
C–S@PANI composite with a polymer spherical network structure for high performance lithium–sulfur batteries†
Junkai
Wang
a,
Kaiqiang
Yue
a,
Xiaodan
Zhu
b,
Kang L.
Wang
b and
Lianfeng
Duan
*ab
aKey Laboratory of Advanced Structural Materials, Ministry of Education, and Department of Materials Science and Engineering, Changchun University of Technology, Changchun 130012, China
bDevice Research Laboratory, Department of Electrical Engineering, University of California, Los Angeles, California 90095, USA. E-mail: duanlf@ccut.edu.cn
First published on 5th November 2015
Abstract
A unique C–S@PANI composite with a conductive polymer spherical network (PSN) has been successfully designed and synthesized by a simple processing approach. The PSN framework is formed at the surface of the oxidized carbon black by conductive polymer self-assembly and grafting, followed by pouring elemental sulfur into the pores of the polymer matrix. As the cathode material for lithium–sulfur batteries, the C–S@PANI composite delivered a high specific capacity of 1453 mA h g−1 at a 0.1 C current rate and a stable cycling performance of 948 mA h g−1 after 200 cycles. The composite also demonstrated high capacities of 922 and 581 mA h g−1 at 50 °C and 0 °C, respectively, after 200 cycles. The conductive PANI coatings were connected with the C–S core–shell composites to form a three-dimensional conducting network, which improves the utilization of the active mass and dual conduction of Li+ and electrons, while at the same time encapsulating sulfur into the PANI hollow spherical network. The structure effectively inhibits the dissolution and migration of polysulfides into the electrolyte, while improving the cycling stability and the coulombic efficiency of the electrode at high current rates, especially the low temperature electrochemical properties of Li–S batteries.
Introduction
With growing interest in the development of a low-cost, high performance and long-lasting rechargeable storage element, how to further improve the electrochemical performance of Lithium-Ion Batteries (LIBs) has emerged as a key technological challenge.1–4 The rechargeable lithium–sulfur (Li–S) battery is considered to be a promising candidate to supplement conventional lithium-ion batteries for energy storage devices, because of its significantly lower material cost, environmentally friendly process and higher theoretical energy density.5–8 Based on the reversible oxidization-reduction reaction between sulfur and lithium (16Li + S8 → 8Li2S), Li–S batteries can reach a high specific capacity of 1672 mA h g−1, with a theoretical energy density of up to 2600 W h kg−1.9–11 However, several challenges need to be overcome before the S-based cathodes can be adopted for practical applications, especially under low temperature conditions.12–14 First, during the disproportionation reactions, the “shuttle effect” is generated by the dissolved polysulfides (Li2Sx 3 ≤ x ≤ 8), causing unstable cycling behavior, high self-discharge rates, low coulombic efficiencies and irreversible capacity loss. Second, the poor conductivity of elemental sulfur (5 × 10−30 S cm−1 at 25 °C) and the discharge products such as Li2S/Li2S2 (∼10−30 S cm−1) could render part of the sulfur phase not electroactive, which leads to a low sulfur utilization (typically 50–70%) and a small electrode capacity. Third, the process of elemental sulfur (ρ1 = 2.03 g cm−3 at room temperature for α-S) converting to Li2S (ρ2 = 1.66 g cm−3 cell structure of the anti-fluorite structure) is accompanied by a large volume expansion (∼80%), causing damage to the electrode structure.15–19In recent years, much more effort has been made to solve the above-mentioned problems in order to realize high performance Li–S batteries. The nanostructured polymer-modified mesoporous C–S (carbon–sulphur) composites were investigated, with capacities of up to 1320 mA h g−1. The composite materials reported can supply nearly 80% of the theoretical capacity of sulphur (1672 mA h g−1), which demonstrated the benefits of confinement effects at a small length scale.20,21 Cui's group focused on solving the volumetric expansion of sulphur during lithiation and designed the TiO2 yolk–shell nano-architecture with internal void space to accommodate the volume expansion of sulfur, resulting in an initial specific capacity of 1030 mA h g−1 at 0.5 C, and a volume expansion rate as small as 0.033% per cycle.22 Focused on creating fine structures, the polymer/S–C composites utilize interactions between lithium ions in solution and the functional group uniformly distributed along the chain backbone of a polymer precursor (e.g. polyacrylonitrile), to control the distribution of lithium sulfide in the host material23,24 similar to vulcanization methods. In other aspects, some additives, especially LiNO3, can enhance the coulombic efficiency via self-sacrifice, but these additives gradually lose their function with repeated cycles.25 The results of synthesis processes with templates (AAO) were reported to overcome the ‘shuttle effect’ of cathodes.26 From these reports, four critical signatures of an ideal sulfur electrode can be identified: (1) a closed structure to encapsulate polysulfides efficiently, improving the sulfur–electrolyte contact; (2) a loose structure to provide sufficient space to accommodate sulfur volumetric expansion and preserve the morphology of electrodes during the transportation of electrons and Li ions; (3) a carbon composite material to improve the conductivity of the cathode and suppress the dissolution of polysulfides simultaneously; (4) sulfur wrapped with conducting polymers to promote a short transport pathway for both electrons and Li ions to achieve high capacity at a high power rate and suitable electrolyte additives to passivate the lithium surface to minimize the shuttle effect. It is very challenging to realize sulfur electrodes with high specific capacity and long cycle life because of the self-conflicting structure designs. Although these advanced electrode structures have led to remarkable progress in the cycle life of Li–S cells, it is more important to synthesise specially designed cathodes in order to improve reversible capacity, promote excellent cycling stability and a good rate capability for lithium–sulfur batteries, especially at lower and higher temperature using simple and environmental friendly processes.
