
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemo-dynamical characterization of polypyrrole actuators coated on gold electrodes†
J. G.
Martinez
 ab,
T. F.
Otero
a and
E. W. H.
Jager
*b
ab,
T. F.
Otero
a and
E. W. H.
Jager
*b
aUniversidad Politécnica de Cartagena, ETSII, Center for Electrochemistry and Intelligent Materials (CEMI), Paseo Alfonso XIII, Aulario II, 30203 Cartagena, Spain
bLinköping University, Department of Physics, Chemistry and Biology, Biosensors and Bioelectronics Centre, 58183 Linköping, Sweden. E-mail: edwin.jager@liu.se
First published on 18th November 2015
Abstract
Polypyrrole coated gold wires were subjected to consecutive square current waves in LiClO4 aqueous solutions using the same constant anodic and cathodic charge. Parallel in situ diameter variations were followed using a laser scan micrometer. The procedure was repeated by changing one experimental variable every time: applied current, electrolyte concentration or working temperature to perform electrochemodynamical characterization of the system. On average, the diameter follows a linear variation of the consumed charge, as expected for any faradaic system, although a high dispersion was attained in the data. Such deviations were attributed to the presence of irreversible hydrogen evolution at the gold/polypyrrole interface at cathodic potentials more than 0.0 V vs. Ag/AgCl, detected and quantified from separated coulovoltammetric responses. Despite this parallel hydrogen evolution the consumed energy during reactions is a robust sensor of the working conditions. In conclusion a gold support, the metal most used for technological applications of conducting polymers, should be avoided when a device is driven by current flow in the presence of aqueous solutions, water contamination or moisture: a fraction of the charge will be consumed by hydrogen generation with possible degradation of the device.
Introduction
Artificial muscles from conducting polymers (CPs)1–16 constitute an emerging technology in the actuator field. They are biomimetic actuators having a similar composition (reactive macromolecules, ions and water) and working in a similar way to natural muscles: when an electrical pulse arrives to the conducting polymer (CP), a chemical reaction occurs varying the chemical composition and the physical properties of the material including the material volume.17–23 In natural muscles, an ionic pulse arrives from the nerve axon, liberates calcium ions inside the sarcomere triggering the ATP reaction whose energy is translated to conformational changes in the myosin motor proteins resulting in a macroscopic movement.24,25The driving electrochemical reaction in CPs exchanging anions during reactions can be expressed as:6
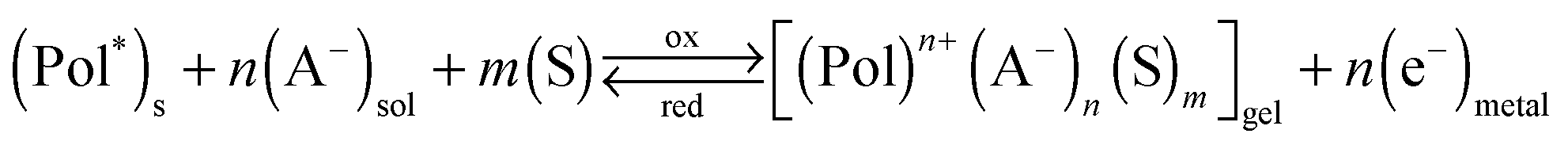 | (1) |
Any physical or chemical variable acting on the reaction rate (temperature, electrolyte concentration, active centre concentration in the polymeric film) influences the swelling/shrinking rates and the actuating properties of the material.8,26–28 The effect of the electrolyte concentration on the electrochemical response has been used to build concentration sensors.27,29–37 The influence of the electrolyte concentration and temperature on the actuation is being studied by different authors trying to get a deeper understanding and improving the actuation process.38–47
When the voltammetric results from CP film coating metals are represented as coulovoltammetric responses, the irreversible generation of hydrogen at the polymer/metal interface was detected and quantified.48,49 This important consumption of charge decreases the efficiency (as this charge is not used for actuation) and promotes a parallel degradation of the actuating polymer supported by a metal.49
The synthesis conditions of polypyrrole (PPy) have been well studied, including the effect of the pyrrole concentration, the type of counter-ion present and its concentration, the solvent, and the potential or current applied.50–58
Many actuators use sputtered or thermally evaporated gold films as supports of the conducting polymer films.13,16,59–71 Here, the effect of different experimental variables (temperature, applied current and electrolyte concentration) on the evolution of the PPy dimensions (electro-chemo-mechanical or electro-chemo-dynamical characterization) supported by gold wires will be followed using a Laser Scan Micrometer (LSM).72 The presence of some parallel hydrogen evolution will also be checked and quantified and its strong influence on the actuation results is discussed here.
