
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conversion of light-energy into molecular strain in the photocycle of the photoactive yellow protein†
Ana P.
Gamiz-Hernandez
and
Ville R. I.
Kaila
*
Department Chemie, Technische Universität München (TUM), Lichtenbergstraße 4, D-85747 Garching, Germany. E-mail: ana.gamiz@tum.de; ville.kaila@ch.tum.de
First published on 18th December 2015
Abstract
The Photoactive Yellow Protein (PYP) is a light-driven photoreceptor, responsible for the phototaxis of halophilic bacteria. Recently, a new short-lived intermediate (pR0) was characterized in the PYP photocycle using combined time-resolved X-ray crystallography and density functional theory calculations. The pR0 species was identified as a highly contorted cis-intermediate, which is stabilized by hydrogen bonds with protein residues. Here we show by hybrid quantum mechanics/classical mechanics (QM/MM) molecular dynamics simulations, and first-principles calculations of optical properties, that the optical shifts in the early steps of the PYP photocycle originate from the conversion of light energy into molecular strain, stored in the pR0 state, and its relaxation in subsequent reaction steps. Our calculations quantitatively reproduce experimental data, which enables us to identify molecular origins of the optical shifts. Our combined approach suggests that the short-lived pR0 intermediate stores ∼1/3 of the photon energy as molecular strain, thus providing the thermodynamic driving force for later conformational changes in the protein.
1. Introduction
The photoactive yellow protein (PYP) is a small (14-kDa) water-soluble protein found in purple photosynthetic bacteria that live in halophilic environments.1 PYP functions as a blue-light receptor, in which the light-absorption triggers a signaling cascade that switches the rotational direction of the bacterial flagellum, and directs the bacteria to swim away from potential harmful high energy light.2,3 PYP has become a key photobiological model system for studying light-absorption induced changes in proteins, due to its small size but relatively complex photocycle.4 The photoactive center of PYP comprises a p-coumaric acid (pCA) chromophore that is covalently linked to a cysteine residue (Cys-69) via a thioester bond (Fig. 1). The pCA is stabilized by hydrogen bonds from Tyr-42, Glu-46 and Thr-50, whereas the backbone of Cys-69 stabilizes the chromophore in early intermediates of the photocycle. Arg-52 shields the chromophore from the surroundings, and may electrostatically stabilize the anionic chromophore. In the PYP dark state (pG), shown in Fig. 1, the C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C3 double bond of pCA is in a trans-conformation, whereas light-absorption triggers a trans-to-cis isomerization that leads to subsequent formation of redshifted (pR) and blueshifted (pB) photocycle intermediates on picosecond–millisecond time scales.5–9
C3 double bond of pCA is in a trans-conformation, whereas light-absorption triggers a trans-to-cis isomerization that leads to subsequent formation of redshifted (pR) and blueshifted (pB) photocycle intermediates on picosecond–millisecond time scales.5–9
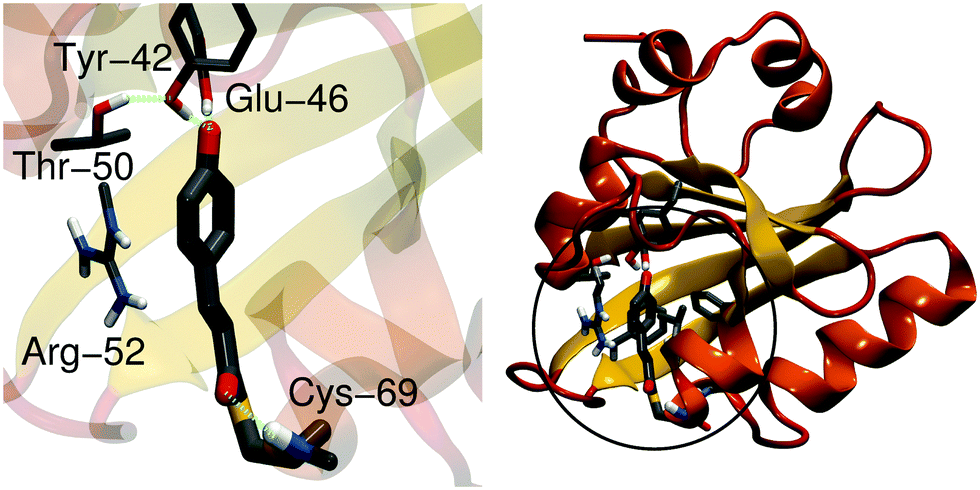 | ||
| Fig. 1 The structure of the pCA chromophore in the PYP dark state (pG). Left: pCA and its hydrogen bonding partners. Right: The structure of PYP showing the chromophore-binding region, modeled at the QM level of theory. | ||
Recently, Schotte et al.10 characterized a new photocycle intermediate using picosecond time-resolved X-ray crystallography and density functional theory (DFT) calculations.10,11 This new short-lived state, pR0, appeared on a 150 ps timescale (Fig. 2B), and was characterized as a highly strained cis-species with the double bond ca. 30° out of plane. The pR0 state was found to further relax into pR1, pR2 and pB states,10,12 that were found to be structurally and kinetically consistent with previously refined structural intermediates obtained from time-resolved X-ray crystallography.12–15 Recently Jung et al.16 also characterized a new short-lived species, IT, forming on time-scales similar to the pR0 state.
