and H. K. Yasuda, J. Appl. Polym. Sci., 1993, 49, 39–51 CrossRef.
- H. Biederman, I. H. Boyaci, P. Bilkova, D. Slavinska, S. Mutlu, J. Zemek, M. Trchova, J. Klimovic and M. Mutlu, J. Appl. Polym. Sci., 2001, 81, 1341–1352 CrossRef CAS.
- D. E. Robinson, A. Marson, R. D. Short, D. J. Buttle, A. J. Day, K. L. Parry, M. Wiles, P. Highfield, A. Mistry and J. D. Whittle, Adv. Mater., 2008, 20, 1166–1169 CrossRef CAS.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- S. Pedersen, J. L. Harek and A. H. Zewail, Science, 1994, 266, 1359–1364 CAS.
- A. Michelmore, D. A. Steele, J. D. Whittle, J. W. Bradley and R. D. Short, RSC Adv., 2013, 3, 13540–13557 RSC.
- H. Wincel and J. A. Herman, J. Chem. Soc., Faraday Trans., 1973, 69, 1797–1810 RSC.
- D. Briggs, Surface analysis of polymers by XPS and static SIMS, Cambridge University Press, Cambridge, 1998 Search PubMed.
- A. O. Christie and D. J. Crisp, J. Appl. Chem., 1967, 17, 11–14 CrossRef CAS.
- R. M. Stephenson, J. Chem. Eng. Data, 1993, 38, 625–629 CrossRef CAS.
- K. Vasilev, A. Michelmore, H. J. Griesser and R. D. Short, Chem. Commun., 2009, 3600–3602 RSC.
- R. T. Chen, B. W. Muir, L. Thomsen, A. Tadich, B. C. C. Cowie, G. K. Such, A. Postma, K. M. McLean and F. Caruso, J. Phys. Chem. B, 2011, 115, 6495 CrossRef CAS PubMed.
- M. J. Druyvesteyn and F. M. Penning, Rev. Mod. Phys., 1940, 12, 87–174 CrossRef CAS.
- G. Padron-Wells, B. C. Jarvis, A. K. Jindal and M. J. Goeckner, Colloids Surf., B, 2009, 68, 163–170 CrossRef CAS PubMed.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, New York, 1953 Search PubMed.
- A. Michelmore, C. Charles, R. W. Boswell, R. D. Short and J. D. Whittle, ACS Appl. Mater. Interfaces, 2013, 5, 5387–5391 CAS.
- M. A. Lieberman and A. J. Lichtenberg, Principles of Plasma Discharges and Materials Processing, John Wiley and Sons, Hoboken, NJ, 2005 Search PubMed.
- A. Choukourov, H. Biederman, D. Slavinska, L. Hanley, A. Grinevich, H. Boldryeva and A. Mackova, J. Phys. Chem. B, 2005, 109, 23086–23095 CrossRef CAS PubMed.
- P. N. Brookes, S. Fraser, R. D. Short, L. Hanley, E. Fuoco, A. Roberts and S. Hutton, J. Electron Spectrosc. Relat. Phenom., 2001, 121, 281–297 CrossRef CAS.
- D. C. Jacobs, Annu. Rev. Phys. Chem., 2002, 53, 379–407 CrossRef CAS PubMed.
- A. Nelson, B. W. Muir, J. Oldham, C. Fong, K. M. McLean, P. G. Hartley, S. K. Oiseth and M. James, Langmuir, 2006, 22, 453–458 CrossRef CAS PubMed.
- A. Choukourov, I. Gordeev, D. Arzhakov, A. Artemenko, J. Kousal, O. Kylián, D. Slavínská and H. Biederman, Plasma Processes Polym., 2012, 9, 48–58 CrossRef CAS.
- D. Hegemann, E. Korner, N. Blanchard, M. Drabik and S. Guimond, Appl. Phys. Lett., 2012, 101, 211603 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp05850c |
| This journal is © the Owner Societies 2016 |