
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protein docking using an ensemble of spin labels optimized by intra-molecular paramagnetic relaxation enhancement†
Jesika
Schilder
a,
Wei-Min
Liu
a,
Pravin
Kumar
b,
Mark
Overhand
a,
Martina
Huber
b and
Marcellus
Ubbink
*a
aLeiden Institute of Chemistry, Leiden University, Gorlaeus Laboratories, Einsteinweg 55, 2333 CC Leiden, The Netherlands. E-mail: m.ubbink@chem.leidenuniv.nl
bDepartment of Physics, Huygens-Kamerlingh Onnes Laboratory, Leiden University, Niels Bohrweg 2, 2333 CA Leiden, The Netherlands
First published on 3rd September 2015
Abstract
Paramagnetic NMR is a useful technique to study proteins and protein complexes and the use of paramagnetic relaxation enhancement (PRE) for this purpose has become wide-spread. PREs are commonly generated using paramagnetic spin labels (SLs) that contain an unpaired electron in the form of a nitroxide radical, with 1-oxyl-2,2,5,5-tetramethyl-2,5-dihydropyrrol-3-ylmethyl methane thiosulfonate (MTSL) being the most popular tag. The inherent flexibility of the SL causes sampling of several conformations in solution, which can be problematic as over- or underestimation of the spatial distribution of the unpaired electron in structural calculations will lead to errors in the distance restraints. We investigated the effect of this mobility on the accuracy of protein–protein docking calculations using intermolecular PRE data by comparing MTSL and the less mobile 3-methanesulfonilthiomethyl-4-(pyridin-3-yl)-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-1-yloxyl (pyMTSL) on the dynamic complex of cytochrome c and cytochrome c peroxidase. No significant differences were found between the two SLs. Docking was performed using either single or multiple conformers and either fixed or flexible SLs. It was found that mobility of the SLs is the limiting factor for obtaining accurate solutions. Optimization of SL conformer orientations using intra-molecular PRE improves the accuracy of docking.
Introduction
Paramagnetic NMR is a convenient approach for determining the binding site and orientation of proteins within low-affinity complexes that undergo minimal structural changes upon binding. Various types of paramagnetic restraints can be used, such as pseudocontact shifts (PCS), residual dipolar couplings (RDC) induced by the alignment caused by the paramagnetic centre, or paramagnetic relaxation enhancement (PRE).1,2 PRE is the most popular choice due to the simplicity of introducing relaxation centres to proteins, mainly in the form of site-specific tags, of which spin labels (SLs) are the most common. SLs are small organic compounds that contain an unpaired electron in the form of a nitroxide radical and are generally quite stable under non-reducing conditions.3 The electron spin can be observed directly using electron paramagnetic resonance (EPR) or indirectly by measuring the PRE effects on nearby nuclei via nuclear magnetic resonance (NMR) spectroscopy. The observed nuclear relaxation rates can either be used directly or after conversion into distances for structural modelling.4,5To convert the observed relaxation rates into distances, the correlation time (τc) of the vector r that connects the paramagnetic centre and the nucleus is required. τc depends of the rotational correlation time of this vector (τr) as well as the longitudinal electronic relaxation time (τs), according to τc−1 = τr−1 + τs−1.4,5 For spin labels, the contribution of τs is small and τc is dominated by τr. Any motions that change the vector will have an effect on τc, such as protein tumbling, SL mobility, local protein dynamics, and, for intermolecular PREs measured in a complex, the motions of one protein relative to the other.6 Thus, it is not straight forward to determine τc but, fortunately, due to the sixth power dependence of the PRE on the distance r between spin label and the nucleus, errors in τc result in only small errors in the distances.7,8
The differences between the free energies of SL conformations are often smaller than the thermal energy in the sample, meaning that the position of the unpaired electron is spatially distributed over an area determined by the occupied SL conformer orientations.6 Over- or underestimation of the spatial distribution of the free electron will lead to errors in the apparent mean distance, [〈r−6〉−1/6].7 To solve this problem, the SL can be treated as an ensemble of non-self-interacting conformers during simulated annealing calculations.7 In this case, the number of conformers required to make up the ensemble depends on the actual spatial distribution of the SL in solution as well as the precision required to match the experimental data for nearby nuclei, which will experience the strongest PRE. However, using an ensemble could potentially generate worse results if non-realistic conformer orientations are used, leading to an inaccurate structure of the protein complex.6 This can be overcome by first determining the most favourable conformer orientations experimentally using intra-molecular PRE data, followed by fixing the SL conformers in those positions during docking calculations based on inter-molecular PRE data. This has yielded good results using as few as one SL conformer for multi-domain proteins8,9 and has also been used to study DNA–protein interactions.10
Due to its wide commercial availability, as well as the availability of a suitable diamagnetic control, the most commonly used SL is 1-oxyl-2,2,5,5-tetramethyl-2,5-dihydropyrrol-3-ylmethyl methane thiosulfonate (MTSL). However, MTSL has a rather long linker consisting of five single bonds from the peptide Cα atom to the 3-pyrolline ring (Fig. 1B), resulting in substantial flexibility and dynamics.