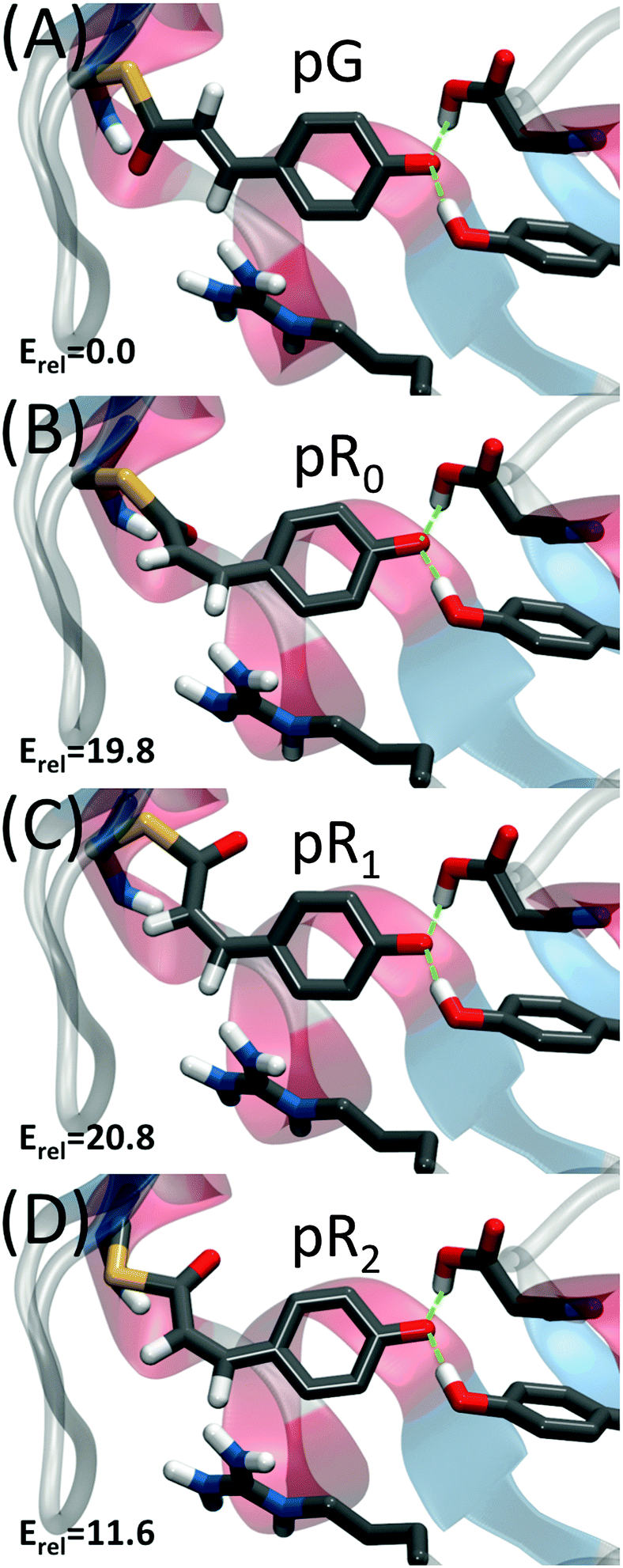 | ||
| Fig. 2 Structure of photocycle intermediates. The relative energy (Erel in kcal mol−1) stored within the pCA chromophore is also shown. The C2C3 double bond in pCA is trans-in the pG state and cis in all pR states. This double bond is strained in pR0, which upon breaking the hydrogen bond to Cys-69 converts to pR1, which is much more planar and less strained. The transition from pR1 to pR2 involves a rotation of the Cβ–Sγ–C1–C2 dihedral angle, which transforms from syn–syn to anti–anti, and further reduces strain in the pCA. | ||
In contrast to Schotte et al.,10 however, the refined species was suggested to be a transition state structure between pG and later intermediates, with the C2C3 double bond refined to ca. 90° out of plane. Jung et al. also used DFT calculations to study the likelihood of their species, but in contrast to their refined X-ray structure, the obtained DFT-structure was in close structural agreement with the pR0 species described by Schotte et al.10
Differences between these two structures were recently discussed by Kaila et al.11 and Jung et al.17 However, the focus of this study concerns optical tuning effects in the PYP photocycle studied by QM/MM calculations.
Proteins can tune the color of their biochromophores by electrostatic polarization and/or by imposing strain effects on the chromophore.18–23 By stabilizing the electronic ground state or excited state, the absorption can be shifted towards the blue (higher energy) or red (lower energy) regime of the spectrum. The strain energy stored in the recently discovered pR0 species is expected to lead to a destabilization of the electronic ground state, which in turn would further redshift the optical properties. Moreover, relaxation of the strain in the subsequent photocycle steps would be expected to blueshift the absorption.
Computation of optical properties of photobiological systems from first-principles is extremely challenging due to the complexity of the biological “active site” region, which normally limits the application of accurate correlated ab initio methods.19,20,24–28 The effect of the protein environments can be accounted for by employing e.g. hybrid quantum mechanical/molecular mechanics (QM/MM)19,20,24,27–31 or frozen density embedding approaches.22,32,33 Previous studies also suggest that nearby environmental effects must explicitly be considered at a quantum chemical level of theory34,35 or by accurate polarizable models36 to obtain quantitatively accurate results.
To study the structure and dynamics of the early photocycle intermediates in PYP, we use here a combination of QM/MM molecular dynamics simulations, first-principles computation of optical properties at second-order approximate coupled cluster (CC2) and time-dependent density functional (TDDFT) theory levels to elucidate how the photon energy is converted into molecular strain in the PYP photocycle, and to characterize the dynamics of the subsequent intermediates.
2. Computational methods
Time-resolved X-ray structures of the pG, pR0, pR1, and pR2 states (PDB ID: 2ZOH, 4B90, 4BBT and 4BBU) were obtained from the Brookhaven Protein Databank and used as starting points for the QM/MM simulations. We also performed QM/MM simulations on the refined IT structure by Jung et al.16 (PDB ID: 4I38) to study the X-ray model dependence of our data. The QM region (143 atoms) included the pCA chromophore, Tyr-42, Glu-46, Thr-50, Arg-52, Phe-62, Val-66, Ala-67, Pro-68, Cys-69, Thr-70 and Phe-96, and was described at the D-BP86/def2-SVP level of theory. The MM region, comprising the surrounding protein residues and water molecules, was described using the CHARMM27 force field.37 Each state was structure optimized, followed by 5 ps QM/MM MD simulations using a time step of 1 fs at 310 K. From the QM/MM trajectories, we extracted 100–500 snapshots for each state along the MD trajectories for calculation of ground state and optical properties. The absorption spectrum of the early PYP photocycle states was computed at the time-dependent density functional theory (TDDFT) level using CAM-B3LYP/def2-SVP, and at the second-order approximate coupled cluster level (CC2/def2-TZVP). The first six vertical excitation energies (VEEs) at TDDFT level of theory were computed for each snapshot leading to evaluation of 5000 VEEs per method (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 VEEs in total), whereas at the CC2 level, we computed the first two VEEs. To speed up the evaluation of the VEEs at the CC2 level, we employed the reduced virtual space (RVS) approach38 where virtual orbitals above 70 eV from the highest-occupied molecular orbital (HOMO) were disregarded from the correlation calculations, leading to a RVS38 cutoff error of 0.02 eV relative to frozen core CC2 calculations (Fig. S1, ESI†). For the isolated chromophore, the CC2 and CAM-B3LYP produced similar VEE profiles, but CAM-B3LYP was found to systematically blueshift the absolute excitation energies relative to CC2 (Fig. S2, ESI†). All CAM-B3LYP profiles were therefore redshifted by a 0.37 eV offset-factor obtained from the CC2 fit. For calculation of optical properties, we reduced the size of the QM region to explicitly include the pCA chromophore, Cys-69, Tyr-42, Glu-46 and Arg-52, for the TDDFT calculations, and the pCA chromophore for CC2 calculations, while the remaining protein and water atoms were described using point charges obtained from the QM/MM trajectories. For the estimation of electrostatic-induced shifts, we calculated the optical spectra of the chromophore along the QM/MM trajectories, by removing all protein residues around the chromophore but by keeping the chromophore structure unchanged. Strain-induced optical shifts were calculated as the difference between the absorption maximum of the isolated (trans or cis) chromophore structures obtained from gas-phase and the protein simulations, without considering the electrostatic polarization of the protein environment. The absorption spectra were calculated as an average of 500 VEEs obtained from the MD trajectories and weighted with their respective oscillatory strengths for each VEE calculation. The distribution of weighted VEE along the trajectories was fitted with a Gaussian width of 100 hc/Eh eV, where Eh is the excitation energy in eV, and combined to give a spectra by scaling the maximum intensity to 1 or by matching to the maximum experimental intensities. The two first excited states were included in the calculations due to larger oscillator strengths at TDDFT level. The spectra of each state were also calculated by computing histograms of the observed VEEs along the MD trajectories, and by normalizing the probability distribution. We obtain very similar spectra using both the Gaussian fitting and the histogram approaches, showing that the calculated line shapes arise from the statistical sampling of the ground state dynamics. Both approaches were found to give similar absorption spectra. QM/MM simulations were performed using the Q-Chem/CHARMM.37,39 TDDFT and CC2 (RVS) calculations were performed with Q-Chem v4.140 and TURBOMOLE v6.3,41 respectively.