Herein, we design a conductive polymer spherical network (PSN) as the cathode materials for lithium–sulfur batteries using an in situ synthesis technique. Oxidized carbon black particles are the core structure of the composite positive electrode material. A conductive polymer grows from the surface of oxidized carbon black to form the spherical network framework. Compared to the carbon material, conductive polymer PANI, which is attractive in absorbing and holding sulfur, has a more flexible structure (as shown in Fig. 1). Due to the flexibility of the polymer material, C–S@PANI composites with a PSN structure as the cathode material show high specific capacity, excellent cycle performance and stable electrochemical properties at low temperature for the Li–S battery.
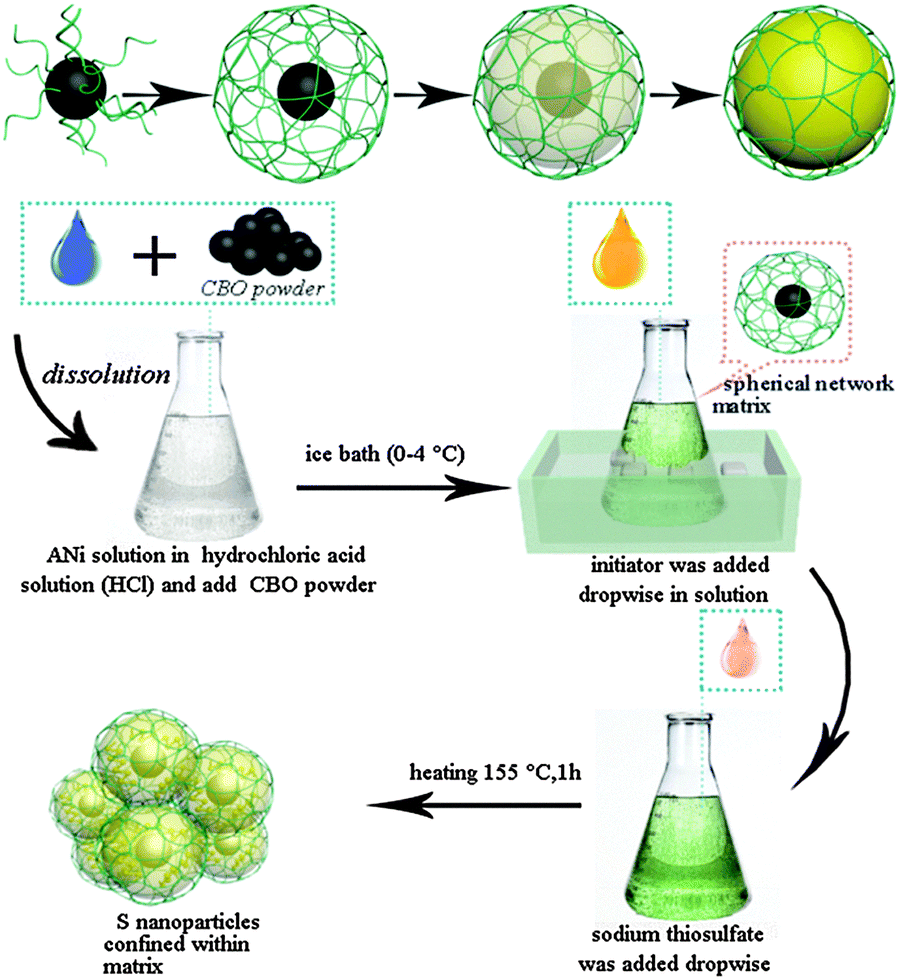 | ||
| Fig. 1 The design of the PSN matrix (C–S@PANI composite) as the cathode material for lithium–sulfur batteries (green: PANI, yellow: sulfur, and black: acetylene black), and schematic illustration of the synthesis process. | ||
Results and discussion
A new scheme is proposed to prepare composites based on lithium sulfide uniformly dispersed in Polymer Spherical Networks by the in situ synthesis technique. During the co-heating process, the molten sulfur can penetrate into the PANI spherical network and the network shows strong absorption ability towards sulfur. The morphology of the C–S@PANI composite is presented in Fig. 2. The FESEM image of the composite shows a matrix structure containing nanowire-like features (Fig. 2a). TEM images (Fig. 2b and c) show clearly the typical matrix structure of the C–S@PANI composite, with C–S particles reunited to form a spherical particle with sizes of 80–100 nm, and the whole particle congeries completely enclosed and pinned by the PANI network matrix of nearly 20 nm in thickness. Fig. 2d shows EDX elemental mappings of the spherical particle, which show that sulfur and carbon can maintain uniform distribution on a single spherical particle. Meanwhile, sulfur is uniformly encapsulated within the PANI matrix after heating. In this way, the PANI layer connects to the C–S particles to form a micro-reactor with many pores. The PANI sphere matrix shows that a simple physical confinement and absorption process utilizes the capillary action of small pores to absorb polysulfide. The pores provide channels for electrolyte infiltration and ion diffusion, and at the same time providing spaces for volume expansion produced by the sulfur discharge products. On the other hand, the chemical bond is generated due to the chemical effect, the interaction between the functional groups of the conductive polymer surface and the reaction products of the polysulfide.27,28 This structure could be better for sulfur wrapped. | ||
| Fig. 2 (a) FESEM images and (b and c) TEM images of C–S@PANI composites and (d) EDX elemental mappings of carbon, sulfur and nitrogen. | ||
