
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShielding the chemical reactivity using graphene layers for controlling the surface properties of carbon materials†
A. E.
Sedykh
,
E. G.
Gordeev
,
E. O.
Pentsak
and
V. P.
Ananikov
*
Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky Prospect 47, Moscow, 119991, Russia. E-mail: val@ioc.ac.ru
First published on 22nd January 2016
Abstract
Graphene can efficiently shield chemical interactions and gradually decrease the binding to reactive defect areas. In the present study, we have used the observed graphene shielding effect to control the reactivity patterns on the carbon surface. The experimental findings show that a surface coating with a tiny carbon layer of 1–2 nm thickness is sufficient to shield the defect-mediated reactivity and create a surface with uniform binding ability. The shielding effect was directly observed using a combination of microscopy techniques and evaluated with computational modeling. The theoretical calculations indicate that a few graphene layers can drastically reduce the binding energy of the metal centers to the surface defects by 40–50 kcal mol−1. The construction of large carbon areas with controlled surface reactivity is extremely difficult, which is a key limitation in many practical applications. Indeed, the developed approach provides a flexible and simple tool to change the reactivity patterns on large surface areas within a few minutes.
Introduction
Graphene systems represent two-dimensional materials consisting of a small number (1 to <10) of stacked carbon layers of extended lateral dimension.1 In the past decade, biomedical applications of graphene systems have emerged and are rapidly developing.2–4 The surface structure, chemical reactivity and properties of graphene are of paramount importance to the development of biomedical applications.3 It has been shown that the defects of graphene sheets, particularly chemically reactive defects, can bind biological molecules and cells, catalyze undesirable biochemical reactions and affect the biological activity of the molecules.4 Therefore, the development of methods to detect defects and regulate their activities is of key importance to the field. Moreover, the state-of-the-art level recalls not only for the characterization of the defects, but also for a possibility to shield the internal environment of a living organism from notably reactive graphene defects while maintaining the mechanical and other important properties of the carbon material. Thus, controlling the surface reactivity of carbon materials is a highly demanding task for biomedical applications of 2D carbon materials.Defects on a graphene surface are primary interaction centers with increased chemical reactivity. Graphene sheets may contain different types of defects: topological defects, single vacancies, line defects, Stone–Wales defects, etc.5 Carbon defects have been extensively investigated in the framework of developing efficient catalytic systems,6 sensors for gas molecules and biologically active compounds,7 nanoelectronic devices,8 and energy storage systems.9 Several outstanding applications of graphene-based materials in chemical technologies originated from the ability of graphene to undergo chemical and physical adsorption of atoms and molecules on the defect sites. The ability of graphene to establish covalent and van der Waals' interactions makes graphene an efficient adsorbent of aromatic compounds10 with possible application in purification systems to remove toxic impurities. Not surprisingly, the adsorption of gas molecules,11 organic compounds10,12 and metal atoms13 on graphene and related carbon materials14 was the subject of many experimental and theoretical studies.
Recently, we reported an efficient approach for the imaging of reactive centers on the 2D carbon surface by adsorbing palladium nanoparticles: metal clusters that are formed from Pd2dba3 in a chloroform solution selectively attached to the defects of graphene layers and served as clearly visible markers.15 This methodology opened an excellent opportunity to make a molecular-level insight into the reactivity of the carbon surface.15
In the present study, we revealed the possibility of shielding the chemical reactivity of defect centers. The surface defects can be efficiently shielded by applying a coating with a thin layer of carbon. The reported experimental and theoretical studies in this article have shown that pristine graphene has strong shielding properties, and even a single carbon layer can noticeably mask the chemical reactivity.
Results and discussion
Using scanning electron microscopy, the boundaries of various types of surface defects can be easily observed on a graphite sample. Although the chemical reactivity of the defects cannot be directly characterized, it can be visualized using a notably sensitive procedure based on the attachment of palladium nanoparticles to the reactive sites.15 To map the chemical reactivity, we used Pd2dba3 as a contrast agent in the imaging procedure. Under mild conditions, palladium nanoparticles attach to the defect sites and serve as markers for the reactive centers of the carbon surface (see the Experimental part for details).A typical carbon material with visible domains on the surface and palladium nanoparticles attached to them is shown in Fig. 1A. For a clear representation, we visualized the defect boundaries, which were observed using microscopy (green lines), and the attached palladium nanoparticles (red dots); the resulting map is shown in Fig. 1B. As observed in the nanoscale map, nanoparticles are preferentially located along the green lines. As expected, the surface defects are the areas of increased chemical reactivity, where the palladium nanoparticles were preferably bound.
 | ||
Fig. 1 Microscopy characterization of Pd/C materials at ×100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 magnification with the corresponding nanometer scale bars in each image: (A) attachment of Pd nanoparticles to the visible defects on the carbon surface; (B) map of the defect boundaries (green lines) and attached Pd nanoparticles (red dots); (C) attachment of Pd nanoparticles to the carbon surface with “shielded” reactivity of the defect areas; and (D) the corresponding map of the defect boundaries (green lines) and attached Pd nanoparticles (red dots). 000 magnification with the corresponding nanometer scale bars in each image: (A) attachment of Pd nanoparticles to the visible defects on the carbon surface; (B) map of the defect boundaries (green lines) and attached Pd nanoparticles (red dots); (C) attachment of Pd nanoparticles to the carbon surface with “shielded” reactivity of the defect areas; and (D) the corresponding map of the defect boundaries (green lines) and attached Pd nanoparticles (red dots). | ||
Amazingly, we found another type of carbon material with clearly visible surface defects and domains, where the chemical reactivity of the defect centers was somehow shielded (Fig. 1C). A representative visualization of the defect boundaries and attached nanoparticles is shown in Fig. 1D. Although the domain boundaries are clearly present (green lines), the metal nanoparticles (red dots) are now uniformly distributed on the surface instead of locating along the defects. We became interested in this significantly visible difference and studied these unusual areas in more details.