000 VEEs in total), whereas at the CC2 level, we computed the first two VEEs. To speed up the evaluation of the VEEs at the CC2 level, we employed the reduced virtual space (RVS) approach38 where virtual orbitals above 70 eV from the highest-occupied molecular orbital (HOMO) were disregarded from the correlation calculations, leading to a RVS38 cutoff error of 0.02 eV relative to frozen core CC2 calculations (Fig. S1, ESI†). For the isolated chromophore, the CC2 and CAM-B3LYP produced similar VEE profiles, but CAM-B3LYP was found to systematically blueshift the absolute excitation energies relative to CC2 (Fig. S2, ESI†). All CAM-B3LYP profiles were therefore redshifted by a 0.37 eV offset-factor obtained from the CC2 fit. For calculation of optical properties, we reduced the size of the QM region to explicitly include the pCA chromophore, Cys-69, Tyr-42, Glu-46 and Arg-52, for the TDDFT calculations, and the pCA chromophore for CC2 calculations, while the remaining protein and water atoms were described using point charges obtained from the QM/MM trajectories. For the estimation of electrostatic-induced shifts, we calculated the optical spectra of the chromophore along the QM/MM trajectories, by removing all protein residues around the chromophore but by keeping the chromophore structure unchanged. Strain-induced optical shifts were calculated as the difference between the absorption maximum of the isolated (trans or cis) chromophore structures obtained from gas-phase and the protein simulations, without considering the electrostatic polarization of the protein environment. The absorption spectra were calculated as an average of 500 VEEs obtained from the MD trajectories and weighted with their respective oscillatory strengths for each VEE calculation. The distribution of weighted VEE along the trajectories was fitted with a Gaussian width of 100 hc/Eh eV, where Eh is the excitation energy in eV, and combined to give a spectra by scaling the maximum intensity to 1 or by matching to the maximum experimental intensities. The two first excited states were included in the calculations due to larger oscillator strengths at TDDFT level. The spectra of each state were also calculated by computing histograms of the observed VEEs along the MD trajectories, and by normalizing the probability distribution. We obtain very similar spectra using both the Gaussian fitting and the histogram approaches, showing that the calculated line shapes arise from the statistical sampling of the ground state dynamics. Both approaches were found to give similar absorption spectra. QM/MM simulations were performed using the Q-Chem/CHARMM.37,39 TDDFT and CC2 (RVS) calculations were performed with Q-Chem v4.140 and TURBOMOLE v6.3,41 respectively.
3. Results and discussion
Ground state dynamics of PYP photocycle intermediates
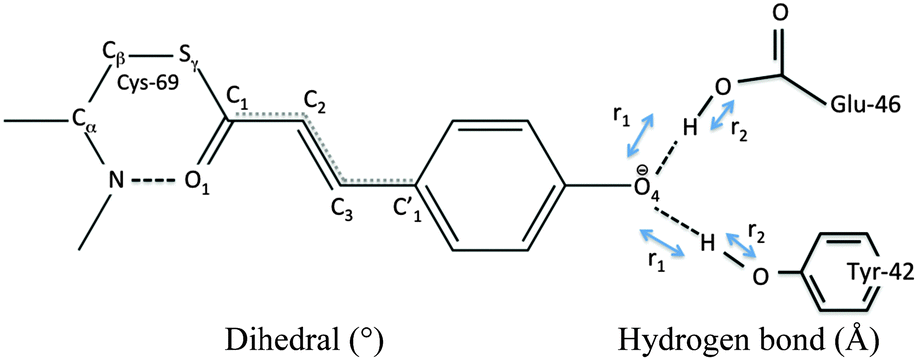
Our QM/MM molecular dynamics simulations suggest that all studied photo-cycle intermediates; pG, pR0, pR1, and pR2 remain stable on picosecond time-scales. Fig. 2 shows how the energy stored in the dihedral bonds of the chromophore is released during the pR0 → pR2 transition, in which the dihedral angle around the C2C3 double bond relaxes from a ∼30° strained cis-conformation in pR0 to planar species in pR1 and pR2. The pR0 state stores ca. 20 kcal mol−1 of strain energy relative to the pG state, which corresponds to about 1/3 of the photon energy (446 nm = 64 kcal mol−1). The pR0 state is stabilized by a hydrogen bond between the carbonyl group of pCA and the backbone of Cys-69 (Fig. 2), as suggested by Schotte et al.10 Consistent with time-resolved X-ray crystallographic experiments, we also observe a transient formation of a planar pR1-like species during the trajectory, suggesting that the pR0/pR1 species may reside in (quasi)-equilibrium during turnover (see Movie S1(C and D), ESI†). Although the strain energy of the pR1 state is lower relative to pR0, the intrinsic energy of the chromophores in the two states are similar, due to dissociation of the backbone hydrogen bond between pCA and Cys-69 (Fig. 2). In the pR2 state, we observe ca. 5–10 kcal mol−1 release of strain energy relative pR1 by further relaxing the Cβ–Sγ–C1–C2 dihedral angle (Table 1).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| C1C2C3C1′ | O1C1C2C3 | SγC1C2C3 | Y42-O4′ | E46-O4′ | T50-Y42 | C69-O1 | |
| a Hydrogen bond broken. | |||||||
| pGC | 168 | −10 | 176 | 2.52 | 2.56 | 2.91 | 2.79 |
| pGQ | 165 ± 8 | 0 ± 10 | 182 ± 11 | 2.52 ± 0.08 | 2.64 ± 0.10 | 2.73 ± 0.10 | 3.00 ± 0.26 |
| pRC0 | 33 | 29 | −154 | 2.59 | 2.73 | 2.95 | 3.05 |
| pRQ0 | 28 ± 18 | 29 ± 12 | −153 ± 10 | 2.55 ± 0.09 | 2.69 ± 0.11 | 2.82 ± 0.15 | 2.85 ± 0.15 |
| pRC1 | −8 | 4 | −174 | 2.57 | 2.64 | 2.92 | 4.91a |
| pRQ1 | 11 ± 13 | 15 ± 10 | −159 ± 9 | 2.53 ± 0.08 | 2.68 ± 0.13 | 2.73 ± 0.09 | 4.10 ± 0.26 |
| pRC2 | 2 | 4 | −175 | 2.47 | 2.61 | 2.92 | 3.88 |
| pRQ2 | 16 ± 11 | 5 ± 11 | −178 ± 9 | 2.54 ± 0.08 | 2.69 ± 12 | 2.78 ± 0.11 | 3.49 ± 0.32 |
We also performed QM/MM MD simulations on the IT structure of Jung et al.16 to study how the dynamics depends on the employed X-ray model. Consistent with our earlier observations11 we find that the refined IT structure is unstable and the dihedral angle around the C2C3 rapidly relaxed from 80° to 34°, and stays in this conformation during the 5 ps QM/MM simulation trajectory (Fig. S4, ESI†). The two X-ray models thus result in very similar conformational dynamics.