X-ray diffraction (XRD) analyses of the C–S@PANI composite after heat treatment are shown in Fig. 3a. PANI exhibits an amorphous structure without sharp crystalline peaks. By comparing the XRD patterns, the C–S@PANI composite (40% sulfur) exhibits significantly lower peak intensities than the sulfur, indicating the amorphous nature of the sulfur within the composite. FTIR measurements were further conducted to characterize the presence of chemical bonds and functional groups between the PANI matrix and sulfur (Fig. 3b). After heating, a slight in situ vulcanization occurs but the polyaniline retains its stable structure. There are two characteristic peaks at 1545.7 and 1482.1 cm−1 which are attributed to quinoid and benzenoid-ring vibrations, respectively, indicating the oxidation state of the emeraldine.29 The bands in the 1200–1400 cm−1 range come from the C–N stretching modes of the aromatic amine. The spectrum of the C–S@PANI composite is significantly different from that of the PANI. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration at 1497 cm−1 assigned to benzenoid rings shifts to lower wave numbers, which could be coming from the substitution of H atoms on benzenoid rings by S atoms. The C–N stretching vibrational bands at 1298.4 and the C–H vibrational band in the vicinity of 1128.7 cm−1 weaken significantly, further confirming the replacement of H atoms on aromatic rings by S atoms.30,31 The different contents of sulfur loaded in the C–S@PANI composite were determined by thermogravimetric analysis (Fig. S1, ESI†). Considering the low content of PANI in this composite, the weight loss of inner PANI could be ignored during sulfur evaporation, therefore we conclude that the sulfur loading is about 40% for the red curve. With increasing sulfur content, the composite loses its weight by 59.17% before 360 °C, which indicates that the sulfur loading was about 60%. In fact, with increasing sulfur content, the sulfur cannot be completely integrated into the polymer network structure. So the actual sulfur content within the network structure is about 40%. The PSN provides strong physical and chemical confinement to the elemental sulfur and the resident polysulfide.
C stretching vibration at 1497 cm−1 assigned to benzenoid rings shifts to lower wave numbers, which could be coming from the substitution of H atoms on benzenoid rings by S atoms. The C–N stretching vibrational bands at 1298.4 and the C–H vibrational band in the vicinity of 1128.7 cm−1 weaken significantly, further confirming the replacement of H atoms on aromatic rings by S atoms.30,31 The different contents of sulfur loaded in the C–S@PANI composite were determined by thermogravimetric analysis (Fig. S1, ESI†). Considering the low content of PANI in this composite, the weight loss of inner PANI could be ignored during sulfur evaporation, therefore we conclude that the sulfur loading is about 40% for the red curve. With increasing sulfur content, the composite loses its weight by 59.17% before 360 °C, which indicates that the sulfur loading was about 60%. In fact, with increasing sulfur content, the sulfur cannot be completely integrated into the polymer network structure. So the actual sulfur content within the network structure is about 40%. The PSN provides strong physical and chemical confinement to the elemental sulfur and the resident polysulfide.