For detailed characterization, high-resolution scanning electron microscopy (SEM) and scanning transmission electron microscopy (STEM) images were recorded (Fig. 2). At low magnification, the SEM image showed the micrometer-sized graphite particle with a uniform distribution of palladium nanoparticles on the entire surface (Fig. 2A). In the bright-field scanning transmission electron microscopy (BF-STEM) image, the carbon layer boundaries are observed – the graphite particle is thicker where the image is darker (Fig. 2B). The annular dark-field scanning transmission electron microscopy (ADF-STEM) image has a better S/N ratio and provides more information about the particles on both sides of the surface (Fig. 2C).
 | ||
| Fig. 2 Microscopy characterization of the Pd/C material at ×40000 (A–C) and ×500000 (D–G) magnifications with corresponding micrometer or nanometer scale bars in each image: (A) and (D) – SEM images; (B and E) – BF-STEM images; (C and F) – ADF-STEM images; and (G) – three EDX maps. | ||
At higher magnifications, the palladium nanoparticles are observed with excellent resolution. Surprisingly, in this image, we found that the graphite surface was covered with a thin net-like material (Fig. 2D). The BF-STEM image reveals a uniform distribution of palladium nanoparticles (Fig. 2E).
In the ADF-STEM image, some additional nanoparticles are clearly visible (Fig. 2F); they are on the backside of this graphite particle and also uniformly distributed. The EDX analysis confirmed that this area contained only carbon and palladium (Fig. 2G). According to the EDX data, the net-like material on the graphite surface is carbon. It has become clear that this carbon on the graphite surface prevents the organization of palladium nanoparticles along the defect boundaries. A notably thin carbon coating appears to shield the chemical reactive sites.
We have also studied the next phase of the nanoparticles attachment after the defect boundaries were fully covered. For this purpose, the concentration of the palladium complex in solution was varied during nanoparticles deposition on the carbon surface. At higher concentrations of the metal precursor in solution (after attachment to the boundaries), it was found that palladium nanoparticles uniformly covered the graphite surface (Fig. S16, ESI†).
To independently confirm that a thin carbon layer could shield the active sites on a graphite surface, we performed a theoretical computational modeling with quantum-chemical calculations. As a model of a graphene sheet, the molecules of coronene (C24H12) and circum-coronene (C54H18) were selected and studied at DFT and semi-empirical levels.
To reliably represent the stacked graphene system, the dispersion interactions were described according to Grimme's D3 corrections.16 Previously it has been shown that the DFT and semi-empirical levels used in the present study provide a reliable description of the graphene materials, which is consistent with experimental findings.17
First, we calculated the binding of the palladium marker to the defect site of the carbon material (Fig. 3A). Two types of carbon defects were studied: (1) missing carbon atom (i.e., one carbon atom was removed with 4+ charge on the remaining part) and (2) organic cation Ph3C6(3+), which was obtained by removing three hydride anions from 1,3,5-triphenylbenzene.
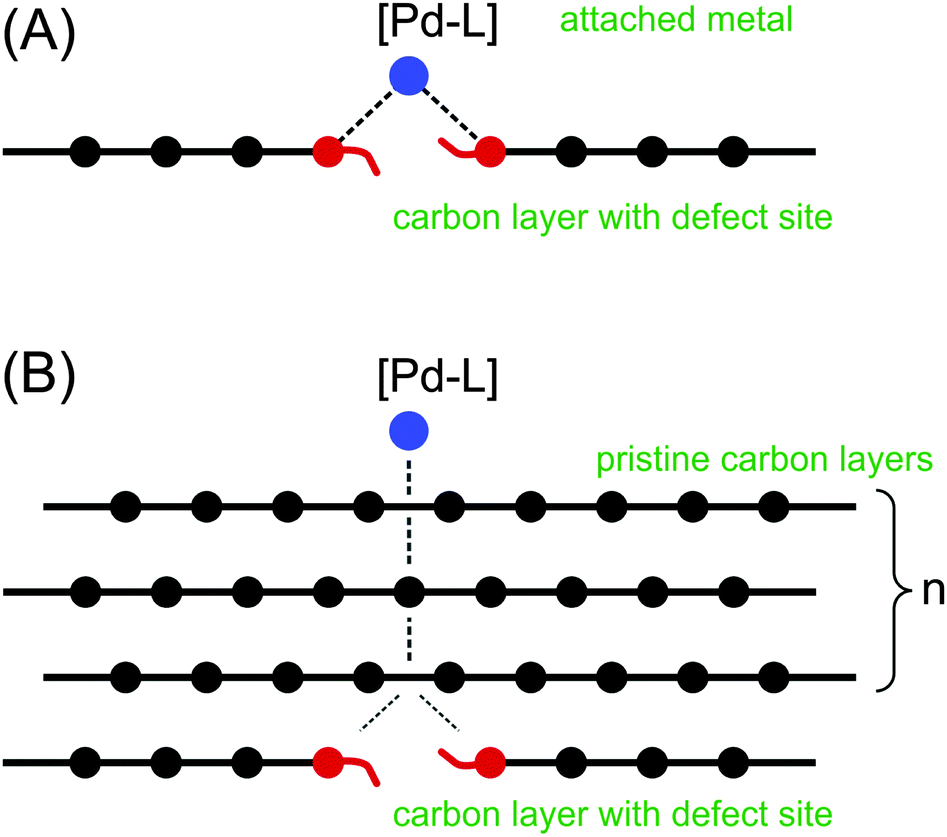 | ||
| Fig. 3 Modelling of shielding by graphene layers: (A) – binding of the [PdL] to the defect site; (B) – shielding of the [PdL]–defect by pristine graphene layers. | ||
A Pd–L unit was used as a marker for the metal binding. With L = CO, useful information is provided by the Pd–LC distance and LC charge. It has been shown that the PdCO model is notably sensitive to the changes in electron density at the palladium center.18 Next, we estimated the shielding of the reactive center by the graphene layers (Fig. 3B). In both cases (Fig. 3A and B), the binding energy was calculated according to the equation: ΔE = E(complex) − E(PdCO) − E(carbon material).