The hydrogen bonding distances between pCA, and Tyr-42 and Glu-46, are shown in Fig. 3 for the four studied photocycle intermediates. We observe short hydrogen-bonding distances (2.5 Å, Table 1) and partial proton transfer between Tyr-46 and the pCA, which becomes more pronounced in the pR1 and pR2 states as strain energy is released from the chromophore. A possible reason is that the proton affinity of the pCA increases with the decrease in strain due to flow of electron density from the double bond to the phenol ring (Fig. S3, ESI†). The hydrogen bond between Glu-46 and the pCA is also short (2.6 Å), but in contrast to Tyr-42, we do not observe proton transfer between the two former residues.
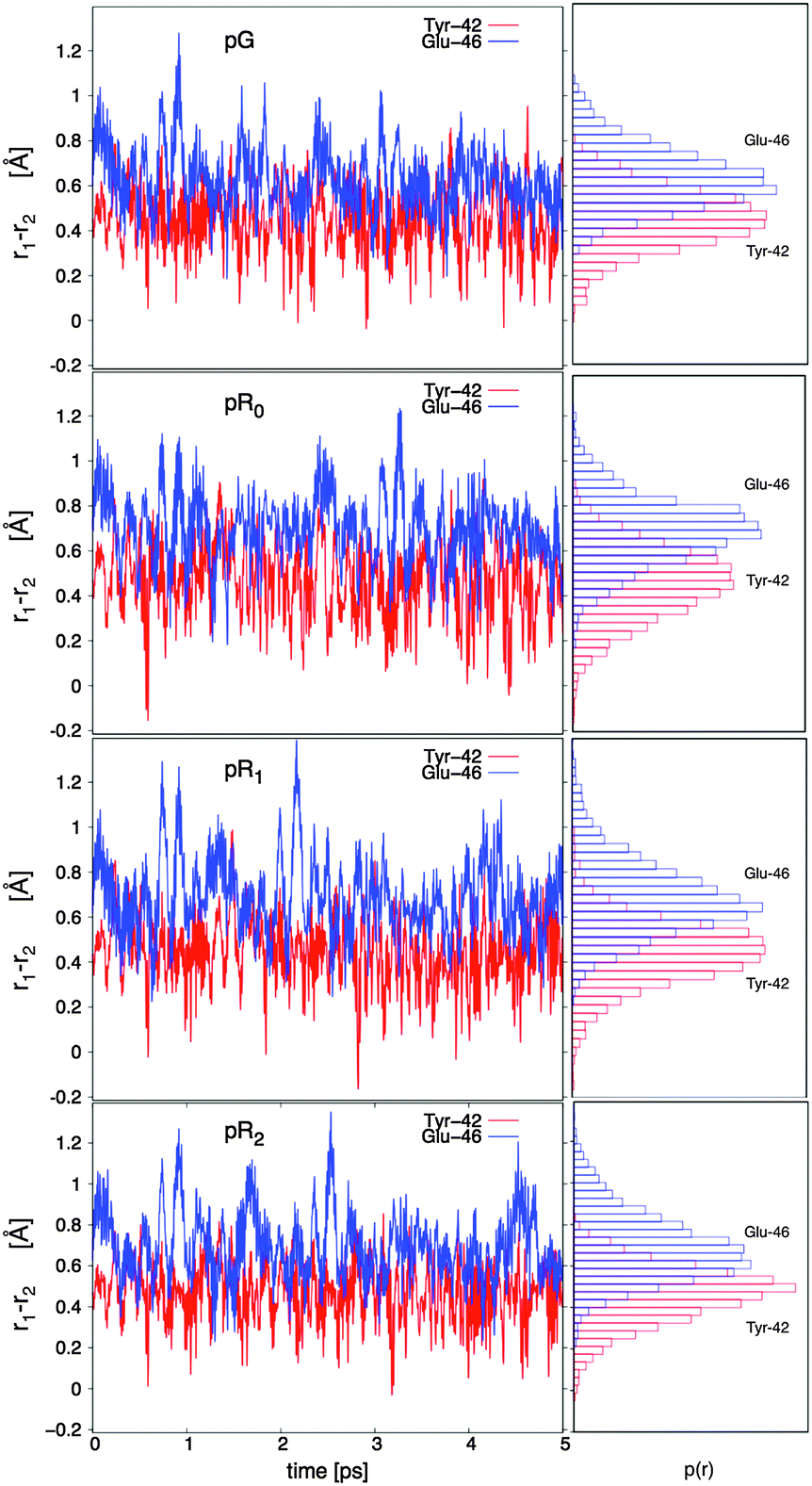 | ||
| Fig. 3 Dynamics of hydrogen bonding distances in the pG, pR0, pR1, and pR2 states. The proton coordinate r = r1(TyrO-H/GluO-H) – r2(pCA:O⋯H) (in Å), with the Tyr-42 and Glu-46 bonds shown in red and blue, respectively. r < 0 indicates that the proton is closer to pCA than to Glu-42/Tyr-46. Right inset: Probability distributions of r, showing that the Tyr-42 is shorter relative to the Glu-42 bond. | ||
Yamaguchi et al.42 refined the position of hydrogen (deuterium) atoms in the pG state using neutron diffraction experiments of deuterated samples, and observed an unusually short hydrogen (deuterium) bond of 1.38 Å between the Glu-42 and the pCA. Saito and Ishikita43,44 studied the energetics of the proton transfer between Glu-42 and pCA by hybrid QM/MM optimizations, and observed short distances between the residues only upon dissociation of Tyr-42 from pCA. They also found a slightly uphill proton transfer barrier between Tyr-42 and the pCA, which seems consistent with our dynamics simulations, where the proton transiently fluctuates towards the pCA (Fig. 3). To study the distance distribution of hydrogen bonds observed in the different photocycle states, we computed the correlation between the proton donor (D – phenolic/carboxylic oxygen of Tyr or Glu) and proton acceptor (A – phenolic oxygen of pCA) distances, with the A–H (acceptor–proton) and D–H (donor–proton) distances. This correlation plot, shown in Fig. 4, suggests that short hydrogen bonding distances, indicated by blue dotted lines, are observed for Tyr-42, but not for Glu-46. Our simulations further suggest the short distances are observed both for the D–H (closed circles) and A–H (open circles) bonds when the A–D distance is <2.5 Å. It is, however, possible that the experimentally observed short distances between the pCA and Glu-46 could arise from dynamically averaged fluctuations of multiple positions relative to the static heavy atom positions obtained from the X-ray structure in the neutron diffraction refinement. Such short bonds have, e.g., been observed in high resolution X-ray structures of aquaporin.45
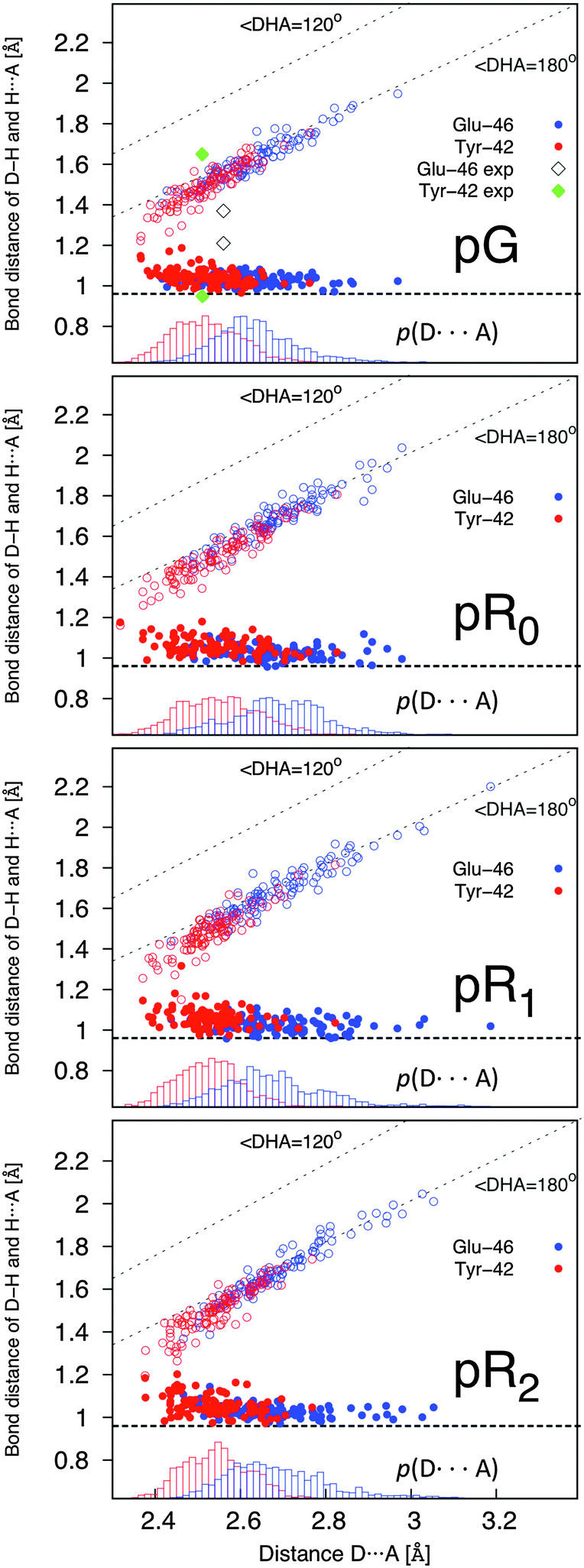 | ||