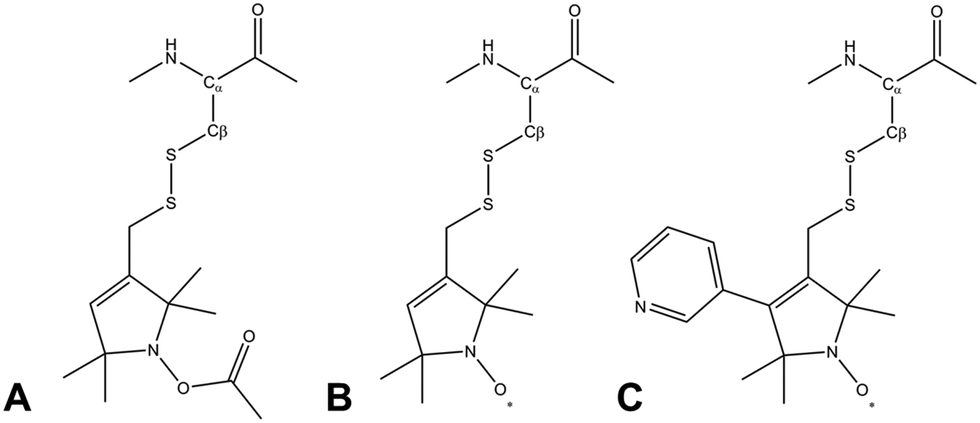 | ||
| Fig. 1 Molecular structures of three methanesulfonothioate based tags attached to a cysteine side chain. (A) MTS (diamagnetic control), (B) MTSL and (C) pyMTSL.11 | ||
In order to limit tag dynamics, the pyrolline ring of MTSL can be modified to increase the tag rigidity.6 This was successfully done via the addition of a pyridyl group to position 4 of the pyrolline ring resulting in 3-methanesulfonilthiomethyl-4-(pyridin-3-yl)-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-1-yloxyl (pyMTSL; also called HO-3606) (Fig. 1C). It was reported that this addition restricts the SL movement, such that the PRE data could be fit using just one conformer.11
The yeast cytochrome c (Cc) and cytochrome c peroxidase (CcP) complex is a well-studied, highly dynamic electron transfer complex. CcP catalyzes the reduction of hydrogen peroxide to water using two electrons it receives from Cc.12 The crystal structure was determined in 1992, showing how Cc is docked on CcP.13 The orientation of Cc relative to CcP in the complex in solution was determined by Volkov et al. in 2006 by attaching MTSL at five positions on CcP. After confirming that the presence of the SLs did not interfere with complex formation, the PRE effects were measured for nuclei in Cc. The orientation of Cc was determined using rigid body docking with an ensemble of four MTSL conformers per position in orientations selected to represent the width of the ensemble of sterically allowed conformers, yielding a position of Cc that was close to the one observed in the X-ray crystal structure (RMSD = 2.2 Å for the Cα atoms of Cc).14 The data also provided evidence for the presence of an encounter complex, in which Cc assumed other orientations relative to CcP. In 2010 Bashir et al., described similar rigid body docking calculations in which the MTSL conformers at each position were treated as an ensemble of non-self-interacting conformers that could move freely during the docking, as could the nearby amino acid side chains. The result was a less precise protein complex ensemble, in which extreme positions for Cc were found within the ensemble when all the MTSL conformers were simultaneously orientated to one side or another; this effect appeared to have been averaged out in the previous study when the conformers were fixed in four dispersed orientations.15
In this work, we revisit the Cc–CcP complex using MTSL and pyMTSL tags at three positions close to the binding interface on the surface of CcP. The aim of this work is to establish whether PRE data alone are sufficient for accurate rigid body docking of two proteins that form a dynamic complex. The Cc–CcP complex spends approximately 30% of the time in an encounter state;14,16–19 therefore, the PRE represent not only the well-defined stereospecific state but also the encounter state. We investigate the role of SL mobility by comparing MTSL to pyMTSL, as well as by comparing the use of a single SL conformer to the use of an ensemble of conformers when the orientations are either