Results
Different applied currents
Before each experiment the state of the PPy electrode was checked by a voltammetric control between −0.7 V and 0.3 V at 20 mV s−1 in 0.1 M LiClO4 aqueous solution, stopping every time the potential sweep at the cathodic potential limit. Then the electrode is subjected to three consecutive square current waves consuming 8 mC during each anodic or cathodic step in order to get stationary chronopotentiometric responses. The experimental procedure was repeated by changing one experimental variable every time. By applying square current waves of different currents while keeping constant the rest of the experimental variables (consumed anodic and cathodic charge, polymer, electrolyte concentration and temperature) the normalized chronopotentiometric (potential/time) responses (the initial potential was set to zero every time) are depicted by Fig. 1a (anodic) and Fig. 1b (cathodic) (see ESI,† results for the different variables). By consuming the same anodic and cathodic charge for every experiment the actuator should be expected to move between the same initial and final redox state of the constituent PPy film. In order to consume the same charge (q = it) with every experimental current (i) the period (t) of the square wave was varied in accordance with the applied current.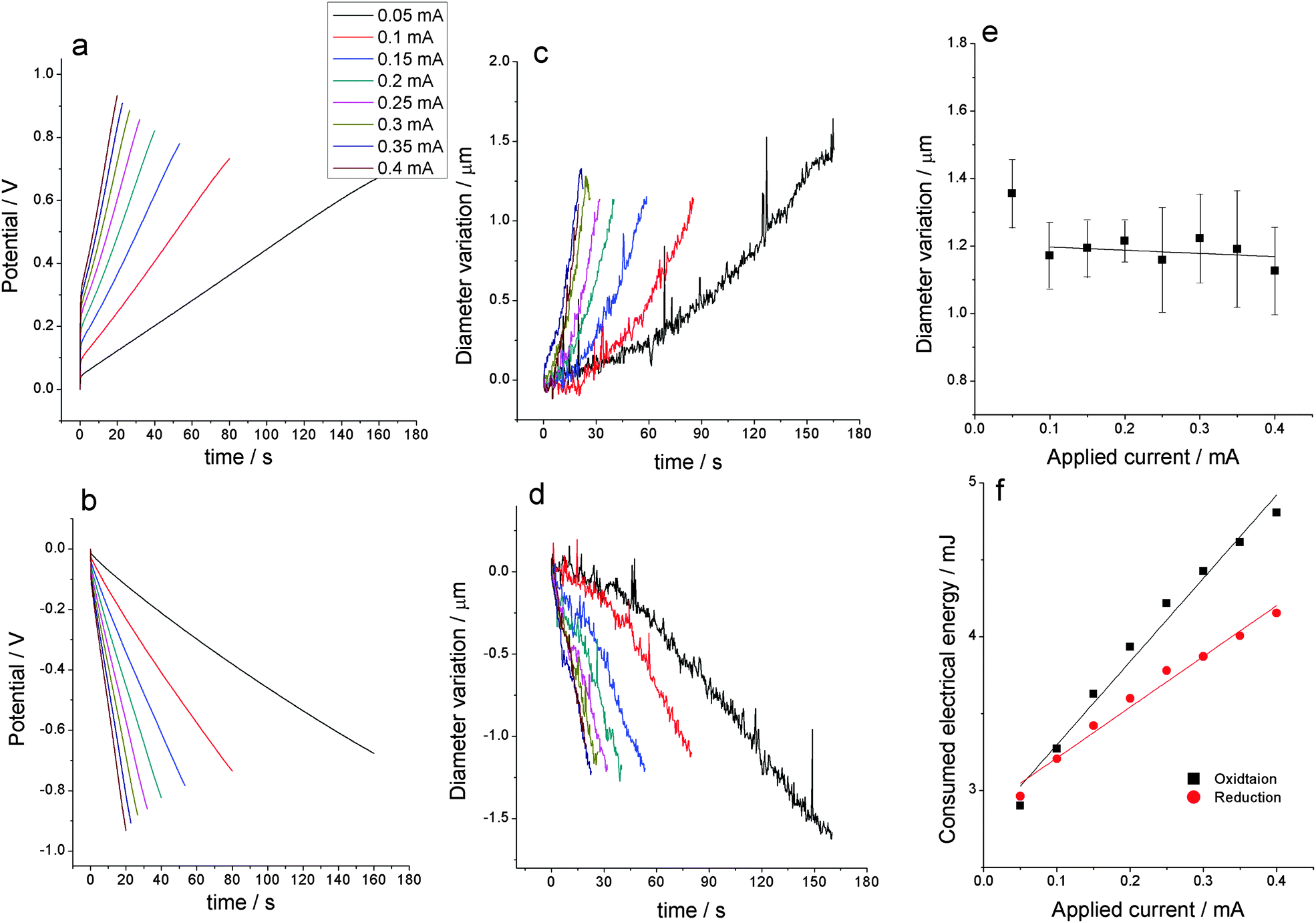 | ||
| Fig. 1 Normalized anodic (a) and cathodic (b) chronopotentiometric responses obtained from a PPy gold coated wire in 0.1 M LiClO4 aqueous solution submitted to consecutive square current waves for the same oxidation and reduction charges (0.8 mC) attained by flow of different currents (from 0.045 to 0.4 mA), indicated in the figure, at room temperature (21 °C). Simultaneous variation of the electrode diameter (average from five measurements) during the anodic (c), or the cathodic (d) processes. (e) Average final electrode diameter after consumption of a constant charge of 8 mC every time. (f) Consumed electrical energy as a function of the applied current: sensing calibration curves. | ||
The diameter variation of the PPy coated gold electrode during oxidation is shown in Fig. 1c and during reduction in Fig. 1d. As expected from reaction (1), during the oxidation reaction, the electrode diameter swells by insertion of counter ions (anions) and solvent. During reduction counter ions and solvent are expelled to the electrolyte, the PPy electrode shrinks and the diameter decreases. A higher applied current driving the reaction results in a faster movement. The rate of the dimensional variation is not fully linear, being slower at the beginning of the processes and faster at the end. This non-linearity has been attributed49 to a different number of water molecules per exchanged ion (required for osmotic balance inside the film)43,73–75 at the beginning and at the end of the process and a faster solvent draining (electro-osmotic effect) from a shrunken film.