The energy of the PdCO coordination with a pure “unshielded” defect was approximately −100.0 kcal mol−1 (Table 1) at the semi-empirical and DFT levels. The value is consistent with the literature data.15,19 The direct binding of PdCO with the defect corresponds to the smallest Pd–C(sheet) interatomic distance. All atoms in the PdCO marker are positively charged according to Loewdin scheme (RI DFT), and the positive charges of the Pd, C and O atoms are the largest at n = 0 (Table 1). In accordance with the PM6-D3H4 Mulliken approach the oxygen atom is negatively charged. Both Loewdin and Mulliken approaches corresponded to each other with respect to the decrease in the total positive charge of the PdCO marker upon increasing the number of layers (see Table 1, Table S1, ESI† and Fig. 4).
| Number of carbon layers, n | 0 | 1 | 2 | 3 | |
|---|---|---|---|---|---|
| a For the C24 system, the RI PBE def2-SVP ZORA level was used; for the C54 system, the PM6-D3H4 level was used. b The calculated values of Loewdin charges (for Mulliken charges see Table S1, ESI, and Fig. 4). For the coronene systems, geometry optimization was performed at the RI PBE level; for the circum-coronene systems, geometry optimization was performed at the PM6-D3H4 level. c The sum of charges of the Pd, C and O atoms. d The shortest Pd–C(sheet) interatomic distances. | |||||
| ΔE, kcal mol−1a |
C54 | −107.4 | −50.7 | −46.0 | −44.0 |
| C24 | −98.9 | −58.7 | −50.1 | −60.6 | |
C(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) chargeb O) chargeb |
C24 | +0.105 | +0.093 | +0.079 | +0.072 |
| O charge | C24 | +0.252 | +0.177 | +0.153 | +0.138 |
| Pd charge | C24 | +0.307 | +0.225 | +0.165 | +0.138 |
| Total PdCO chargec | C24 | +0.664 | +0.495 | +0.397 | +0.348 |
| Pd–C(CO) distance, Å |
C54 | 2.004 | 1.956 | 1.959 | 1.959 |
| C24 | 2.097 | 1.957 | 1.957 | 1.957 | |
| Pd–C(sheet) distance, Åd | C54 | 2.081 | 2.106 | 2.118 | 2.113 |
| C24 | 1.971 | 2.127 | 2.127 | 2.127 | |
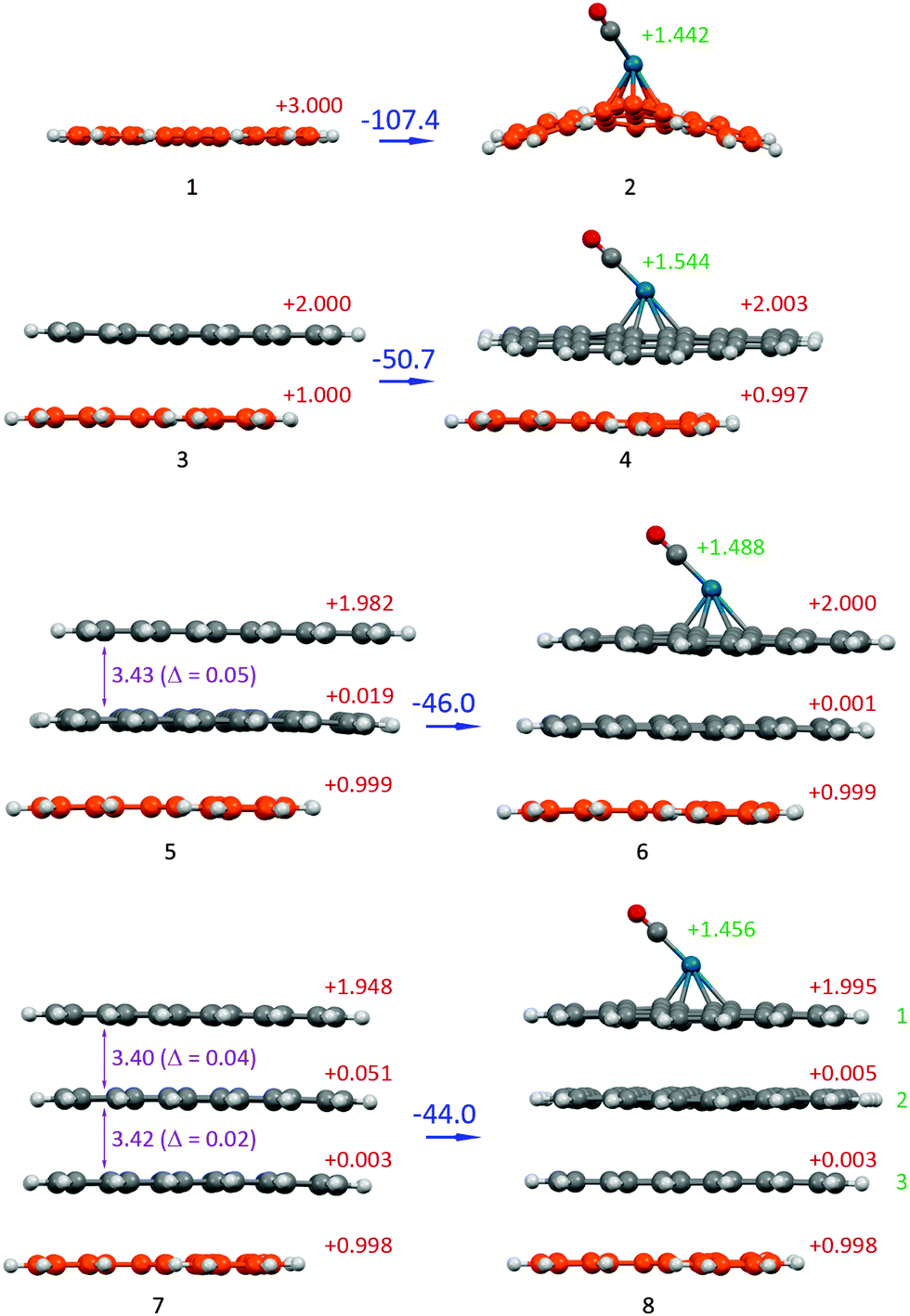 | ||
| Fig. 4 Optimized molecular structures of the Pd/C model system, which reflects the shielding effect of graphene layers: n = 0 (1 and 2), n = 1 (3 and 4), n = 2 (5 and 6) and n = 3 (7 and 8) at the PM6-D3H4 level (see Fig. 3 for definition of n; the carbon defect layers are highlighted in orange; the total charge of the system is 3+). The binding energy (ΔE, kcal mol−1) is shown in each case (blue values). The total Mulliken charges of each layer are denoted in red. The total Mulliken charges of the PdCO group are denoted in green. The interlayer distances (Å) are denoted in violet. | ||
 | ||
| Fig. 5 (A) Pd nanoparticles localized on the defect areas (if uncoated carbon surface is used); (B) coating of the graphite surface by a thin carbon layer results in a uniform distribution of Pd nanoparticles. | ||
It is interesting to note that an increase in the number of sheets in a stack leads to a gradual reduction in the positive charge of the defect-containing layer and an increase of positive charges on the central sheets of the stack (see Fig. S1, ESI†). Simultaneously, the charge of the surface sheet that was connected with the PdCO marker decreased. This effect was more pronounced for the Loewdin charge distribution (Fig. S1, ESI†). However, despite the charge transfer to the central sheets, the major part of the entire positive charge is localized on the outer sheets. This phenomenon may be used for designing nanoelectronic devices with multilayer graphene parts.