| Fig. 4 Correlation of the D–H and A–H bond with the D–A distance (in Å) in the pG, pR0, pR1, and pR2 states. The D, H, and A, refer to the phenolic/carboxylic oxygen of Tyr-42/Glu-46; the proton between these residues and the pCA; and the phenolic proton of the pCA, respectively. Distances obtained for the Glu and Tyr bonds are shown in blue and red, respectively, with bonds originating from the D–H distance (in open circles) and A⋯H distance (in closed circles). Lower inset in each subfigure shows probability distribution of the D–A distance. The hydrogen (deuterium) distances for the pCA refined by Yamaguchi et al.42 are shown as black (Glu) and green diamonds (Tyr). The correlation plots suggest that short hydrogen bonds are observed between Tyr-42 and the pCA, when the heavy atom distance is <2.5 Å. | ||
Optical properties of PYP photocycle intermediates
Fig. 5 shows the dynamically averaged absorption spectra computed at the TDDFT and CC2 levels of theory for the pG, pR0, pR1, and pR2 states. The computed absorption maxima are compared to experimental spectra,9 summarized in Table 2. We find an excellent agreement between the experimental and computational data. The expected absolute error of the employed electronic structure methods is around 0.2 eV (or 40 nm in the 450–500 nm regime) based on benchmarking calculations, which may lead to small discrepancies between the first-principles computed and measured spectra. The quantitative agreement allows us to computationally characterize the likely reason for the observed optical shifts.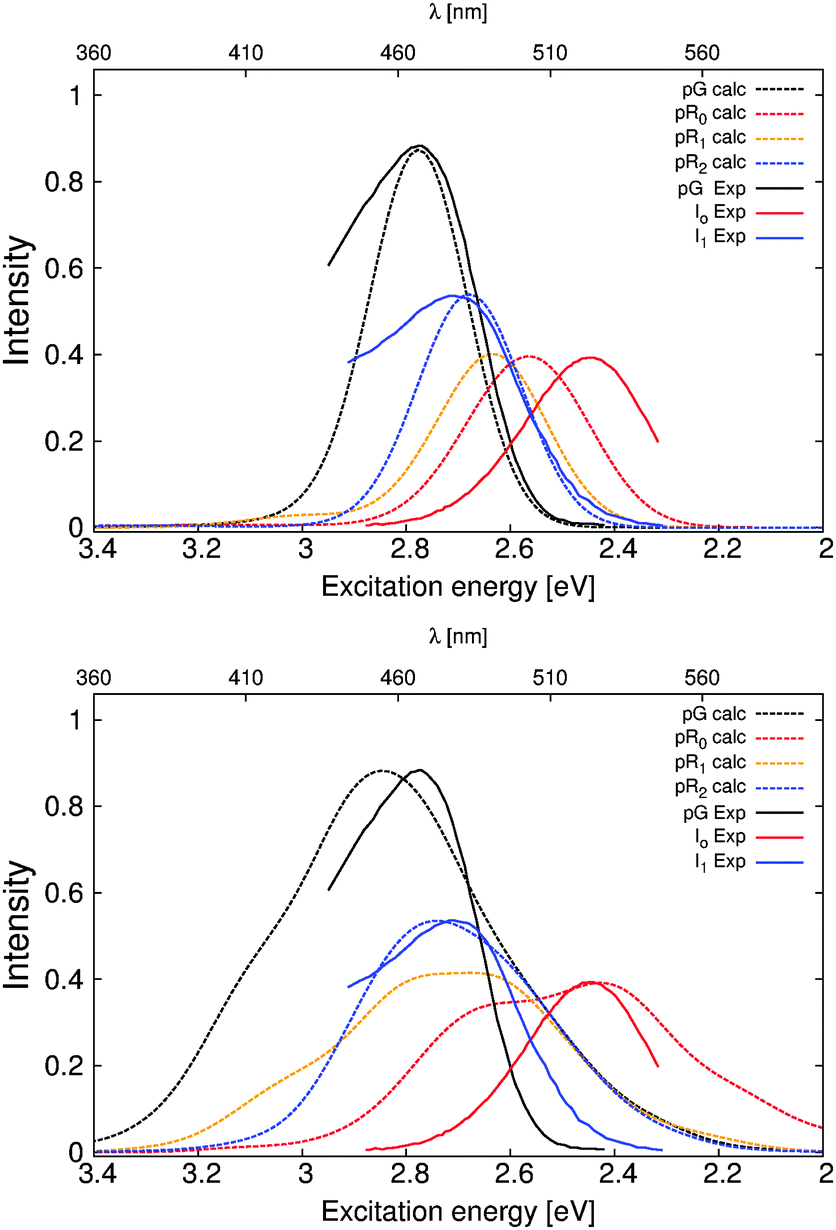 | ||
| Fig. 5 Comparison of the calculated and experimental absorption spectra obtained from ref. 9 of the PYP photocycle intermediates. TDDFT (CAM-B3LYP/def2-SVP) and CC2 (RVS/CC2/def2-TZVP) values are shown in the upper and lower figures, respectively. | ||
| Protein (nm) | Experiment (nm) | Isolated (nm) | Strain (eV) | Electrostatics (eV) | ||
|---|---|---|---|---|---|---|
| pG | TDDFT | 446 | 452 | 458 | 0.12 | 0.08 |
| CC2 | 428 | 465 | 0.18 | 0.13 | ||
| pR0 | TDDFT | 485 | 506 | 512 | 0.41 | 0.16 |
| CC2 | 483 | 544 | 0.61 | 0.18 | ||
| pR1 | TDDFT | 472 | 506 | 491 | 0.29 | 0.12 |
| CC2 | 437 | 501 | 0.37 | 0.16 | ||
| pR2 | TDDFT | 460 | 464 | 475 | 0.19 | 0.10 |
| CC2 | 443 | 479 | 0.26 | 0.06 |
The dynamically averaged absorption spectra obtained for the pG, pR0, pR1, and pR2 states are shown in Fig. 6 relative to the absorption properties of the pCA chromophore in gas-phase (dotted line) at the TDDFT/CAM-B3LYP and RVS/CC2 levels of theory. To separate strain and electrostatic polarization effects, we computed the absorption properties of the pCA chromophore in its protein conformation by removing the electrostatically polarizing protein environment around the chromophore (red, isolated), and by computing the absorption properties of the chromophore embedded in its native protein environment (blue, protein).
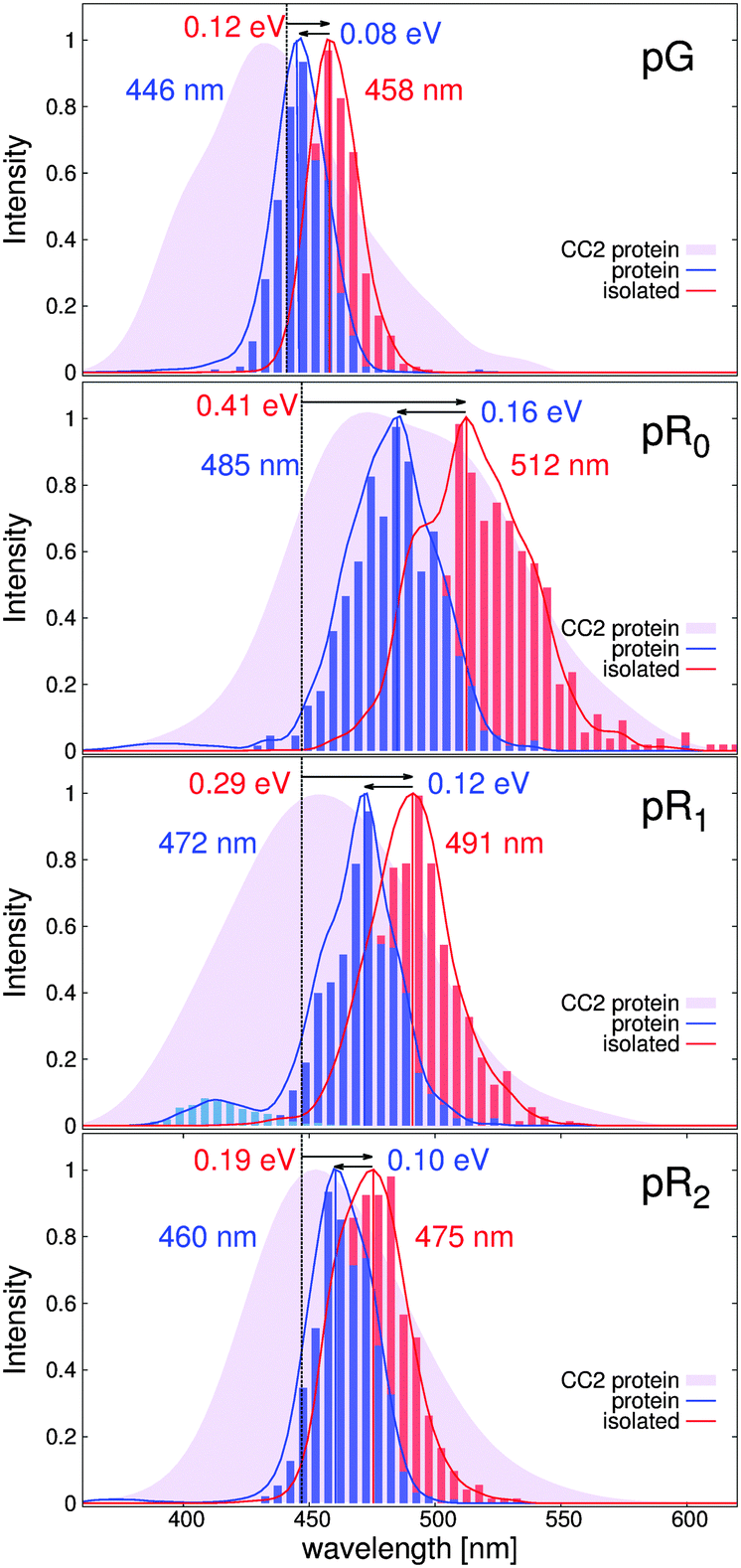 | ||