The total diameter variation of the PPy coated gold wire as a function of the applied current can be seen in Fig. 1e. A large dispersion on the measured diameter can be observed. Such dispersion and the non-constancy of the diameter variation, as expected from the constant consumed charge, could be mainly related to the possible presence of a parallel hydrogen release and some kinetic effect of the osmotic pressure. Applying higher currents here has a similar effect to applying a higher frequency as reported by Jafeen et al.76
By integration of Fig. 1a and b the consumed electrical energy ( ) during the actuator movement is attained. Fig. 1f indicates that the consumed energy is a linear function of the applied current. This means that the energy consumed by the actuator senses the driving current, i.e. the actuator simultaneously acts as a sensor. The lines in Fig. 1f are the calibrating lines of the sensor for both, anodic and cathodic processes. Any physical or chemical perturbation affecting reaction (1) should promote a shift in the reaction energy to fit the new imposed energetic requirements (Otero's principle).8,27 The current is such a variable determining the reaction rate, so the energy (
) during the actuator movement is attained. Fig. 1f indicates that the consumed energy is a linear function of the applied current. This means that the energy consumed by the actuator senses the driving current, i.e. the actuator simultaneously acts as a sensor. The lines in Fig. 1f are the calibrating lines of the sensor for both, anodic and cathodic processes. Any physical or chemical perturbation affecting reaction (1) should promote a shift in the reaction energy to fit the new imposed energetic requirements (Otero's principle).8,27 The current is such a variable determining the reaction rate, so the energy ( ) required to move between the same initial and final oxidation states is a linear function (a sensor) of the applied current (Fig. 1f). A detailed theoretical description of the origin of the current sensing properties has been described previously by Otero et al.8,27 Similar results have been obtained with different bending actuators based on conducting polymers.8,27,38,77–81
) required to move between the same initial and final oxidation states is a linear function (a sensor) of the applied current (Fig. 1f). A detailed theoretical description of the origin of the current sensing properties has been described previously by Otero et al.8,27 Similar results have been obtained with different bending actuators based on conducting polymers.8,27,38,77–81
Different temperatures
Normalized chronopotentiometric responses obtained at different temperatures while keeping the rest of the experimental variables (oxidation and reduction time, applied current, electrolyte concentration and applied current) constant are shown in Fig. 2a for the anodic and Fig. 2b for the cathodic processes. As in the previous case, the consumed charge is constant, during both, oxidation and reduction processes: constant anodic/cathodic currents were applied, at every temperature, for a constant time.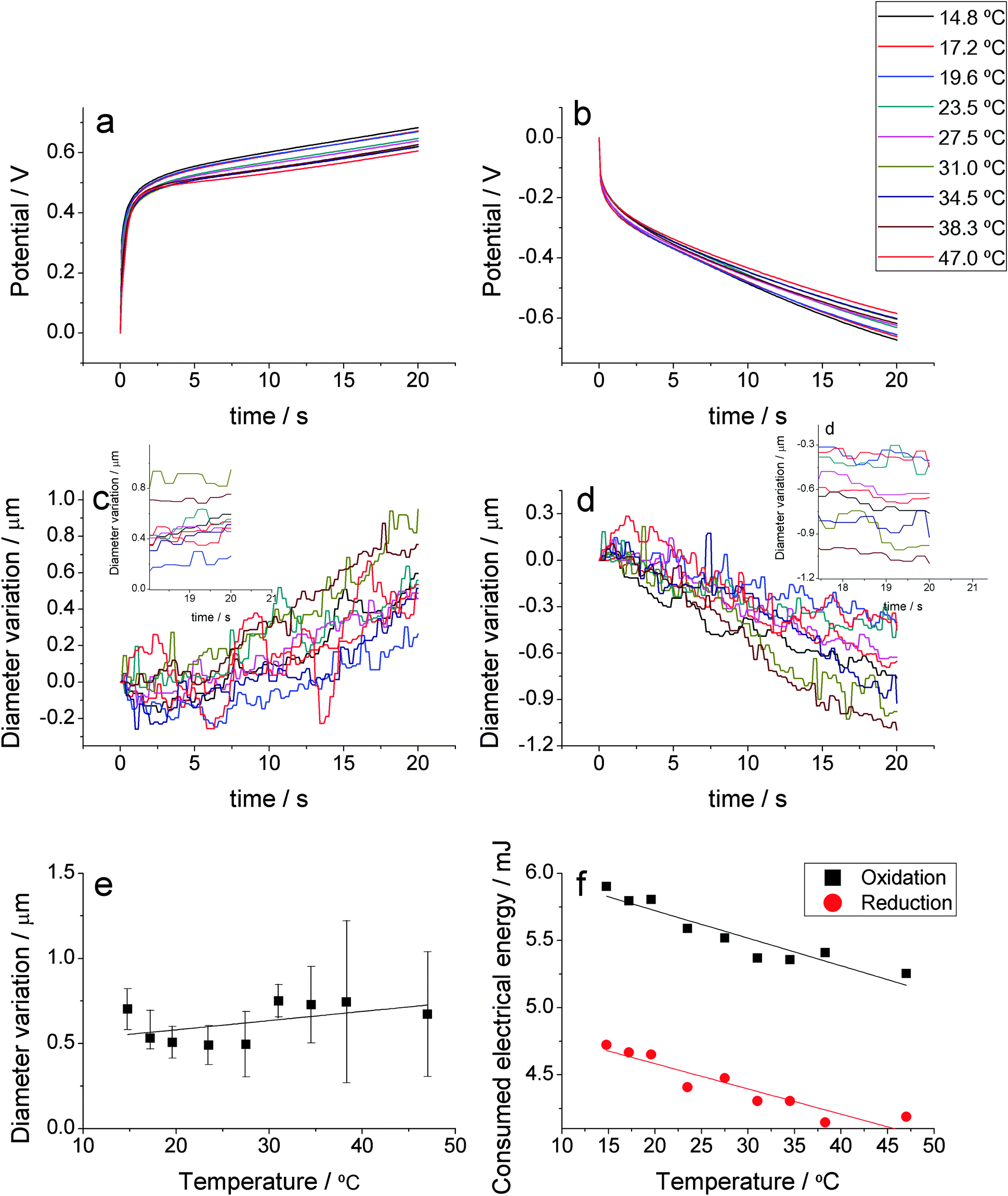 | ||
| Fig. 2 Normalized anodic (a) and cathodic (b) chronopotentiometric responses obtained from a PPy gold coated wire in 0.1 M LiClO4 aqueous solution submitted to consecutive square current waves of ±0.5 mA, at different temperatures, for the consumption of the same oxidation and reduction charges. Parallel evolution variation of the electrode diameter during the anodic (c), or cathodic (d), processes. (e) Average electrode diameter (five different repetitions) after consumption of 10 mC every time. (f) Consumed electrical energy as a function of the working temperature: sensing calibration curves. | ||
The diameter variation of the PPy coated gold wire is shown in Fig. 2c and d for the oxidation and reduction processes, respectively. As above for different currents (Fig. 1), during oxidation the PPy film swells by insertion of balancing counter ions (anions) and solvent, as expected from reaction (1), causing a diameter increase. During reduction, counter ions and solvent are expelled to the electrolyte and PPy shrinks, causing a decrease of the diameter. In fact some small deviations from the linearity are observed during the oxidation process: a slower increase of the diameter at the beginning of the actuation and faster at the end of the current flow.
Here, the apparent noise (dispersion of the measured diameter) is even greater than for the results attained applying different currents. This fact points again to hydrogen evolution at the gold-conducting polymer interface.