fixed or mobile during the calculations. The position of Cc in the X-ray crystal structure has been used as the benchmark, under the assumption that the well-defined state of the complex in solution is very similar to the one observed in the crystalline state. It is concluded that the results of PRE based docking are highly dependent on the choice of SL conformers. Thus, the SL flexibility appears to be the limiting factor for obtaining accurate results. SL ensembles of conformers optimized by intra-molecular PRE yield the best results.
Experimental
Protein sample preparation
The genes for the yeast CcP C128A containing mutations NC38, NC200 and TC28814 were sub-cloned in the pET28aCcP plasmid and were expressed to produce CcP, which was purified as described previously.16 The yields were 40 mg L−1 for NC38 [2H15N], 20 mg L−1 for NC200 [15N] and 120 mg L−1 for TC288 [2H15N] in minimal media. A pUC19 based plasmid containing the S. cerevisiae iso-1-cytochrome c gene was used to produce Cc, which was purified according to published procedures.20,21 The yield was 20 mg L−1 [15N] in minimal media. The concentrations of CcP and Cc were determined using UV-Vis spectroscopy with ε408nm = 98 mM−1 cm−1 and ε410nm = 106.1 mM−1 cm−1, respectively.21,22Spin label preparation
MTS and MTSL tags were obtained from Toronto Research Chemicals (North York, ON, Canada) and pyMTSL was synthesized according to the published protocol.11 The SLs were stored as 100 mM stocks dissolved in DMSO at 4 °C prior to use. The CcP mutants were tagged with MTS, MTSL or pyMTSL, as described previously.14,16 The tagging efficiency was determined by mass spectroscopy to be essentially 100% and SLs at these positions have previously been shown not to interfere with Cc–CcP complex formation.14Continuous wave-EPR experiments
The SL mobility was determined using X-band continuous wave (cw) EPR measurements. These were performed using an ELEXSYS E680 spectrometer (Bruker, Rheinstetten, Germany) with a rectangular cavity. All the measurements were done at room temperature (20 °C), using 0.6346 mW (0.5 mW for N38C) microwave power, 100 kHz modulation frequency and 0.08 mT (0.05 mT for N38C) modulation amplitude. The total measurement time was between 1 and 2 h per spectrum.The cw-EPR spectra were simulated using Matlab version 7.14.0.739 (Natick, Massachusetts, USA) and the EasySpin package version 4.5.5.23 For all simulations, the following spectral parameters were used: g = [2.00906, 2.00687, 2.00300]24 and the hyperfine tensor parameters AXX = AYY = 13 MHz. Isotropic rotation was assumed in all cases, to reduce the number of simulation parameters and get a better overall picture of the relative changes in rotational properties of the spin labels. Usually a superposition of more than one component was required to simulate the spectra. One of these components has a rotation correlation time of several tens of ps and a contribution of less than 0.5% to the total simulation and is therefore assigned to a small fraction of residual free spin label in the sample.