 | ||
| Fig. 3 (a) XRD patterns of sublimed S, PANI and C–S@PANI composite 40% of sulfur and (b) FTIR spectra of the PANI and C–S@PANI composite. | ||
The electrochemical performances of the C–S@PANI composite with PSN structure were evaluated by galvanostatic charge/discharge cycling. The discharge curves of the composite at different cycle numbers are shown in Fig. 4a. Two voltage plateaus are observed at 2.3 V and 2.0 V, corresponding to charging platforms at 0.1 C (1 C = 1672 mA h g−1, according to the theoretical capacity of sulfur). The elemental sulfur capacity is calculated by subtracting the amount of carbon black and polyaniline from the PSN/S. The high voltage plateau corresponds to the long polysulfide chains being produced during the first reduction step (2.4–2.2 V vs. Li+/Li), such as S82− and S62−, as well as S42− being produced during the second reduction step (2.15–2.1 V vs. Li+/Li).32,33 The low voltage plateau is attributed to the gradual decrease of the polysulfide chain lengths. Finally, short polysulfide species, such as S32−, S22− and S2−, are produced at the end of the reduction process.31 The electrode exhibits a discharge capacity of 608 mA h g−1 and a charge capacity of 628 mA h g−1 at 0.1 C for the first cycle. And then, after 20 cycles, the discharge and charge capacity increased to 1238 mA h g−1 and 1453 mA h g−1 (Fig. 4a). Fig. 4b shows the exceedingly stable cycling performance of C–S@PANI composites at different current densities 0.1 C, 0.2 C and 0.5 C, respectively. The initial discharge capacity of the composites increases significantly up to about 20 cycles, and then remains relatively stable at a value as high as 948 mA h g−1 after 200 cycles at 0.1 C. A similar trend is observed for current densities of 0.2 C and 0.5 C, reaching a specific capacity of 758 mA h g−1 and 743 mA h g−1 after 200 cycles, respectively. This is a result of the active material having sufficient contact with the liquid electrolyte, leading to the high utilization rate of sulfur. When the C–S@PANI composites electrochemically react with lithium, additional energy is consumed to break the complex bonds, particularly during the first discharge. The replacement of H atoms in the aromatic rings by S atoms and the generation of a C–S bond could be confirmed during heat treatment.27 A small amount of elemental sulfur reacts with polyaniline to form a cross-linked PANI network.32 During the process of discharge, this sulfur leaves the network. The bonds broken include C–S, S–S and so on, which are stronger than van der Waals forces.23 The discharge capacity exceeds the initial discharge capacity due to the fact that during the previous discharge process, the internal active material in contact with the electrolyte and activation is not sufficient.33 After 200 cycles, the specific capacity remains at 900 mA h g−1, with a capacity retention rate of nearly 75% at 0.1 C (Fig. 4b). In addition, as seen in Fig. 4c, the electrode was cycled from low current rates to high current rates and then reversed back to low rates. The discharge capacity of the composite cathode returns to 1012 mA h g−1 from 727 mA h g−1, when the rate decreases from 0.5 C to 0.1 C. Compared to other active materials such as Li–S batteries, the as-prepared C–S@PANI composite exhibits superior capacity retention, excellent cycling performance and rate stability. Fig. 4d shows the electrochemical impedance spectra (EIS) of the composite electrode after 50 cycles. The Nyquist plot is composed of two depressed semicircles in the middle and high frequency region and an oblique line in the low frequency region. The semicircle from high to medium frequency represents the SEI layer resistance. The SEI layer formed by LiNO3 was adopted to protect the lithium anode and improve coulombic efficiency. The intermediate frequency semicircle represents charge transfer resistance between the electrolyte and the cathode electrode. The oblique line represents diffusion impedance. For EIS of the composite electrode after 50 cycles, the charge-transfer resistance remains small which serves as an indication of the structure's stability.