The insertion of the model graphene sheet between the defect and the PdCO molecule resulted in a considerably reduced complexation energy value (see values for n = 1 in Table 1). For example, in the coronene complexes at the DFT levels, the insertion of the first additional coronene sheet decreased the complexation energy from −98.9 to −58.7 kcal mol−1, i.e., the “shielding effect” of one coronene sheet was 40.2 kcal mol−1 (and 56.7 kcal mol−1 at the PM6-D3H4 level).
An increase in the number of sheets in the graphene stack leads to a gradual reduction in binding energy (see ΔE in Table 1 for the C54 system). In addition, a larger number of shielding layers (n = 1, 2, 3; Fig. 3B) correspond to smaller positive charges of the C, O and Pd atoms and the PdCO marker and a larger Pd–C (sheet) interatomic distance (Table 1 and Fig. 4). For the C54 system the sum of the positive charges of the PdCO marker is decreased from n = 1 (1.544) to n = 3 (1.456). Such a charge variation may account for a shielding effect.
The Pd–C(CO) distance is sensitive to the insertion of the first graphene sheet and noticeably decreased after such insertion (this distance shortened to 0.14 Å at the DFT level for the C24 system). Further increases in the number of sheets (n = 2 and n = 3) did not change the Pd–C(CO) distance at the DFT and semi-empirical levels.
Changing the interlayer distance is also sensitive to the size of the system. For system 5 (n = 2), the interlayer distance was 3.43 Å and its deviation from the equilibrium interlayer distance of “unperturbed” (without cation effect) bilayer system was Δ = 0.05 Å (Fig. 4). Adding a layer in system 7 resulted in a decrease of this change (Δ = 0.04 and 0.02). Indeed, the observed shielding effect led to a dissipation of the influence of a defect site.
For both model systems, the most significant changes in binding energy, atomic charges and interatomic distances occurred after the insertion of the first shielding graphene layer. The insertion of the second and third layers resulted in a similar trend in the changes of the studied parameters but with a less pronounced difference.
An example of the molecular arrangement of the studied system is shown in Fig. 4. The direct binding of the PdCO marker to the defect layer caused the cation structure to distort and transform from the planar form to the convex form. The insertion of only one shielding graphene sheet between the cation and the PdCO particle eliminated the distortion, and the arrangement became planar.
The theoretical calculations show the excellent shielding properties of graphene layers. Indeed, even a single layer can efficiently shield the chemical reactivity of the defect areas, which was observed in the experiment (Fig. 1).
These findings open an outstanding opportunity to control the surface reactivity of carbon materials and change the arrangement of metal nanoparticles on the carbon surface. To verify this possibility, we performed a dedicated experiment. A carbon material with reactive centers on the surface was coated with a carbon layer, which consisted of ∼95% sp2 sites and formed from disordered graphitic islands of 15–20 Å20 with subsequent attachment of palladium nanoparticles (Fig. 5). If our assumption is correct, the nanoparticle arrangement along the defect boundaries should disappear after a notably thin carbon layer is deposited.
The assumption was completely confirmed in the experiment. We observed a uniform distribution of palladium nanoparticles on the surface (Fig. 6B and D), which indicates the shielding of the originally presented defect boundaries (Fig. 5B). For comparison, the attachment of palladium nanoparticles to an unmodified sample led to an alignment along the defect boundaries (Fig. 6A and C), as we described for this approach (Fig. 5A).
 | ||
| Fig. 6 FE-SEM images of Pd/C. (A and C) – Palladium nanoparticles on the graphite; (B and D) – palladium nanoparticles on the graphite that was coated with 1.4 nm of carbon. Microscopy characterization of the precipitates at ×100000 (A and B) and ×200000 (C and D) magnifications with corresponding nanometer scale bars in each image. | ||
Thus, the experimental findings have completely confirmed the idea. It should be emphasized that even a notably thin layer of carbon was sufficient to shield the chemical reactivity of the defects. Indeed, the theoretical calculations have shown that a single graphene layer can significantly reduce the susceptibility of nanoparticles to the defect centers. After coating with 1.4 nm of carbon, the defect boundaries remain clearly visible, whereas the preferential palladium binding to the defect areas disappeared (Fig. 6B and D). The images with higher magnification (Fig. 6C and D) show that the nanoparticles on the modified graphite are smaller (3.8 ± 0.7 nm, versus 5.8 ± 0.9 nm on the clear graphite), which also confirms the change in the nature of the carbon support.
To confirm that coating only shielded the defects and did not create new active sites, BET (Brunauer–Emmett–Teller) specific surface area analysis was carried out. In these measurements, nitrogen gas was adsorbed on the material surface, particularly on the defects. The surface area of the unmodified graphite was measured as 4.127 m2 g−1. After coating the graphite with a carbon layer of 2.8 nm thickness, the measured BET response significantly decreased to 1.047 m2 g−1. Most likely, a thin coating on a nearly planar 2D surface cannot dramatically change the physical area of the sample; therefore, a change in the BET response indicates a decrease in the number of chemically reactive defect sites after coating.