Paramagnetic NMR spectroscopy
For the intra-molecular PRE measurements, NMR samples contained 300 μM unlabelled Cc wt and 300 μM double labelled [15N, 2H] NC38 or TC288 or 15N-labelled NC200 CcP with either MTS, MTSL or pyMTSL tags attached in 20 mM NaPi, 100 mM NaCl, 6% D2O, pH 6.0. 2D BEST-TROSY-HSQC experiments25 were recorded on a Bruker AVIII HD spectrometer equipped with a 1H[13C/15N] TCI-cryoprobe operating at a Larmor frequency of 850 MHz at 293 K with 1024 and 100 complex points in the 1H and 15N dimensions, respectively. For the inter-molecular PRE measurements, NMR samples contained 300 μM 15N-labelled Cc wt and 300 μM unlabelled NC38, NC200 or TC288 CcP with either MTS, MTSL or pyMTSL tags attached in the same buffer solution. 2D HSQC experiments were recorded on the same spectrometer and at the same temperature with 512 and 64 complex points in the 1H and 15N dimensions respectively. All data were processed using Topspin 3.2 (Bruker, Karlsruhe, Germany) and analyzed using CCPN Analysis 2.1.5.26The 1H and 15N resonance assignments were obtained from previous studies for Cc27–29 and from our previous work for CcP.16 In the early stages of this work for the intra-molecular PREs of TC288-(py)MTSL, we noticed that residue 120 experienced a very strong PRE (the paramagnetic peak disappeared from the spectra) while residues 123 and 124 did not experience any PREs. It was concluded that residues 123 and 124 had been swapped with those of residues 101 and 102. BMRB entry 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 884 containing these backbone resonance assignments has been updated.
884 containing these backbone resonance assignments has been updated.
Data analysis
The intensity ratio of the amide resonances in the spectra of the paramagnetic (MTSL- or pyMTSL-tagged) and diamagnetic (MTS-tagged) samples (Ipara/Idia) was measured and normalized as described previously.18 The paramagnetic contribution to the transverse relaxation rate, R2,para, was calculated as described previously (Tables S1 and S2, ESI†).5,16,18 The average R2,dia value was used with a large error margin for those amides for which an Ipara/Idia could be measured but for which the line width of the diamagnetic peak could be not obtained. For the amide peaks that disappeared in the paramagnetic spectrum, an upper limit for Ipara was set to two standard deviations of the noise level of the spectrum.16The calculated R2,para values were then converted into distances using (eqn (1)):18