The total diameter variation of the PPy coated gold wire as a function of the electrolyte temperature can be seen in Fig. 2e. Despite consumption of the same charge every time, the diameter increment is not constant, as would be expected for a faradaic process. Dispersion of the diameter and noise during measurements points to the simultaneous presence of gas evolution from the electrode during experimental measurements.
By integration of Fig. 2a and b the consumed electrical energy ( ) during the actuator movement is attained. Fig. 2f indicates that the consumed energy is a linear function of the working temperature. This means that, despite the observed dispersion of the final diameter variation, the energy consumed by the actuator senses (is a linear sensor) the working temperature. Fig. 2f presents the sensing calibration curves for both, anodic and cathodic processes. Again the energy of the driving reaction adapts to, and senses, the thermal working conditions: higher available thermal energies require the consumption of lower electrical energies for the actuation. Similar results have been obtained with different bending actuators based on conducting polymers.8,26,38,77–79,82
) during the actuator movement is attained. Fig. 2f indicates that the consumed energy is a linear function of the working temperature. This means that, despite the observed dispersion of the final diameter variation, the energy consumed by the actuator senses (is a linear sensor) the working temperature. Fig. 2f presents the sensing calibration curves for both, anodic and cathodic processes. Again the energy of the driving reaction adapts to, and senses, the thermal working conditions: higher available thermal energies require the consumption of lower electrical energies for the actuation. Similar results have been obtained with different bending actuators based on conducting polymers.8,26,38,77–79,82
Different electrolyte concentrations
Normalized chronopotentiograms obtained at different electrolyte concentrations while keeping the other experimental variables (oxidation and reduction time, electrolyte concentration, applied current and temperature) constant are shown in Fig. 3a and b for the oxidation and reduction processes, respectively. Again, the consumed charge was kept constant: the polymer should actuate between the same initial and final redox states. | ||
| Fig. 3 Normalized anodic (a) and cathodic (b) chronopotentiometric responses obtained from a PPy gold coated wire in different concentrations of LiClO4 aqueous solution submitted to consecutive square current waves of ±0.2 mA, at 21 °C, for the same oxidation and reduction charges. Parallel variation of the electrode diameter during the anodic (c), or the cathodic (d), process. (e) Average electrode diameter (five different repetitions) after consumption of a constant charge of 6 mC every time. (f) Consumed electrical energy as a function of the electrolyte concentration: sensing calibration curves. | ||
The diameter variation of the PPy coated gold wire for the chronopotentiometric experiments is shown in Fig. 3c for the oxidation and in Fig. 3d for the reduction processes. As presented above for the study of the applied current and temperature the diameter increases during oxidation (swelling) and decreases during reduction (shrinking), as expected from reaction-driven (reaction (1)) actuation. Again noise and dispersion (Fig. 3e) of the final diameter variation point to an important influence of some parallel hydrogen generation at the gold-conducting polymer interface. Despite that, the total movement is almost constant in every case, as expected from a reaction-driven actuation.
As in previous studies45,49 there seems to be an indication of a maximum expansion at an optimum electrolyte concentration. However the error in this particular study is too large to draw any solid conclusions.
By integration of Fig. 3a and b the electrical energy consumed during the actuator movement is attained. Fig. 3f indicates that the consumed energy is a semi-logarithmic function of the working concentration. This means that the energy consumed by the actuator to achieve a constant volume change is a sensor of the electrolyte concentration, the sensor calibration curves for both, anodic and cathodic processes are shown in Fig. 3f. Again the energy consumed during a constant (in average) diameter variation adapts (senses) the chemical energy (electrolyte concentration) of the ambient: rising available chemical energies requires the consumption of lower electrical energies during actuation. Similar results have been obtained with different actuators based on conducting polymers and checked at different electrolyte concentrations.27,79
Voltammetric and coulovoltammetric control