Conclusions
To summarize, a fundamental difference in the binding of palladium nanoparticles to surface defects was established using electron microscopy (FE-SEM, BF-STEM, and ADF-STEM), theoretical calculations (DFT and PM6-D3H4) and BET surface area analysis. As expected, the specific binding of palladium nanoparticles to the defect areas was observed on regular carbon surfaces. However, on the carbon surface that was covered with a thin carbon layer (∼1–2 nm), a uniform distribution of the deposited palladium nanoparticles was found. The theoretical calculations have shown that even a single graphene layer significantly reduces the binding energy of metal nanoparticles to the chemically reactive defect sites (by ∼40 kcal mol−1).Thus, coating by a single pristine graphene layer or a few layers provides an excellent opportunity to shield the chemical reactivity. A notably sensitive test based on the attachment of metal nanoparticles has clearly shown the loss of reactivity of the visible defect areas on the carbon surface. The deposition of thin carbon layers is a simple and technically feasible procedure, which creates a flexible tool to control the reactivity of carbon materials and their interaction with other molecules. The proposed experimental technique is an efficient method to regulate the activity of graphite defects and design new nanostructured hybrid carbon materials. Using this carbon coating approach, one can switch the chemical-reactivity pattern of a carbon surface from defect-oriented specific binding to a uniform binding across the carbon surface.
Experimental
General methods and materials
Pd2dba3·CHCl3 was synthesized according to the reported procedure.21 Unless otherwise noted, the experiments were performed in screw-cap glass tubes, which were equipped with a magnetic stir bar. Graphite powder of high purity (90% w/w of particles with a size of less than 90 μm) was purchased from a commercial source.The electron microscopy images were recorded using a Hitachi SU8000 field-emission scanning electron microscope (FE-SEM). For the FE-SEM measurements, the samples were fixed with a conductive silver paint on a 25 mm aluminum specimen stub. The images were acquired in a secondary electron mode at an accelerating voltage of 30 kV and a working distance of 8–10 mm.
The high-resolution electron microscopy images were recorded using a Hitachi HD-2700 scanning transmission electron microscope (STEM), which was equipped with a CEOS GmbH spherical aberration (Cs) corrector. All the images were recorded at an accelerating voltage of 200 kV. The analysis time for the EDX-elemental mapping was 30 minutes.
The BET measurements were performed on the specific surface analyzer Catacon Sorbtometer-M and obtained from multipoint BET calculations.
Coating of graphite with carbon
Graphite powder (average 60 mg) was placed in a Petri dish (inner diameter: 88 mm) and uniformly distributed on the glass to create a nearly monolayer. A Petri dish with the carbon material was carefully placed into the Cressington Carbon Coater 208c. At a pressure of 1.5 × 10−4 mbar, the sample was coated with a layer of carbon. The thickness was measured in situ using the Cressington Thickness Monitor MTM-10.Deposition of Pd on carbon materials
The procedure was performed as previously reported.15 Pd2dba3·CHCl3 (1.2 mg, 1.16 μmol) was dissolved in CHCl3 (2.4 mL). Parts of this solution were added to the carbon materials to obtain 0.5% Pd/C material. To the unmodified graphite (12.2 mg), 0.61 mL of a complex solution (containing 0.305 mg of Pd2dba3·CHCl3) was added. To coated with carbon graphite (11.1 mg), 0.56 mL of a complex solution (containing 0.280 mg of Pd2dba3·CHCl3) was added. The complex was decomposed at 50 °C for 2 h. After the completion of the reaction, the solutions became colorless. The value was consistent with the literature data. The carbon powders were washed with acetone and CHCl3 and subsequently dried.Computational procedures
For all RI PBE22 calculations, the def2-SVP ZORA23 basis set and def2-SVP/J auxiliary basis set were used (Orca 3.0.324 program package). The RI (resolution of identity) approximation allows in particular to speed up the DFT calculations using pure (non-hybrid) functionals.The ZORA (zeroth-order regular approximation) is applied for the description of the relativistic effects in DFT methods. Relativistic effects may be important for molecular systems containing atoms of heavy elements such as palladium.25
The D3BJ16 dispersion correction was applied to more accurately describe the dispersion interactions between graphene sheets within RI DFT calculations.
The MOPAC 2012 program package26 was used for the semi-empirical PM6-D3H427 calculations with parameter GNORM = 0.2.
The visualization of optimized molecular structures was performed by using the Mercury 3.5.1 program package.28
Acknowledgements
The project was supported by the Russian Science Foundation (RSF Grant 14-13-01030). The authors thank Hitachi High-Technologies (Japan) and Interlab LLC (Russia) for providing access to the advanced microscopy hardware. The authors acknowledge Kurnakov Institute of General and Inorganic Chemistry, Moscow, for the BET analysis.Notes and references
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- (a) Y. Chen, C. Tan, H. Zhang and L. Wang, Chem. Soc. Rev., 2015, 44, 2681 RSC; (b) C. Chung, Y. K. Kim, D. Shin, S. R. Ryoo, B. H. Hong and D. H. Min, Acc. Chem. Res., 2013, 46, 2211 CrossRef CAS PubMed; (c) A. Jankovic, S. Erakovic, M. Vukasinovic-Sekulic, V. Miskovic-Stankovic, S. J. Park and K. Y. Rhee, Prog. Org. Coat., 2015, 83, 1 CrossRef CAS; (d) X. Hu and Q. Zhou, Chem. Rev., 2013, 113, 3815 CrossRef CAS PubMed; (e) A. E. Jakus, E. B. Secor, A. L. Rutz, S. W. Jordan, M. C. Hersam and R. N. Shah, ACS Nano, 2015, 9, 4636 CrossRef CAS PubMed; (f) D. Nuvoli, V. Alzari, R. Sanna, S. Scognamillo, J. Alongi, G. Malucelli and A. Mariani, J. Nanopart. Res., 2013, 15, 1512 CrossRef; (g) C. M. Santos, J. Mangadlao, F. Ahmed, A. Leon, R. C. Advincula and D. F. Rodrigues, Nanotechnology, 2012, 23, 395101 CrossRef PubMed; (h) H. Shen, L. Zhang, M. Liu and Z. Zhang, Theranostics, 2012, 2, 283 CrossRef CAS PubMed; (i) J. K. Wang, G. M. Xiong, M. Zhu, B. Özyilmaz, A. H. Castro Neto, N. S. Tan and C. Choong, ACS Appl. Mater. Interfaces, 2015, 7, 8275 CrossRef CAS PubMed; (j) Z. Wang, H. Zeng and L. Sun, J. Mater. Chem. C, 2015, 3, 1157 RSC; (k) K. Yang, L. Feng, H. Hong, W. Cai and Z. Liu, Nat. Protoc., 2013, 8, 2392 CrossRef CAS PubMed; (l) Y. Yang, A. M. Asiri, Z. Tang, D. Du and Y. Lin, Biochem. Pharmacol., 2013, 16, 365 CAS; (m) P. T. Yin, S. Shah, M. Chhowalla and K.-B. Lee, Chem. Rev., 2015, 115, 2483 CrossRef CAS PubMed.