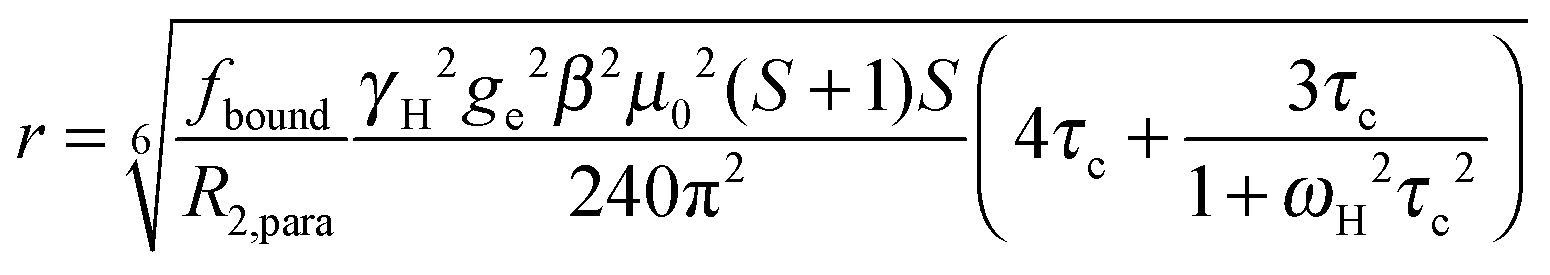 | (1) |
Optimisation of the SL orientations
The protein coordinates for CcP were taken from 1ZBY for CcP.30 The addition of surface cysteine mutations and the introduction of the SLs to the CcP structure were done in silico as described previously.14 The initial positions for the four conformers of MTSL were generated by systematic rotation of the SL around the five single bonds that join the pyrolline ring to the Cα atom of the cysteine residue and choosing four of the sterically allowed orientations for each mutant that represented the ensemble well, as described previously.14 The same orientations as reported in that article were used here. For pyMTSL, the four conformers were snapshots of short molecular dynamics runs of the SL in vacuo. Using the intra-molecular PRE data, a set of distance restraints was calculated and used to determine the most favourable SL conformer orientations for a single conformer or an ensemble of four conformers at each mutant position using Xplor-NIH version 2.34.31,32 For localizing the SL orientations on CcP, only PREs from amides within 23 Å of the SL oxygen atom were used, thus eliminating the data that do not yield relevant restraints (class III PRE). When an ensemble of four SL conformers was used, r−6 ensemble averaging was done for the distances r between the four SL positions and a nucleus. The calculations were performed in two steps using side-chain dynamics, in which all atoms were fixed apart from those of the SL and the amino side chains within 10 Å of the SL. During the first step, only van der Waals (vdW) forces were considered, followed by a second step in which both the vdW forces and the distance restraints were used. The vdW forces were defined as repel forces between the SL and the protein atoms but were set to zero between multiple SLs in the ensemble. This was repeated 1000 times and the orientation of the lowest energy was used as the experimentally determined orientation in subsequent docking calculations of Cc to CcP.Protein docking