| Fig. 6 The computed ensemble averaged spectra of the four photocycle intermediates pG, pR0, pR1, and pR2 obtained from hybrid QM/MM MD simulations. The absorption maximum of the isolated and protein embedded chromophore are shown in red and blue, respectively, indicating strain (arrow and red values in eV) and electrostatic (arrow and blue values in eV) induced optical shifts. The vertical dotted line shows the averaged absorption maximum for pCA in the trans (pG), and cis (pR0, pR1, and pR2) conformations. The CAM-B3LYP/def2-SVP spectra are computed as a sum of individual Gaussians, weighted with their respective oscillator strength (solid lines) and as histograms of the VEEs obtained from the MD simulations (vertical bars). The spectra obtained at the RVS/CC2/def2-TZVP level is shown in transparent purple in the background. | ||
The TDDFT (CC2) calculations predict that the absorption spectrum of the pG state at 446 nm (430 nm) is redshifted by 0.12 eV (0.18 eV) due to strain effects relative to the gas-phase absorption of the trans-pCA at 460 nm.46 The effect is counter-balanced by a 0.08 eV (0.13 eV) blueshift due to electrostatic stabilization of the surrounding protein environment. Formation of the pR0 intermediate leads to a large strain-induced redshifted of 0.41 eV (0.61 eV), consistently with the destabilization of the electronic ground state, discussed above. The large strain effect is somewhat counter balanced by the blue-shifting effect of 0.16 eV (0.18 eV) by the protein environment, resulting in absorption peak at around 485 nm (483 nm). Relaxation of this strain energy by 0.1–0.2 eV leads to a gradual blueshift of the predicted spectra in pR1 and pR2. Interestingly, we find that the partial proton transfer between Tyr-42 and pCA leads to a small population of blueshifted intermediates in the pR0 and pR1 states, an effect that becomes more pronounced when the pCA chromophore relaxes. This effect might arise from the increased oscillator strength with decreasing chromophore strain, as suggested by model calculation on the isolated pCA chromophore.
We also computed the absorption spectrum of the 5 ps QM/MM simulation of the IT model by Jung et al.16 As expected from the similar conformational dynamics (Fig. S4, ESI†), we also obtain nearly identical spectra for the IT and pR0 trajectories (Fig. S5, ESI†), further supporting that the early 150 ps intermediate leads to large redshift due to strain-induced effects in the pCA.
Our calculations suggest that the molecular strain redshifts all intermediates, while electrostatic polarization has a blueshifting effect on the absorption properties. However, we observe that in contrast to the electrostatic effect, which is 0.08–0.16 eV in all intermediates, the main spectral shift of the pCA chromophore originates from the molecular strain effects, which has a maximum in the recently characterized pR0 state.
4. Conclusions