- (a) D. Bitounis, H. Ali-Boucetta, B. H. Hong, D. H. Min and K. Kostarelos, Adv. Mater., 2013, 25, 2258 CrossRef CAS PubMed; (b) X. Hu and Q. Zhou, Chem. Rev., 2013, 113, 3815 CrossRef CAS PubMed; (c) H. Y. Mao, S. Laurent, W. Chen, O. Akhavan, M. Imani, A. A. Ashkarran and M. Mahmoudi, Chem. Rev., 2013, 113, 3407 CrossRef CAS PubMed; (d) S. Shi, F. Chen, E. B. Ehlerding and W. Cai, Bioconjugate Chem., 2014, 25, 1609 CrossRef CAS PubMed; (e) Y. Zhang, T. R. Nayak, H. Hong and W. Cai, Nanoscale, 2012, 4, 3833 RSC.
- (a) L. H. Hess, A. Lyuleeva, B. M. Blaschke, M. Sachsenhauser, M. Seifert, J. A. Garrido and F. Deubel, ACS Appl. Mater. Interfaces, 2014, 6, 9705 CrossRef CAS PubMed; (b) L. Kong, A. Enders, T. S. Rahman and P. A. Dowben, J. Phys.: Condens. Matter, 2014, 26, 443001 CrossRef PubMed; (c) C. X. Lim, H. Y. Hoh, P. K. Ang and K. P. Loh, Anal. Chem., 2010, 82, 7387 CrossRef CAS PubMed; (d) H. Wang, X. Bo and L. Guo, Sens. Actuators, B, 2014, 192, 181 CrossRef CAS; (e) Y. Yue, G. Hu, M. Zheng, Y. Guo, J. Cao and S. Shao, Carbon, 2012, 50, 107 CrossRef CAS.
- (a) F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26 CrossRef CAS PubMed; (b) D. W. Boukhvalov and M. I. Katsnelson, Nano Lett., 2008, 8, 4374 CrossRef; (c) L. Cao, M. J. Meziani, S. Sahu and Y.-P. Sun, Acc. Chem. Res., 2013, 46, 171 CrossRef CAS PubMed; (d) O. Cretu, A. V. Krasheninnikov, J. A. Rodríguez-Manzo, L. Sun, R. M. Nieminen and F. Banhart, Phys. Rev. Lett., 2010, 105, 1 CrossRef PubMed; (e) M. F. De Lange, T. J. H. Vlugt, J. Gascon and F. Kapteijn, Microporous Mesoporous Mater., 2014, 200, 199 CrossRef CAS; (f) X. Jia, J. Campos-Delgado, M. Terrones, V. Meunier and M. S. Dresselhaus, Nanoscale, 2011, 3, 86 RSC; (g) A. Mesaros, S. Papanikolaou, C. F. J. Flipse, D. Sadri and J. Zaanen, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 1 Search PubMed; (h) J. Moellmer, E. B. Celer, R. Luebke, A. J. Cairns, R. Staudt, M. Eddaoudi and M. Thommes, Microporous Mesoporous Mater., 2010, 129, 345 CrossRef CAS; (i) M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cançado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276 RSC; (j) D. Wei and J. Kivioja, Nanoscale, 2013, 5, 10108 RSC; (k) S. C. Xu, S. Irle, D. G. Musaev and M. C. Lin, J. Phys. Chem. B, 2006, 110, 21135 CrossRef CAS PubMed; (l) S. C. Xu, S. Irle, D. G. Musaev and M. C. Lin, J. Phys. Chem. C, 2009, 113, 18772 CrossRef CAS; (m) S. C. Xu, S. Irle, D. G. Musaev and M. C. Lin, J. Phys. Chem. C, 2007, 111, 1355 CrossRef CAS.
- (a) B. Dai, K. Chen, Y. Wang, L. Kang and M. Zhu, ACS Catal., 2015, 5, 2541 CrossRef CAS; (b) C. Huang, C. Li and G. Shi, Energy Environ. Sci., 2012, 5, 8848 RSC; (c) S. D. Kushch, N. S. Kujunko and B. P. Tarasov, Russ. J. Gen. Chem., 2009, 79, 706 CrossRef CAS; (d) Z. Lin, M. Song, Y. Ding, Y. Liu, M. Liu and C. Wong, Phys. Chem. Chem. Phys., 2012, 14, 3381 RSC; (e) B. F. Machado and P. Serp, Catal. Sci. Technol., 2012, 2, 54 RSC; (f) L. Wang, A. Ambrosi and M. Pumera, Angew. Chem., Int. Ed., 2013, 52, 13818 CrossRef CAS PubMed; (g) R. Wang, Z. Wu, C. Chen, Z. Qin, H. Zhu, G. Wang, H. Wang, C. Wu, W. Dong, W. Fan and J. Wang, Chem. Commun., 2013, 49, 8250 RSC; (h) T. Xue, S. Jiang, Y. Qu, Q. Su, R. Cheng, S. Dubin, C. Y. Chiu, R. Kaner, Y. Huang and X. Duan, Angew. Chem., Int. Ed., 2012, 51, 3822 CrossRef CAS PubMed; (i) Y. Yao, Z. Yang, D. Zhang, W. Peng, H. Sun and S. Wang, Ind. Eng. Chem. Res., 2012, 51, 6044 CrossRef CAS; (j) T. Zeng, X. Zhang, Y. Ma, H. Niu and Y. Cai, J. Mater. Chem., 2012, 22, 18658 RSC; (k) M. Zhou, A. Zhang, Z. Dai, C. Zhang and Y. P. Feng, J. Chem. Phys., 2010, 132, 1 Search PubMed.