 | ||
| Fig. 4 (a) Charge–discharge profile for the C–S@PANI composite at 0.1 C between 0.1 and 3.0 V (vs. Li/Li+) at room temperature, (b) cycle performance of the C–S@PANI composite at 0.1 C, 0.2 C, and 0.5 C, respectively, and (c) rate performance of the C–S@PANI composite. (d) Electrochemical impedance spectra of the composite electrode after 50 cycles. | ||
As discussed before, the reversible capacity of the C–S@PANI composite cathode shows an increase during the first few cycles. It is an activation. It is due to diffusion and polarization phenomena. At a higher C-rate, the polarization phenomenon is more serious. The diffusion and contact with the electrolyte are more inadequate.32–34 As a result, the activation is not complete.23 So the high C-rate requires more activation time. This could also be explained by considering the PSN/sulfur composites with high conducting carbon materials coated by the insulating sulfur forming a core–shell structure and PANI, while the redox reaction of the sulfur cathode can only occur at the surface of carbon. During the first several cycles, the surface of the composites exposed to the electrolyte gradually increases with the phase change of sulfur from a solid state to the dissolved polysulfide state. These results were consistent with the lower initial discharge capacity.35 The improvement of the cycling performance may be contributed to the well-percolated electrical path and structural stability given by the distributed carbon in the S-coated-PANI cathode. In addition, the C–S@PANI composite cathode shows a better cycle performance than the S–C composite cathode, owing to the 3D network matrix to accommodate the mechanical stress induced by the volume change of the redox reaction of sulfur and maintain the stability of the electrode at high current rates during the discharge–charge cycles. In addition, the PSN, with its high aspect ratio and multiple cross junctions, can effectively hinder the dissolution of polysulfides, which is critical for improving the stability of sulfur as the electrode at high current rates.36–39
The comparison of the cycling performances of the C–S@PANI composite at different temperatures is shown in Fig. 5. It can be seen that the discharge capacity reached 922, 948 and 581 mA h g−1 at 50 °C, 20 °C and 0 °C, respectively, after 200 cycles. The specific capacity of S-coated-CB cathodes at 0 °C is also shown for comparison. Only after 100 cycles, most of its initial discharge capacity is lost to 117 mA h g−1. The discharge reaction of sulfur always consists of stepwise reduction processes and generates various forms of intermediate polysulfides which can dissolve in the electrolyte selected for lithium/sulfur batteries.40,41 Because the redox reaction of the sulfur cathode can only occur at the surface of carbon due to the insulating nature of sulfur and its reduction products, the facile transport of the polysulfides to the carbon matrix is very important for a high sulfur utilization.36 At lower temperature, the electrolyte viscosity increases, which leads to a lower ion diffusion coefficient in the electrolyte and serious concentration polarization, causing performance deterioration. The as-prepared C–S@PANI composite with PSN shows significantly higher specific capacity and cycling performance stability even at low test temperature (0 °C), although the specific capacity is much lower than the sample at room temperature (20 °C).
 | ||
| Fig. 5 The cycling performances of C–S@PANI and C–S composites at different test temperatures (0 °C, 20 °C, and 50 °C). | ||
The charge–discharge reaction in a lithium–sulfur battery can be described in multiple reactions. Sulphur can react with metallic lithium to form Li2S with a large negative free energy change, which can be harnessed in a battery with a two-electron reaction. In the redox reaction, sulfur entirely dissolves into the liquid electrolyte in the form of Li2S8 with the S8 ring as the initial form. The color of the catholyte changed from red to green with decreasing chain length of polysulfides.42 The final product is insoluble Li2S2/Li2S, which is deposited on the electrode surface, blocking further reaction. This process of inclusion complex disproportionation results in the “shuttle effect”.31,43,44 The total reaction is as follows: S8 + 16Li → 8Li2S. The PSN provides access to Li+ ingress/egress for conversion of Li2S into Li2Sx (as illustrated in Fig. S4, ESI†). The higher specific capacity and excellent cycle stability are achieved simultaneously, which can be attributed to the special microstructure of the C–S@PANI composite. Firstly, the conductive PANI coatings connected with the C–S core–shell composites form a three-dimensional conducting network (Fig. S4a, ESI†). It greatly improves the utilization of the active mass and dual conduction of Li+ and electrons, which is beneficial for the enhancement of the rate capability of the sulfur cathode. Secondly, in the encapsulation of sulfur into the PANI hollow spherical network, the S–C bonds generated during heating treatment act as the bridge between the PANI and sulfur or polysulfides. The structure of hyperbranched, high aspect ratio, and multiple cross junctions can inhibit effectively the dissolution and migration of polysulfides into the electrolyte, thus effectively enhancing the cycling stability and the coulombic efficiency of the electrode at high current rates.45–47 Thirdly, the designed void space in the PANI hollow network supplies sufficient space to buffer the large volume expansion of sulfur during the charge–discharge process, resulting in the stable structure of the cathode, and therefore the long-term stability of the lithium sulfur battery (Fig. S4b, ESI†). In addition, the primary C–S@PANI particles increase the electrochemically active surface area, beneficial for the infiltration of the electrolyte as well as for electron transfer and ion diffusion, which improves the cathode activity at low temperature effectively. For the structure of the C–S@PANI composite with the PSN, the majority of the sulfur is trapped by the cross-linked stereo PANI-S network and the C–S shell, promoting superior specific capacity, excellent cycling performance, rate stability and especially low temperature electrochemical properties.