In conclusion, we have studied here the structure and dynamics of early photocycle intermediates of PYP, and identified how strain energy, originating from light-absorption is stored in the pCA chromophore using a combination of QM/MM molecular dynamics simulations, and first principles prediction of optical properties. Our data suggest that the recently identified pR0 state is stable on picosecond timescales, and that the highly strained species is stabilized by hydrogen bonds to the protein environment, which allows the intermediate to store ca. 1/3 of the photon energy as molecular strain. Optically, we find that this strained intermediate leads to formation of redshifted species, followed by gradual blueshifting of the spectra with relaxation of the strain. Our simulations also identified short hydrogen bonds and partial proton transfer between Tyr-42 and the pCA chromophore, leading to a small population of blueshifted species in both the computed and experimental optical spectra. While reproducing well the observed optical shifts, our work cannot be used to unambiguously assign the structures responsible for the early steps of the PYP photocycle (ref. 10, 11, 16 and 17) due to the intrinsic computational errors involved in the employed computational methodology. Our work, nevertheless, clearly shows how the strain effects contribute to the optical tuning in the early PYP photocycle intermediates, providing a powerful methodology to study elementary energy conversion steps in the photocycle of PYP.Abbreviations
| pCA | p-Coumaric acid |
| PYP | Photoactive yellow protein |
| CC2 | Second-order approximate coupled cluster |
| RVS | Reduced virtual space |
| QM/MM | Quantum mechanics/molecular mechanics |
| GS | Ground state |
| ES | Excited state |
| VEE | Vertical excitation energy |
Acknowledgements
We thank Dr Philip Anfinrud, Dr Friedrich Schotte and Dr Hyun-Sun Cho at National Institutes of Health (NIH) for stimulating discussion on the PYP photocycle. The Finnish IT Center for Science (CSC) and the Leibniz Supercomputing Centre (LRZ). We thank the Jane and Aatos Erkko foundations for financial support.Notes and references
- T. E. Meyer, Biochim. Biophys. Acta, 1985, 806, 175–183 CrossRef CAS.
- E. Hustede, M. Liebergesell and H. G. Schlegel, Photochem. Photobiol., 1989, 50, 809–815 CrossRef.
- W. W. Sprenger, W. D. Hoff, J. P. Armitage and K. J. Hellingwerf, J. Bacteriol., 1993, 175, 3096–3104 CAS.
- K. J. Hellingwerf, J. Hendriks and T. Gensch, J. Phys. Chem. A, 2003, 107, 1082–1094 CrossRef CAS.
- T. E. Meyer, E. Yakali, M. A. Cusanovich and G. Tollin, Biochemistry, 1987, 26, 418–423 CrossRef CAS PubMed.
- T. E. Meyer, G. Tollin, J. H. Hazzard and M. A. Cusanovich, Biophys. J., 1989, 56, 559–564 CrossRef CAS PubMed.
- T. E. Meyer, G. Tollin, T. P. Causgrove, P. Cheng and R. E. Blankenship, Biophys. J., 1991, 59, 988–991 CrossRef CAS PubMed.
- W. D. Hoff, I. H. van Stokkum, H. J. van Ramesdonk, M. E. van Brederode, A. M. Brouwer, J. C. Fitch, T. E. Meyer, R. van Grondelle and K. J. Hellingwerf, Biophys. J., 1994, 67, 1691–1705 CrossRef CAS PubMed.
- L. Ujj, S. Devanathan, T. E. Meyer, M. A. Cusanovich, G. Tollin and G. H. Atkinson, Biophys. J., 1998, 75, 406–412 CrossRef CAS PubMed.