In order to clarify the origin of the noise in the experimental responses, the dispersion of the results for each studied variable and the poor linearity between the consumed charge and the diameter variation, as should be expected for a faradaic process, the voltammetric and the coulovoltammetric behaviour of both, uncoated and PPy coated gold electrodes, were studied. Fig. 4a and b show the voltammetric responses in 0.1 M LiClO4 aqueous solution from the uncoated and coated, respectively, electrodes from different cathodic potential limits to the same anodic potential limit of 0.25 V at 20 mV s−1. From bare gold wires only cathodic currents flow through the electrode at cathodic potentials more than 0 V. Voltammetric responses under analogous conditions from the PPy coated gold wires show typical voltammograms from PPy films.6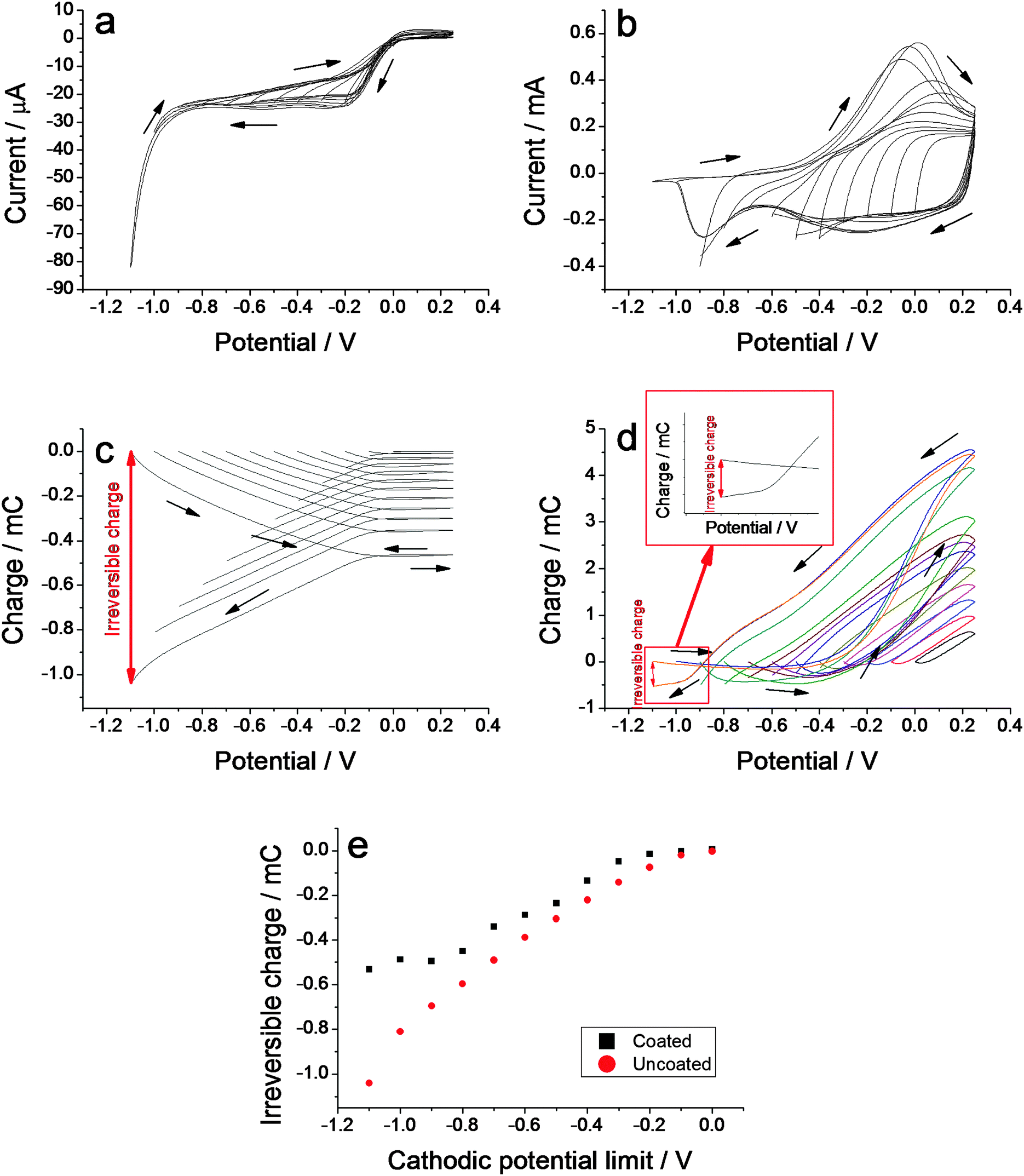 | ||
| Fig. 4 (a) Cyclic voltammograms obtained from an uncoated gold wire from different cathodic potential limits to the same anodic potential limit of 0.3 V at 20 mV s−1 in 0.1 M LiClO4 aqueous solution at room temperature. (b) Similar voltammetric responses using a PPy coated gold wire electrode. (c) Evolution of the consumed charge (coulovoltammograms) during the voltammetric responses of Fig. 3a, or (d), during responses from Fig. 3c. (e) Irreversible charge consumed during voltammetric experiments at different cathodic potential limits for both, uncoated and PPy coated gold wires. | ||
Coulovoltammetric responses, obtained by the integration of the voltammograms, are powerful tools to reveal, detect and quantify reversible and irreversible redox processes occurring in conducting polymer coating metals.48,49 Here the voltammetric responses from Fig. 4a and b were integrated to get the coulovoltammograms shown in Fig. 4c and d for uncoated and coated PPy gold wires, respectively. Responses from the PPy–gold electrode up to cathodic potential limits less cathodic than −0.0 V (versus Ag/AgCl) give closed coulovoltammetric loops indicating that only reversible oxidation/reduction reactions are present in the film: oxidation charges equal reduction charges. For more cathodic potential limits every coulovoltammogram presents two well differentiated parts: a closed loop on the right side whose charge difference from the minimum to the maximum accounts for the reversible redox processes inside the polypyrrole film, and an open part on the left side of this loop. The charge difference from the initial point to the final point of this open part determines the charge consumed by an irreversible reduction reaction (Fig. 4d inset, for the most cathodic potential limit).
For the uncoated electrode only the irreversible open part is present (Fig. 4c), no matter the cathodic potential limit. The irreversible charge consumed during the voltammetric experiments for the different cathodic potential limits from both, coated and uncoated gold electrodes is depicted in Fig. 4e. The polypyrrole film coating the gold electrode protects the metal surface from the direct contact of the electrolyte thereby reducing the irreversible reactions and thus giving lower irreversible charges than those attained for the same process on the uncoated gold electrode. Hydrogen evolution at the gold-conducting polymer interface was proposed as the origin of the irreversible charge: self-supported polypyrrole films do not give any hydrogen evolution up to −2.5 V.48
The formation, growth and migration of hydrogen bubbles from the gold-conducting polymer interface should originate those important noises observed by the experimental diameter variation (Fig. 1c, d, 2c, d, 3c and d). The charge consumed by the hydrogen evolution has low reproducibility under similar experimental conditions. The result is a lower reproducibility of either the charge fraction consumed by polypyrrole reactions or the concomitant diameter variation and a great dispersion of the results for different values of each studied variable (Fig. 1e, 2e, 3e and 4e).
Under those circumstances the result is surprisingly that the consumed energy (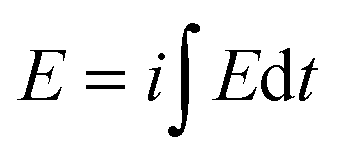 ),which means the area under the chronopotentiometric experimental responses multiplied by the applied current, still is a very robust sensor (Fig. 1f, 2f and 3f) of the studied variables. The energy consumed for the film oxidation is always different than that required for film reduction due to the asymmetry of the structural reaction-driven changes.48
),which means the area under the chronopotentiometric experimental responses multiplied by the applied current, still is a very robust sensor (Fig. 1f, 2f and 3f) of the studied variables. The energy consumed for the film oxidation is always different than that required for film reduction due to the asymmetry of the structural reaction-driven changes.48
The charge consumed by irreversible hydrogen evolution at the gold-conducting polymer interface and the formation of the concomitant hydrogen bubbles and their diffusion across the PPy film can explain the empirical dispersion from the expected constant variation of the diameter. Qualitatively, higher experimental currents lead to a higher cathodic potential (Fig. 1a) and, consequently, more charge consumed by the irreversible hydrogen evolution (according to Fig. 4e). This means that a lower fraction of the charge is consumed by the PPy reaction giving volume and diameter variations as depicted by Fig. 1e: the diameter variation decreases, in average, for rising currents.