Experimental
The C–S@PANI composite with the conductive PSN has been synthesized by a grafting method. Fig. 1 shows a schematic illustration of the synthesis process.Acetylene black oxidation treatment
The suspension of CBO (oxidized carbon black) was prepared by dispersing a stoichiometric amount of acetylene black (AC) in concentrated nitric acid (HNO3 69.2 wt%). For the typical preparation of 20 M AC suspension, 0.2 g of AC were added to 10 mL of HNO3. The obtained suspension was stirred for 5 min and heated at 170 °C for 1 h. Finally, it is washed with deionized water.In situ synthesis of PANI composites
5 mL of 2 M hydrochloric acid solution (HCl) was prepared and mixed with 0.01 g of CBO, followed by the addition of 0.46 mL of aniline (ANi) under ice cold (0–4 °C) conditions with magnetic stirring. Then the 1 M initiator (the molar amount of ammonium persulfate (APS) and ferric chloride (FeCl3) is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 1:1) was added dropwise while stirring the mixed suspension of CBO/ANi (speed < 0.5 mL min−1). The solution was stirred for 10 min, sealed, reacted for t1 (18 h) at 0–4 °C, and t2 (24 h) at room temperature (20–30 °C), ultrasonicated for 10 min, washed with acetone and deionized water, ultrasonicated after each wash and centrifuged to obtain the conductive polymer (CBO-PANI).
:0 to 1:1) was added dropwise while stirring the mixed suspension of CBO/ANi (speed < 0.5 mL min−1). The solution was stirred for 10 min, sealed, reacted for t1 (18 h) at 0–4 °C, and t2 (24 h) at room temperature (20–30 °C), ultrasonicated for 10 min, washed with acetone and deionized water, ultrasonicated after each wash and centrifuged to obtain the conductive polymer (CBO-PANI).
Pouring the sulfur element
The sulfur of composite materials was filled by the liquid phase deposition method, first the mixture (CBO-PANI) is added into 1 M HCl solution and stirred for 5 min. Sodium thiosulfate (Na2S2O3·5H2O 0.2 M) (rate v2 <1 mL min−1) was added dropwise with magnetic stirring into the conductive frame (CBO-PANI). After stirring for 10 min, the solution is allowed to stand for 1 h, followed by heating at 155 °C for 18 h. The obtained solution was washed with deionized water and alcohol, with ultrasonication after each wash, and the composite (C–S@PANI) was obtained through centrifugation.Materials characterization
The fabricated phases from the above solutions were identified by means of X-ray diffraction (XRD) using a Rigaku D/max 2500 pc X-ray diffractometer with Cu Kα radiation (λ = 1.54156 Å) at a scan rate of 0.04 deg s−1. The Fourier transform infrared (FTIR) spectra were recorded on a NICOLET AVA-TAR 360 FTIR spectrometer with KBr pellets. The morphologies and compositions of the as-prepared products were characterized using a JEOL JSM-6700F field-emission scanning electron microscope (FESEM) equipped with an energy dispersive X-ray spectroscope (EDS). Transmission electron microscopy (TEM) was carried out on a JEOL 2100F with an emission voltage of 200 kV. The amount of sulfur in the composites was determined by thermogravimetric (TG) analysis using a Perkin-Elmer TGA 7 thermogravimetric analyzer from 50 °C to 500 °C at a heating rate of 10 °C min−1 in flowing Ar. Multi-point Brunauer–Emmett–Teller (BET) surface area measurements were carried out using a volumetric sorption analyzer (NOVA 2000, Quantachrome) using physical adsorption/desorption of nitrogen gas at liquid-nitrogen temperature. Pore size distributions were calculated according to the Barrett–Joyner–Halenda (BJH) method. The micropore surface area and volume were estimated by the t-plot method. EIS data were obtained using a Solartron Impedance Analyzer (SI 1260 + SI 1287) from 10 kHz to 10 MHz at room temperature with an AC voltage amplitude of 5 mV at the open-circuit voltage of the cells with the Li metal foil as both auxiliary and reference electrodes.Electrochemical properties
Positive electrodes were comprised of 80 wt% PSN/S composite, 15 wt% Acetylene black (AC) and 5 wt% Polyacrylic acid (PAC-10) binder. The slurry was evenly coated with a coating machine scraper on the aluminum foil. Water as the solvent used to make the slurry. The cathode was prepared by coating a slurry on aluminum foil. After being dried at 90 °C for 24 h in a vacuum oven, the electrode disks were punched, tabletted and weighed. The cathode materials were evaluated in 2025 coin cells (MTI) in a glove box under an argon atmosphere. The lithium metal (99.99%, Sigma-Aldrich) cell assembly was performed in an Ar filled glovebox. Li metal (10 mm diameter) was used as the anode and the reference electrode. The separator was a commercial micro-porous polypropylene product (Celgard 2400). Aluminum foil was used as the cathode current collector, and 1.0 M LiN(CF3SO2)2 (1 M) (99.95%, trace metals basis, Sigma-Aldrich) salt dissolved in a mixture of dioxolane (DOL) (99.8%, Sigma-Aldrich) and 1,2-dimethoxyethane (DME) (99.5, Sigma-Aldrich) in a volume ratio of 1:1 containing LiNO3 (1 wt%) was used as the electrolyte. Based on sulfur content specific capacity calculations, galvanostatic charge/discharge tests (0.1, 0.2, and 0.5 C) were performed to evaluate the electrochemical capacity and cycle life of the electrodes at 0 °C, 20 °C and 50 °C using a LAND-CT2001A instrument (Wuhan, China).