- (a) A. Ambrosi, C. K. Chua, A. Bonanni and M. Pumera, Chem. Rev., 2014, 114, 7150 CrossRef CAS PubMed; (b) R. Bogue, Assem. Autom., 2008, 28, 211 CrossRef; (c) M. Carbone, L. Gorton and R. Antiochia, Electroanalysis, 2015, 16 CrossRef CAS; (d) J. Chang, G. Zhou, E. R. Christensen, R. Heideman and J. Chen, Anal. Bioanal. Chem., 2014, 406, 3957 CrossRef CAS PubMed; (e) X. Deng, H. Tang and J. Jiang, Anal. Bioanal. Chem., 2014, 406, 6903 CrossRef CAS PubMed; (f) J. D. Fowler, M. J. Allen, V. C. Tung, Y. Yang, R. B. Kaner and B. H. Weiller, ACS Nano, 2009, 3, 301 CrossRef CAS PubMed; (g) T. Gan and S. Hu, Microchim. Acta, 2011, 175, 1 CrossRef CAS; (h) Q. He, S. Wu, Z. Yin and H. Zhang, Chem. Sci., 2012, 3, 1764 RSC; (i) Y. Liu, X. Dong and P. Chen, Chem. Soc. Rev., 2012, 41, 2283 RSC; (j) F. Schedin, A. K. Geim, S. V Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652 CrossRef CAS PubMed; (k) Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. Lin, Electroanalysis, 2010, 22, 1027 CrossRef CAS.
- (a) M. Freitag, Nat. Nanotechnol., 2008, 3, 455 CrossRef CAS PubMed; (b) S. Ghosh, I. Calizo, D. Teweldebrhan, E. P. Pokatilov, D. L. Nika, A. A. Balandin, W. Bao, F. Miao and C. N. Lau, Appl. Phys. Lett., 2008, 92, 1 Search PubMed; (c) G. Iannaccone, G. Fiori, M. MacUcci, P. Michetti, M. Cheli, A. Betti and P. Marconcini, Tech. Dig. - Int. Electron Devices Meet., 2009, 245 Search PubMed; (d) E. A. Il'ichev, A. E. Kuleshov, R. M. Nabiev, G. N. Petrukhin, G. S. Rychkov, O. A. Sakharov and E. S. Chernyavskaya, Tech. Phys. Lett., 2013, 39, 808 CrossRef; (e) R. S. Sundaram, M. Steiner, H. Y. Chiu, M. Engel, A. a. Bol, R. Krupke, M. Burghard, K. Kern and P. Avouris, Nano Lett., 2011, 11, 3833 CrossRef CAS PubMed; (f) X. Zheng, M. Zhang, X. Shi, G. Wang, L. Zheng, Y. Yu, A. Huang, P. K. Chu, H. Gao, W. Ren, Z. Di and X. Wang, Adv. Funct. Mater., 2015, 25, 1805 CrossRef CAS.
- (a) L. Dai, Acc. Chem. Res., 2013, 46, 31 CrossRef CAS PubMed; (b) B. Dawoud, E. Amer and D. Gross, Int. J. Energy Res., 2007, 31, 135 CrossRef CAS; (c) H. Gwon, H.-S. Kim, K. U. Lee, D.-H. Seo, Y. C. Park, Y.-S. Lee, B. T. Ahn and K. Kang, Energy Environ. Sci., 2011, 4, 1277 RSC; (d) J. Hou, Y. Shao, M. W. Ellis, R. B. Moore and B. Yi, Phys. Chem. Chem. Phys., 2011, 13, 15384 RSC; (e) F. Liu and D. Xue, Mater. Res. Innovations, 2015, 19, 7 CrossRef CAS; (f) P. Martin, Chem. Rec., 2009, 9, 211 CrossRef PubMed; (g) M. Pumera, Energy Environ. Sci., 2011, 4, 668 RSC; (h) J. Zhu, D. Yang, Z. Yin, Q. Yan and H. Zhang, Small, 2014, 10, 1 CrossRef.
- (a) J. Björk, F. Hanke, C. A. Palma, P. Samori, M. Cecchini and M. Persson, J. Phys. Chem. Lett., 2010, 1, 3407 CrossRef; (b) S. D. Chakarova-Käck, A. Vojvodic, J. Kleis, P. Hyldgaard and E. Schröder, New J. Phys., 2010, 12, 013017 CrossRef; (c) M. Corno, A. Rimola, V. Bolis and P. Ugliengo, Phys. Chem. Chem. Phys., 2010, 12, 6309 RSC; (d) Y. J. Dappe, M. Andersen, R. Balog, L. Hornekær and X. Bouju, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 1 CrossRef CAS; (e) L. M. Woods, Ş. C. Bǎdescu and T. L. Reinecke, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 1 CrossRef; (f) R. Zacharia, H. Ulbricht and T. Hertel, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 1 CrossRef; (g) X. Zhang, H. Ji, X. Zhang, Z. Wang and D. Xiao, Anal. Methods, 2015, 7, 3229 RSC; (h) J. Zhao, J. P. Lu, J. Han and C. K. Yang, Appl. Phys. Lett., 2003, 82, 3746 CrossRef CAS.