Experimental
Lithium perchlorate LiClO4 (Acros Organics) was used as received. Pyrrole (Sigma-Aldrich) was distilled before used under vacuum and stored at −18 °C in the refrigerator in a nitrogen atmosphere. Ultrapure water was obtained from Millipore Milli-Q equipment (18.2 MΩ).A polypyrrole coated gold wire was used as a working electrode. It was fabricated employing a gold wire (having an average diameter of 0.5 mm) from Goodfellow. The gold wire was electrically insulated with an electrically insulating heat-shrink polymer, leaving an uncoated length of 10 mm in the middle of the wire. This wire was used as a working electrode for polypyrrole electrodeposition. A cylindrical gold counter electrode was used for the polymerization of the polypyrrole films, ensuring a uniform electrical field around, and thus a uniform coating of the working electrode. This counter electrode was constructed by first thermally evaporating a layer of chromium (30 Å) onto an acetate sheet. This formed an adhesion layer onto which gold (2000 Å) was then thermally evaporated. This flexible material was cut into an appropriate shape to fit the cylindrical electrochemical cell.
For the electrogeneration, a cylindrical electrochemical cell with a diameter of 2 cm was used. The working electrode was set in the center of the cell, surrounded by the counter electrode. A silver/silver chloride (Ag/AgCl) electrode from BASi was used as the reference electrode located very close to the upper part of the working electrode. The polypyrrole coating was obtained in 8 mL of 0.1 M LiClO4 and 0.1 M pyrrole acetonitrile solution by applying a constant potential of 0.872 V versus Ag/AgCl, during the time required to consume a constant charge of 130 mC. After electrogeneration, the polypyrrole films were reduced at −0.322 V versus Ag/AgCl during 300 s. Employing the same procedure every time, different coatings were attained, having the same thickness (as expected it is a function of the consumed charge) and the same mass. In this work, cylindrical films having a 3.81 ± 0.46 μm of thickness and 0.32 ± 0.03 mg of mass were obtained.
After generation the coated electrode was immersed in water for 20 seconds and then was dried for 3 minutes in air, ensuring always the same state. Then the thickness of the polymer films was determined by measuring the difference between the diameter of the coated and uncoated wire, using a Laser Scan Micrometer (LSM), keeping the position of the electrode constant. Next, the polypyrrole coated electrode was weighed and the polypyrrole mass was obtained by mass difference between the coated and the uncoated electrode using a Sartorious BP210D balance (precision 10−5 g).
The electrochemical characterization and simultaneous determination of the induced diameter variation was performed in a transparent glass cell of 50 mL (Starna) with a rectangular cross-section. A flat and rectangular platinized titanium electrode was used as a counter-electrode. The electrolyte was 0.1 M LiClO4 aqueous solution, which was filtrated through a 0.2 μm filter to remove any potential particulate matter that could interfere with the LSM.
Electrochemical experiments were performed using a potentiostat–galvanostat Autolab PGSTAT-20 attached to a personal computer with GPES software.
Diameter variations were measured using a LSM from Mitutoyo (Mitutoyo LSM-501H) controlled by means of a display unit (Mitutoyo LSM-6100). In order to obtain dynamic measurements, the output signal of the LSM (diameter of the working electrode) was fed to the potentiostat where it was recorded simultaneously with the electrochemical experiments (Fig. 5). For more details on the measurement procedure we refer to Melling et al.72
 | ||
| Fig. 5 Laser scan micrometer allowing diameter variation measurements. | ||
Conclusions
The diameter variation of PPy coated gold wire electrodes was followed using a laser scan micrometer parallel to the film oxidation/reduction under square current waves in LiClO4 aqueous solution.The diameter of the PPy coated wire increases during oxidation and decreases during reduction whatever the studied experimental variable: applied current, temperature and electrolyte concentration. Those facts corroborate that the electrochemically induced volume change of this PPy(ClO4) system is driven by the exchange of anions between the film and the electrolyte.
The attained results point to the faradaic nature of the dimension variations for the different studied variables. The charge controls, through reaction (1), the number of exchanged ions, the film volume and the film diameter variation and the current controls, through the same reaction, the rate of the diameter variation.
The important dispersion of the attained results, related to previous characterization of different actuators based on conducting polymers, is due to the important simultaneous evolution of hydrogen at cathodic potentials more than 0.0 V (Ag/AgCl). The charge consumed by this irreversible reaction during the PPy actuation was detected and quantified from the coulovoltammetric responses. Results from the literature show that so high irreversible charges were not detected, for the same potential ranges, when the PPy film was coated on wires from other metals.
In the experimental results presented in this paper the potential evolves up to −0.8 V during the reduction process by flow of a constant cathodic current. This means that the perturbation of the experimental responses by the formation and growth of hydrogen bubbles at the gold–CP interface between 0.0 and 0.8 V and their migration across the polymer film can be the origin of the diameter variation dispersion related to the expected constant faradaic results.
Despite the parallel presence of the irreversible hydrogen evolution the electrical energy consumed during the film actuation maintains the high sensing ability of those dual electrochemical sensing-actuators: a linear or semi-logarithmic variation of the consumed energy with the studied variable. During actuation (oxidation/reduction) the consumed energy is a robust sensor of: the applied current, the working temperature or the ion concentration in the electrolyte.
Gold electrodes should be avoided for technological applications of conducting polymers driven by current flow in the presence of aqueous solutions, water contamination or moisture: an important fraction of the applied charge will be consumed by hydrogen generation.
Acknowledgements
Authors acknowledge the financial support from the Spanish Government (MCINN) project MAT2011-24973, the Seneca Foundation project 19253/PI/14 and the European Science Foundation COST Action MP1003 European Scientific Network for Artificial Muscles (ESNAM), COST-STSM-MP1003-11575 and -11581, Swedish Research Council (VR-2014-3079), and Linköping University. J. G. Martinez acknowledges the Spanish Education Ministry for a FPU grant (AP2010-3460). E. W. H. Jager wishes to express his gratitude to Prof. Anthony Turner (LiU, IFM) for his support.Notes and references
- T. F. Otero, E. Angulo, J. Rodríguez and C. Santamaría, J. Electroanal. Chem., 1992, 341, 369–375 CrossRef CAS
.