Conclusions
A C–S@PANI composite with a conductive PSN has been successfully designed and synthesized by a facile processing approach. As the cathode material for Li–S batteries, the C–S@PANI composite delivered a high specific capacity of 1453 mA h g−1 at a 0.1 C current rate and a stable cycling performance of 948 mA h g−1 after 200 cycles. The composite also demonstrated high capacities of 922 and 581 mA h g−1 at 50 °C and 0 °C, respectively, after 200 cycles. The novel structure improves the utilization of the active mass and dual conduction of Li+ and electrons, which are beneficial for the enhancement of cycling performance, rate stability and low temperature electrical properties of advanced energy storage devices.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21221061), the State Scholarship Fund of China Scholarship Council (Grant No. 201408220025), Science Foundation of Jilin Education Department (Grant No. 2015118) and the Special Funds of Changchun University of Technology.Notes and references
- A. Manthiram, A. V. Murugan, A. Sarkar and T. Muraliganth, Energy Environ. Sci., 2008, 1, 621–628 CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- Y. X. Tang, Y. Y. Zhang, J. Y. Deng, D. P. Qi, W. R. Leow, J. Q. Wei, S. Y. Yin, Z. L. Dong, R. Yazami, Z. Chen and X. D. Chen, Angew. Chem., Int. Ed., 2014, 53, 13488–13492 CrossRef CAS PubMed.
- R. D. Rauh, K. M. Abraham, G. F. Pearson, J. K. Surprenant and S. B. Brummer, J. Electrochem. Soc., 1979, 126, 523–527 CrossRef CAS.
- X. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821–9826 RSC.
- X. W. Yu and A. Manthiram, Phys. Chem. Chem. Phys., 2015, 17, 2127–2136 RSC.
- M. Duduta, B. Ho, V. C. Wood, P. Limthongkul, V. E. Brunini, W. C. Carter and Y. M. Chiang, Adv. Energy Mater., 2011, 1, 511–516 CrossRef CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- H. Wang, Y. Yang, Y. Liang, L. F. Cui, H. S. Casalongue, Y. G. Li, G. S. Hong, Y. Cui and H. J. Dai, Angew. Chem., 2011, 123, 7502–7506 CrossRef.
- N. Li, Z. Chen, W. Ren, L. Feng and H. M. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17360–17365 CrossRef CAS PubMed.
- X. Ji, S. Evers, R. Black and L. F. Nazar, Nat. Commun., 2011, 2, 325–342 CrossRef PubMed.
- J. Shim, K. A. Striebel and E. J. Cairns, J. Electrochem. Soc., 2002, 149, A1321–A1325 CrossRef CAS.
- G. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462–4467 CrossRef CAS PubMed.
- C. Barchasz, J. C. Leprêtre, F. Alloin and P. Sébastien, J. Power Sources, 2012, 199, 322–330 CrossRef CAS.
- A. S. Aricò, P. Bruce, B. Scrosati, J. M. Tarascon and W. V. Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969–A1976 CrossRef CAS.
- H. Yamin, A. Gorenshtein, J. Penciner, Y. Sternberg and E. Peled, J. Electrochem. Soc., 1988, 135, 1045–1048 CrossRef CAS.
- D. Aurbach, E. Pollak, R. Elazari, G. Salitraa, C. S. Kelley and J. Affinito, J. Electrochem. Soc., 2009, 156, A694–A702 CrossRef CAS.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- S. Evers and L. F. Nazar, Chem. Commun., 2012, 48, 1233–1235 RSC.
- Z. W. Seh, W. Li, J. J. Cha, G. Y. Zheng, Y. Yang, M. T. McDowell, P. C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331–1337 CrossRef PubMed.
- J. Wang, Y. S. He and J. Yang, Adv. Mater., 2014, 3, 569–572 Search PubMed.