The protein coordinates for the individual proteins were obtained from PDB 2YCC for Cc33 and 1ZBY for CcP30 and for the complex from PDB 2PCC.13 The docking of Cc to CcP was driven by a set of distance restraints derived from inter-molecular PRE data using Xplor-NIH version 2.34.31,32 This was done using either a single SL conformer or an ensemble of four SL conformers, the orientations of which were either fixed in selected positions, fixed in experimentally determined positions as described above, or were free to move during docking. Cc was docked to CcP using rigid body dynamics with vdW repel forces and the distance restraints contributing to the total energy. The vdW forces were set to zero for interactions between atoms of multiple SLs within the ensemble. Docking was repeated 10000 times and the twenty lowest energy structures were used to generate the protein complex ensemble.
The precision of the ensemble was quantified by the average of the pairwise RMSD values of members of the ensemble and its mean structure. The accuracy was assessed by comparing the mean structure of the calculated structure ensemble to the crystal structure of the complex, using both the root mean squared deviation (RMSD) and distance root mean squared deviation (DRMS) for these two structures, which is defined as (eqn (2)):34
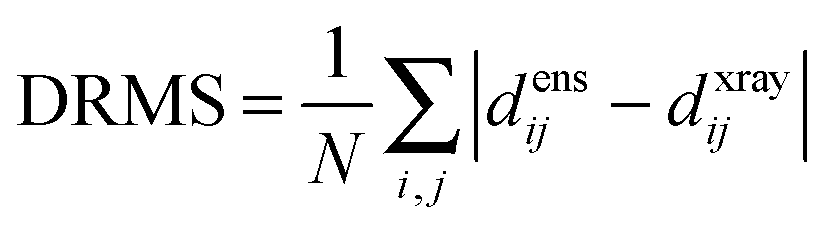 | (2) |
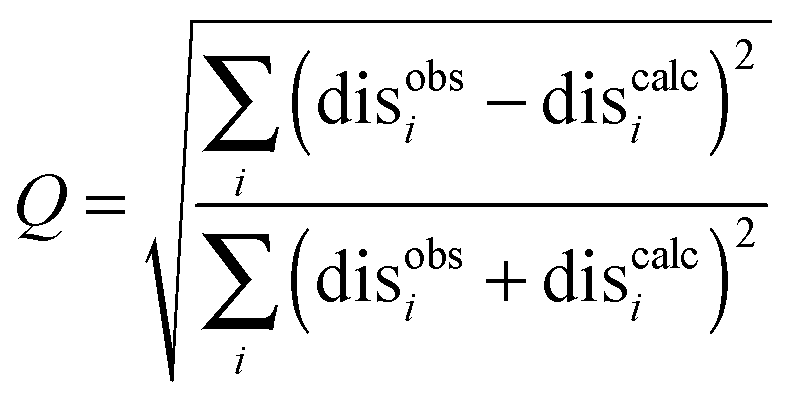 | (3) |
Results and discussion
SL mobility studied by EPR
The inherent flexibility of SL linkers allows a SL such as MTSL to occupy several conformer orientations over time,6 which can be problematic when trying to accurately determine R2,para values.7 Substitution of a bulky side group on the pyrolline ring was shown to restrict the movement of pyMTSL and reduce the number of allowed orientations.11 In order to compare the mobility of MTSL and pyMTSL, these SLs were attached to three positions on the surface of CcP, positions 38, 200 and 288 (Fig. 2).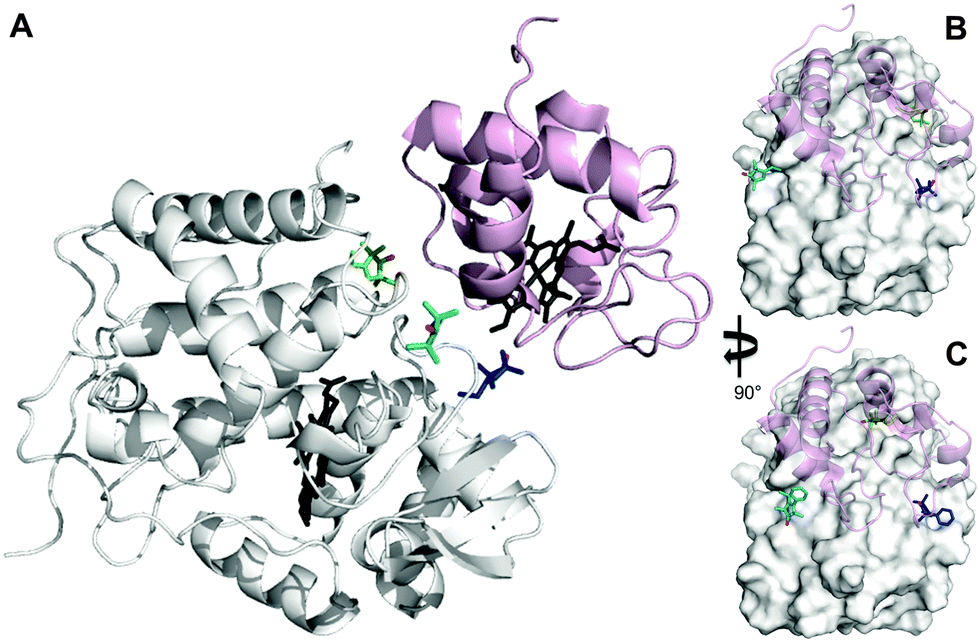 | ||
| Fig. 2 Locations of SLs attached on the surface of CcP at positions 38 (teal), 200 (blue) and 288 (green) (nitroxide oxygen atom in red). (A) The locations of three MTSL tags shown on CcP (white) with respect to the binding site of Cc. The relative orientations of MTSL (B) or pyMTSL (C) around the binding site on the surface of CcP are also shown; Cc is shown as a pink cartoon shadow. The haem groups are shown in black sticks (PDB-entry 2PCC).13 | ||
These three attachment positions ring the stereo-specific binding interface with Cc and have also previously been used to produce significant inter-molecular PRE effects on Cc.14,18 The spin-label mobility at these positions for MTSL and pyMTSL was compared using EPR measured in solution at room temperature (Fig. 3).