- Q. Pei and O. Inganas, Adv. Mater., 1992, 4, 277–278 CrossRef CAS
- G. W. Wang, Y. N. Lu, L. P. Wang, H. J. Wang and J. Y. Wang, J. Nanosci. Nanotechnol., 2014, 14, 596–612 CrossRef CAS PubMed
- T. K. Das and S. Prusty, Polym. – Plast. Technol. Eng., 2012, 51, 1487–1500 CrossRef CAS
- Y.-Z. Long, M.-M. Li, C. Gu, M. Wan, J.-L. Duvail, Z. Liu and Z. Fan, Prog. Polym. Sci., 2011, 36, 1415–1442 CrossRef CAS
- T. F. Otero, J. G. Martinez and J. Arias-Pardilla, Electrochim. Acta, 2012, 84, 112–128 CrossRef CAS
- A. A. Entezami and B. Massoumi, Iran. Polym. J., 2006, 15, 13–30 CAS
- T. F. Otero and J. G. Martinez, Prog. Polym. Sci., 2015, 44, 62–78 CrossRef CAS
- M. T. Cortes and J. C. Moreno, Eur. Polym. J., 2003, 041 Search PubMed
- T. Mirfakhrai, J. D. W. Madden and R. H. Baughman, Mater. Today, 2007, 10, 30–38 CrossRef CAS
- E. Smela, Adv. Mater., 2003, 15, 481–494 CrossRef CAS
- N. K. Guimard, N. Gomez and C. E. Schmidt, Prog. Polym. Sci., 2007, 32, 876–921 CrossRef CAS
- E. W. H. Jager, E. Smela and O. Inganas, Science, 2000, 290, 1540–1545 CrossRef CAS PubMed
- T. F. Otero and J. M. Sansiñena, Adv. Mater., 1998, 10, 491–494 CrossRef CAS PubMed
- G. M. Spinks, L. Liu, G. G. Wallace and D. Z. Zhou, Adv. Funct. Mater., 2002, 12, 437–440 CrossRef CAS
- L. Bay, K. West, P. Sommer-Larsen, S. Skaarup and M. Benslimane, Adv. Mater., 2003, 15, 310–313 CrossRef CAS
- T. F. Otero and J. G. Martinez, J. Mater. Chem. B, 2013, 1, 26–38 RSC
- E. M. Andrade, F. V. Molina, M. I. Florit and D. Posadas, Electrochem. Solid-State Lett., 2000, 3, 504–507 CrossRef CAS
- X. Chen and O. Inganas, Synth. Met., 1995, 74, 159–164 CrossRef CAS
- T. Okamoto, Y. Kato, K. Tada and M. Onoda, Thin Solid Films, 2001, 393, 383–387 CrossRef CAS
- P. R. Singh, S. Mahajan, S. Raiwadec and A. Q. Contractor, J. Electroanal. Chem., 2009, 625, 16–26 CrossRef CAS
- E. Smela and N. Gadegaard, Adv. Mater., 1999, 11, 953–957 CrossRef CAS
- M. F. Suarez and R. G. Compton, J. Electroanal. Chem., 1999, 462, 211–221 CrossRef CAS
- J.-G. Barbara and F. Clarac, Brain Res., 2011, 1409, 3–22 CrossRef CAS PubMed
- G. G. Matthews, Cellular Physiology of Nerve and Muscle, Wiley-Blackwell, Hoboken, NJ, USA, 2009 Search PubMed.
- T. F. Otero, J. J. Sanchez and J. G. Martinez, J. Phys. Chem. B, 2012, 116, 5279–5290 CrossRef CAS PubMed
- J. G. Martinez and T. F. Otero, J. Phys. Chem. B, 2012, 116, 9223–9230 CrossRef CAS PubMed
- J. G. Martinez and T. F. Otero, Sens. Actuators, B, 2014, 195, 365–372 CrossRef CAS
- K. Potje-Kamloth, Crit. Rev. Anal. Chem., 2002, 32, 121–140 CrossRef CAS
- D. Wei, M. J. A. Bailey, P. Andrew and T. Ryhaenen, Lab Chip, 2009, 9, 2123–2131 RSC
- L. Rover, G. D. Neto and L. T. Kubota, Quim. Nova, 1997, 20, 519–527 CrossRef
- C. Dhand, M. Das, M. Datta and B. D. Malhotra, Biosens. Bioelectron., 2011, 26, 2811–2821 CrossRef CAS PubMed
- D. S. Correa, E. S. Medeiros, J. E. Oliveira, L. G. Paterno and L. H. C. Mattoso, J. Nanosci. Nanotechnol., 2014, 14, 6509–6527 CrossRef CAS PubMed
- R. D. McCullough, Adv. Mater., 1998, 10, 93–96 CrossRef CAS
- M. Gerard, A. Chaubey and B. D. Malhotra, Biosens. Bioelectron., 2002, 17, 345–359 CrossRef CAS PubMed
- J. Janata and M. Josowicz, Nat. Mater., 2003, 2, 19–24 CrossRef CAS PubMed
- B. Adhikari and S. Majumdar, Prog. Polym. Sci., 2004, 29, 699–766 CrossRef CAS
- L. V. Conzuelo, J. Arias-Pardilla, J. V. Cauich-Rodríguez, M. A. Smit and T. F. Otero, Sensors, 2010, 10, 2638–2674 CrossRef CAS PubMed
- F. Vidal, C. Plesse, G. Palaprat, A. Kheddar, J. Citerin, D. Teyssie and C. Chevrot, Synth. Met., 2006, 156, 1299–1304 CrossRef CAS
- M. Onoda, Y. Kato, H. Shonaka and K. Tada, Electr. Eng. Jpn., 2004, 149, 7–13 CrossRef
- Y. Kato, K. Tada and M. Onoda, Jpn. J. Appl. Phys., Part 1, 2003, 42, 1458–1461 CrossRef CAS
- C. Laslau, D. E. Williams, B. E. Wright and J. Travas-Sejdic, J. Am. Chem. Soc., 2011, 133, 5748–5751 CrossRef CAS PubMed
- L. Bay, T. Jacobsen, S. Skaarup and K. West, J. Phys. Chem. B, 2001, 105, 8492–8497 CrossRef CAS