- F. Schotte, H. S. Cho, V. R. I. Kaila, H. Kamikubo, N. Dashdorj, E. R. Henry, T. J. Graber, R. Henning, M. Wulff, G. Hummer, M. Kataoka and P. A. Anfinrud, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19256–19261 CrossRef CAS PubMed.
- V. R. I. Kaila, F. Schotte, H. S. Cho, G. Hummer and P. A. Anfinrud, Nat. Chem., 2014, 6, 258–259 CrossRef CAS PubMed.
- H. Ihee, S. Rajagopal, V. Šrajer, R. Pahl, S. Anderson, M. Schmidt, F. Schotte, P. A. Anfinrud, M. Wulff and K. Moffat, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7145–7150 CrossRef CAS PubMed.
- U. K. Genick, G. E. O. Borgstahl, K. Ng, Z. Ren, C. Pradervand, P. M. Burke, V. Šrajer, T.-Y. Teng, W. Schildkamp, D. E. McRee, K. Moffat and E. D. Getzoff, Science, 1997, 275, 1471–1475 CrossRef CAS PubMed.
- B. Perman, V. Šrajer, Z. Ren, T.-Y. Teng, C. Pradervand, T. Ursby, D. Bourgeois, F. Schotte, M. Wulff, R. Kort, K. Hellingwerf and K. Moffat, Science, 1998, 279, 1946–1950 CrossRef CAS PubMed.
- S. Anderson, S. Crosson and K. Moffat, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1008–1016 CrossRef PubMed.
- Y. O. Jung, J. H. Lee, J. Kim, M. Schmidt, K. Moffat, V. Srajer and H. Ihee, Nat. Chem., 2013, 5, 212–220 CrossRef CAS PubMed.
- Y. O. Jung, J. H. Lee, J. Kim, M. Schmidt, K. Moffat, V. Srajer and H. Ihee, Nat. Chem., 2014, 6, 259–260 CrossRef CAS PubMed.
- G. Wald, Nature, 1968, 219, 800–807 CrossRef CAS PubMed.
- M. Wanko, M. Hoffmann, P. Strodel, A. Koslowski, W. Thiel, F. Neese, T. Frauenheim and M. Elstner, J. Phys. Chem. B, 2005, 109(8), 3606–3615 CrossRef CAS PubMed.
- P. Altoe, A. Cembran, M. Olivucci and M. Garavelli, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20172–20177 CrossRef CAS PubMed.
- V. R. I. Kaila, R. Send and D. Sundholm, Phys. Chem. Chem. Phys., 2013, 15(13), 4491–4495 RSC.
- X. Zhou, D. Sundholm, T. Wesołowski and V. R. I. Kaila, J. Am. Chem. Soc., 2014, 136, 2723–2726 CrossRef CAS PubMed.
- A. M. Virshup, B. G. Levine and T. J. Martinez, Theor. Chem. Acc., 2014, 133, 1506 CrossRef.
- S. Sekharan and K. Morokuma, J. Am. Chem. Soc., 2011, 133, 4734–4737 CrossRef CAS PubMed.
- C. M. Isborn, A. W. Götz, M. A. Clark, R. C. Walker and T. J. Martínez, J. Chem. Theory Comput., 2012, 8, 5092–5106 CrossRef CAS PubMed.
- L. González, D. Escudero and L. Serrano-Andrés, ChemPhysChem, 2012, 13, 28–51 CrossRef PubMed.
- C. Filippi, F. Buda, L. Guidoni and A. Sinicropi, J. Chem. Theory Comput., 2012, 8(1), 112–124 CrossRef CAS.
- A. P. Gamiz-Hernandez, A. Neycheva, R. Send, D. Sundholm and V. R. I. Kaila, Angew. Chem., Int. Ed., 2015, 54, 11564–11566 CrossRef CAS PubMed.
- H. M. Senn and W. Thiel, Angew. Chem., Int. Ed., 2009, 48, 1198–1229 CrossRef CAS PubMed.
- F. Shirin, G. Gerrit and A. Dreuw, J. Phys. Chem. B, 2013, 117(35), 10071–10079 CrossRef PubMed.
- P. Campomanes, M. Neri, B. A. C. Horta, U. F. Röhrig, V. Stefano, T. Ivano and U. Röthlisberger, J. Am. Chem. Soc., 2014, 136(10), 3842–3851 CrossRef CAS PubMed.
- T. A. Wesolowski and A. Warshel, J. Phys. Chem., 1993, 97, 8050–8053 CrossRef CAS.
- J. Neugebauer, ChemPhysChem, 2009, 10(18), 3148–3173 CrossRef CAS PubMed.
- V. R. I. Kaila, R. Send and D. Sundholm, Phys. Chem. Chem. Phys., 2013, 4491–4495 RSC.
- R. Send, C.-M. Suomivuori, V. R. I. Kaila and D. Sundholm, J. Phys. Chem. B, 2015, 119(7), 2933–2945 CrossRef CAS PubMed.
- K. Sneskov, T. Schwabe, O. Christiansen and J. Kongsted, Phys. Chem. Chem. Phys., 2011, 13, 18551–18560 RSC.
- B. R. Brooks, C. L. Brooks, A. D. Mackerell, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- R. Send, V. R. I. Kaila and D. Sundholm, J. Chem. Phys., 2011, 134, 214114 CrossRef PubMed.
- H. L. Woodcock, M. Hodošček, A. T. B. Gilbert, P. M. W. Gill, H. F. Schaefer and B. R. Brooks, J. Comput. Chem., 2007, 28, 1485–1502 CrossRef CAS PubMed.
- Y. Shao, L. Fusti Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O'Neill, R. A. Distasio Jr., R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Y. Lin, T. Van Voorhis, S. H. Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C.-P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. M. Rhee, J. Ritchie, E. Rosta, C. D. Sherrill, A. C. Simmonett, J. E. Subotnik, H. L. Woodcock III, W. Zhang, A. T. Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer III, J. Kong, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 3172–3191 RSC.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- S. Yamaguchi, H. Kamikubo, K. Kurihara, R. Kuroki, N. Niimura, N. Shimizu, Y. Yamazaki and M. Kataoka, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 440–444 CrossRef CAS PubMed.
- K. Saito and H. Ishikita, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 167–172 CrossRef CAS PubMed.
- K. Saito and H. Ishikita, Biochim. Biophys. Acta, 2013, 1827, 387–394 CrossRef CAS PubMed.
- U. Kosinska Eriksson, G. Fischer, R. Friemann, G. Enkavi, E. Tajkhorshid and R. Neutze, Science, 2013, 340, 1346–1349 CrossRef CAS PubMed.
- I. B. Nielsen, S. Boyé-Péronne, M. O. A. El Ghazaly, M. B. Kristensen, S. Brøndsted Nielsen and L. H. Andersen, Biophys. J., 2005, 89, 2597–2604 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S5 and Movies S1 and S2. See DOI: 10.1039/c5cp05244k |
| This journal is © the Owner Societies 2016 |