- J. Guo, Z. Yang, Y. Yu, H. D. Abruña and L. A. Archer, J. Am. Chem. Soc., 2012, 135, 763–767 CrossRef PubMed.
- S. S. Zhang and J. A. Read, J. Power Sources, 2012, 200, 77–82 CrossRef CAS.
- B. Zhang, X. Qin, G. R. Li and X. P. Gao, Energy Environ. Sci., 2010, 3, 1531–1537 CAS.
- G. Ma, Z. Wen, J. Jin, L. Yan, X. W. Wu, M. F. Wu and C. H. Chen, J. Mater. Chem. A, 2014, 2, 10350–10354 CAS.
- W. Zhou, Y. Yu, H. Chen, F. J. DiSalvo and H. D. Abruña, J. Am. Chem. Soc., 2013, 135, 16736–16743 CrossRef CAS PubMed.
- A. Kellenberger, E. Dmitrieva and L. Dunsch, Phys. Chem. Chem. Phys., 2011, 13, 3411–3420 RSC.
- M. Trchova, P. Matějka, J. Brodinova, A. Kalendovác, J. Prokešd and J. Stejskal, Polym. Degrad. Stab., 2006, 91, 114–121 CrossRef CAS.
- C. Barchasz, F. Molton, C. Duboc, J. C. Leprêtre, S. Patoux and F. Alloin, Anal. Chem., 2012, 84, 3973–3980 CrossRef CAS PubMed.
- L. Xiao, Y. Cao, J. Xiao, B. Schwenzer, M. H. Engelhard, L. V. Saraf, Z. Nie, G. J. Exarhos and J. Liu, Adv. Mater., 2012, 24, 1176–1181 CrossRef CAS PubMed.
- T. Xu, J. Song, M. L. Gordin, H. S. Sohn, Z. X. Yu, S. R. Chen and D. H. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 11355–11362 CAS.
- S. S. Zhang, D. T. Tran and Z. C. Zhang, J. Mater. Chem. A, 2014, 2, 18288–18292 CAS.
- A. Kawase, S. Shirai, Y. Yamoto, R. Arakawa and T. Takata, Phys. Chem. Chem. Phys., 2014, 16, 9344–9350 RSC.
- G. C. Li, G. R. Li, S. H. Ye and X. P. Gao, Adv. Energy Mater., 2012, 2, 1238–1245 CrossRef CAS.
- L. Yuan, H. Yuan, X. Qiu, L. Q. Chen and W. T. Zhu, J. Power Sources, 2009, 189, 1141–1146 CrossRef CAS.
- H. Chen, W. Dong and J. Ge, Sci. Rep., 2013, 3, 1910–1916 Search PubMed.
- W. Li, G. Zheng, Y. Yang, C. H. Wang, X. D. Wu, W. Lu and L. W. Chen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7148–7153 CrossRef CAS PubMed.
- G. Zheng, Q. Zhang, J. J. Cha, Y. Yang, W. Y. Li, Z. W. Seh and Y. Cui, Nano Lett., 2013, 13, 1265–1270 CrossRef CAS PubMed.
- S. Chen, X. Huang, H. Liu, B. Sun, W. K. Yeoh, K. F. Li, J. Q. Zhang and G. X. Wang, Adv. Energy Mater., 2014, 4, 1301761 Search PubMed.
- D. Marmorstein, T. H. Yu, K. A. Striebel, F. R. McLarnon, J. Hou and E. J. Cairns, J. Power Sources, 2000, 89, 219–226 CrossRef CAS.
- D. H. Han, B. S. Kim, S. J. Choi, Y. J. Jung, J. Kwakd and S. M. Park, J. Electrochem. Soc., 2004, 151, E283–E290 CrossRef CAS.
- M. U. M. Patel, R. Demir-Cakan, M. Morcrette, J. M. Tarascon, M. Gaberscek and R. Dominko, ChemSusChem, 2013, 6, 1177–1181 CrossRef CAS PubMed.
- Q. S. Wang, J. Jin, X. W. Wu, G. Q. Ma, J. H. Yang and Z. Y. Wen, Phys. Chem. Chem. Phys., 2014, 16, 21225–21229 RSC.
- Y. Diao, K. Xie, S. Xiong and X. B. Hong, J. Power Sources, 2013, 235, 181–186 CrossRef CAS.
- J. Song, T. Xu, M. L. Gordin, P. Y. Zhu, D. P. Lv, Y. B. Jiang, Y. S. Chen, Y. H. Duan and D. H. Wang, Adv. Funct. Mater., 2014, 24, 1243–1250 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp05447h |
| This journal is © the Owner Societies 2016 |