- C. Jo, H. E. Naguib and R. H. Kwon, Smart Mater. Struct., 2011, 20, 045006 CrossRef
-
AWAPATENT AB, WO 2009/038501 A1, 2009, 74
- V. Venugopal, H. Zhang, R. Northcutt and V. B. Sundaresan, Sens. Actuators, B, 2014, 201, 293–299 CrossRef CAS
- J. Wu, D. Zhou, C. O. Too and G. G. Wallace, Synth. Met., 2005, 155, 698–701 CrossRef CAS
- T. F. Otero, M. Alfaro, V. Martinez, M. A. Perez and J. G. Martinez, Adv. Funct. Mater., 2013, 23, 3929–3940 CrossRef CAS
- J. G. Martinez, T. F. Otero and E. W. H. Jager, Langmuir, 2014, 30, 3894–3904 CrossRef CAS PubMed
- T. F. Otero, Polym. Rev., 2013, 53, 311–351 CrossRef CAS
- T. F. Otero and J. M. Sansinena, J. Electroanal. Chem., 1996, 412, 109–116 CrossRef
- T. V. Vernitskaya and O. N. Efimov, Russ. Chem. Rev., 1997, 66, 443 CrossRef
- T. Zama, S. Hara, W. Takashima and K. Kaneto, Bull. Chem. Soc. Jpn., 2005, 78, 506–511 CrossRef CAS
- S. Hara, T. Zama, W. Takashima and K. Kaneto, Polym. J., 2004, 36, 151–161 CrossRef CAS
- L. Viau, J. Y. Hihn, S. Lakard, V. Moutarlier, V. Flaud and B. Lakard, Electrochim. Acta, 2014, 137, 298–310 CrossRef CAS
- M. Pyo, J. Reynolds, L. Warren and H. Marcy, Synth. Met., 1994, 68, 71–77 CrossRef CAS
- T. F. Otero and I. Cantero, J. Electrochem. Soc., 1999, 146, 4118–4123 CrossRef CAS
- T. Otero and J. Rodriguez, Electrochim. Acta, 1994, 39, 245–253 CrossRef CAS
- G. Alici, A. Punning and H. R. Shea, Sens. Actuators, B, 2011, 157, 72–84 CrossRef CAS
- W. Zheng, G. Alici, P. R. Clingan, B. J. Munro, G. M. Spinks, J. R. Steele and G. G. Wallace, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 57–63 CrossRef CAS
- G. Y. Han and G. Q. Shi, J. Electroanal. Chem., 2004, 569, 169–174 CrossRef CAS
- P. Du, X. Lin and X. Zhang, Sens. Actuators, A, 2010, 163, 240–246 CrossRef CAS
- B. Shapiro and E. Smela, J. Intell. Mater. Syst. Struct., 2007, 18, 181–186 CrossRef CAS
- M. Christophersen, B. Shapiro and E. Smela, Sens. Actuators, B, 2006, 115, 596–609 CrossRef CAS
- M. Pyo, C. C. Bohn, E. Smela, J. R. Reynolds and A. B. Brennan, Chem. Mater., 2003, 15, 916–922 CrossRef CAS
- X. Y. Cui and D. C. Martin, Sens. Actuators, A, 2003, 103, 384–394 CrossRef CAS
- J. Tangorra, P. Anquetil, T. Fofonoff, A. Chen, M. Del Zio and I. Hunter, Bioinspiration Biomimetics, 2007, 2, S6–S17 CrossRef PubMed
- P. A. Kilmartin, J. Travas-Sejdic, R. Temmer, T. Tamm, A. Aabloo and R. Kiefer, Int. J. Nanotechnol., 2014, 11, 477–485 CrossRef CAS
- S. D. Deshpande, J. Kim and S. R. Yun, Smart Mater. Struct., 2005, 14, 876–880 CrossRef CAS
- E. Smela, M. Kallenbach and J. Holdenried, J. Microelectromech. Syst., 1999, 8, 373–383 CrossRef CAS
- M. J. M. Jafeen, M. A. Careem and S. Skaarup, Ionics, 2014, 20, 535–544 CrossRef CAS
- D. Melling, S. Wilson and E. W. H. Jager, Smart Mater. Struct., 2013, 22, 104021 CrossRef
- T. F. Otero and J. G. Martinez, Chem. Mater., 2012, 24, 4093–4099 CrossRef CAS
- T. F. Otero, J. G. Martinez and B. Zaifoglu, Smart Mater. Struct., 2013, 22, 104019 CrossRef
- L. Valero, T. F. Otero and J. G. Martinez, ChemPhysChem, 2014, 15, 293–301 CrossRef CAS PubMed
- M. J. M. Jafeen, M. A. Careem and S. Skaarup, Ionics, 2010, 16, 1–6 CrossRef CAS
- J. Arias-Pardilla, C. Plesse, A. Khaldi, F. Vidal, C. Chevrot and T. F. Otero, J. Electroanal. Chem., 2011, 652, 37–43 CrossRef CAS
- F. García-Córdova, L. Valero, Y. A. Ismail and T. F. Otero, J. Mater. Chem., 2011, 21, 17265–17272 RSC
- Y. A. Ismail, J. G. Martínez, A. S. Al Harrasi, S. J. Kim and T. F. Otero, Sens. Actuators B Chem., 2011, 160, 1180–1190 CrossRef CAS
- Y. A. Ismail, J. G. Martínez and T. F. Otero, Electrochim. Acta, 2014, 123, 501–510 CrossRef CAS
- Y. A. Ismail, J. G. Martinez and T. F. Otero, J. Electroanal. Chem., 2014, 719, 47–53 CrossRef CAS
- T. F. Otero and M. T. Cortes, Sens. Actuators, B, 2003, 96, 152–156 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp05841d |
| This journal is © the Owner Societies 2016 |