- (a) D. W. Boukhvalov, Surf. Sci., 2010, 604, 2190 CrossRef CAS; (b) H. Chang, J. Do Lee, S. M. Lee and Y. H. Lee, Appl. Phys. Lett., 2001, 79, 3863 CrossRef CAS; (c) O. Leenaerts, B. Partoens and F. M. Peeters, Appl. Phys. Lett., 2008, 92, 243125 CrossRef; (d) O. Leenaerts, B. Partoens and F. M. Peeters, Phys. Rev. B, 2008, 77, 125416 CrossRef; (e) J. Ma, A. Michaelides, D. Alfè, L. Schimka, G. Kresse and E. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 1 Search PubMed; (f) Y. Miura, H. Kasai, W. Diño, H. Nakanishi and T. Sugimoto, J. Appl. Phys., 2003, 93, 3395 CrossRef CAS; (g) S. Peng and K. J. Cho, Nano Lett., 2003, 3, 513 CrossRef CAS; (h) A. Sadrzadeh, A. A. Farajian and B. I. Yakobson, Appl. Phys. Lett., 2008, 92, 12 CrossRef; (i) A. Saffarzadeh, J. Appl. Phys., 2010, 107, 114309 CrossRef; (j) S. Santucci, S. Picozzi, F. Di Gregorio, L. Lozzi, C. Cantalini, L. Valentini, J. M. Kenny and B. Delley, J. Chem. Phys., 2003, 119, 10904 CrossRef CAS; (k) Y. Sun, L. Chen, F. Zhang, D. Li, H. Pan and J. Ye, Solid State Commun., 2010, 150, 1906 CrossRef CAS; (l) C. Thierfelder, M. Witte, S. Blankenburg, E. Rauls and W. G. Schmidt, Surf. Sci., 2011, 605, 746 CrossRef CAS; (m) T. O. Wehling, A. I. Lichtenstein and M. I. Katsnelson, Appl. Phys. Lett., 2008, 93, 1 CrossRef; (n) J. Zhao, A. Buldum, J. Han and J. P. Lu, Nanotechnology, 2002, 13, 195 CrossRef CAS.
- T. Pankewitz and W. Klopper, J. Phys. Chem. C, 2007, 111, 18917 CAS.
- (a) K. T. Chan, J. B. Neaton and M. L. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 1 Search PubMed; (b) X. Fan, W. T. Zheng and J. L. Kuo, ACS Appl. Mater. Interfaces, 2012, 4, 2432 CrossRef CAS PubMed; (c) J. Granatier, P. Lazar, M. Otyepka and P. Hobza, J. Chem. Theory Comput., 2011, 7, 3743 CrossRef CAS PubMed; (d) J. Grantier, P. Lazar, R. Prucek, K. Šafářová and R. Zbořil, J. Phys. Chem. C, 2012, 116, 14151 CrossRef; (e) S. M. Kozlov, F. Viñes and A. Görling, J. Phys. Chem. C, 2012, 116, 7360 CrossRef CAS; (f) A. Smolyanitsky and V. K. Tewary, J. Phys.: Condens. Matter, 2011, 23, 355006 CrossRef CAS PubMed; (g) Z. Xu and M. J. Buehler, J. Phys.: Condens. Matter, 2010, 22, 485301 CrossRef PubMed.
- (a) A. V. Zabula, A. S. Filatov, S. N. Spisak, A. Yu. Rogachev and M. A. Petrukhina, Science, 2011, 333, 1008 CrossRef CAS PubMed; (b) A. V. Zabula, S. N. Spisak, A. S. Filatov and M. A. Petrukhina, Angew. Chem., Int. Ed., 2012, 51, 12194 CrossRef CAS PubMed; (c) A. S. Filatov, A. V. Zabula, S. N. Spisak, A. Yu. Rogachev and M. A. Petrukhina, Angew. Chem., Int. Ed., 2014, 53, 140 CrossRef CAS PubMed; (d) A. S. Filatov, S. N. Spisak, A. V. Zabula, J. McNeely, A. Yu. Rogachev and M. A. Petrukhina, Chem. Sci., 2015, 6, 1959 RSC.
- E. O. Pentsak, A. S. Kashin, M. V. Polynski, K. O. Kvashnina, P. Glatzel and V. P. Ananikov, Chem. Sci., 2015, 6, 3302 RSC.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) C. Steffen, K. Thomas, U. Huniar, A. Hellweg, O. Rubner and A. Schroer, J. Comput. Chem., 2010, 31, 2967 CAS.
- (a) M. A. Vincent and I. H. Hillier, J. Chem. Inf. Model., 2014, 54, 2255 CrossRef CAS PubMed; (b) E. G. Gordeev, M. V Polynski and V. P. Ananikov, Phys. Chem. Chem. Phys., 2013, 15, 18815 RSC.
- (a) A. Dedieu, Chem. Rev., 2000, 100, 543 CrossRef CAS PubMed; (b) M. Filatov, Chem. Phys. Lett., 2003, 373, 131 CrossRef CAS.
- (a) M. V. Polynski and V. P. Ananikov, Computational Modeling of Graphene Systems Containing Transition Metal Atoms and Clusters, Understanding Organometallic Reaction Mechanisms and Catalysis, Wiley-VCH, Weinheim, 2014 Search PubMed; (b) Y. Tang, Z. Yang and X. Dai, J. Chem. Phys., 2011, 135, 224704 CrossRef PubMed; (c) X. Liu, L. Li, C. Meng and Y. Han, J. Phys. Chem. C, 2012, 116, 2710 CrossRef CAS.
- J. Robertson and E. O'Reilly, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 2946 CrossRef CAS.
- S. S. Zalesskiy and V. P. Ananikov, Organometallics, 2012, 31, 2302 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- D. A. Pantazis, X. Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908 CrossRef CAS PubMed.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CrossRef CAS.
- G. Eickerling, R. Mastalerz, V. Herz, W. Scherer, H.-J. Himmel and M. Reiher, J. Chem. Theory Comput., 2007, 3, 2182 CrossRef CAS PubMed.
- J. J. P. Stewart, Stewart Computational Chemistry, MOPAC2012, Version 15.156L, http://HTTP://OpenMOPAC.net Search PubMed.
- (a) P. Hobza, Acc. Chem. Res., 2012, 45, 663 CrossRef CAS PubMed; (b) J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173 CrossRef CAS PubMed.
- Mercury 3.5.1 (Build RC5), CCDC 2001–2014, http://www.ccdc.cam.ac.uk/mercury/.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp05586e |
| This journal is © the Owner Societies 2016 |