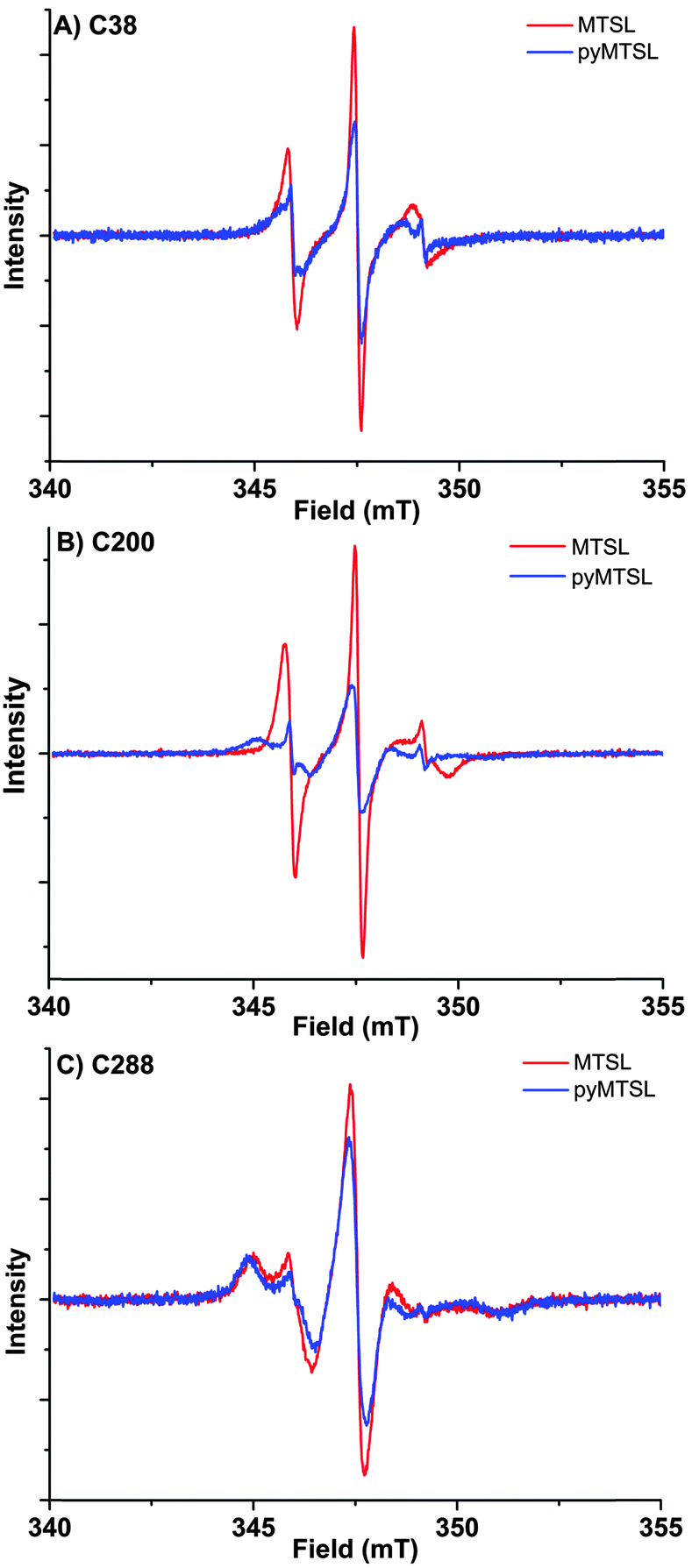 | ||
| Fig. 3 EPR spectra for MTSL (red) and pyMTSL (blue) at positions 38 (A), 200 (B) and 288 (C) on CcP. Spectra are normalized to the number of spins, which emphasises differences in line width; narrow lines have larger amplitudes. | ||
The EPR spectra of the spin labels at the three positions investigated show that the lineshapes of the nitroxides are under conditions of not fully averaged anisotropy, typical for spin-labels attached to proteins. In all cases, the spectrum of the protein labelled with MTSL at a given position has narrower lines than that of the protein labelled with pyMTSL, showing that the rotation of the nitroxide group in pyMTSL is slower than that of MTSL. More details can be seen from the parameters of the spectral simulation (Fig. S1, ESI† and Table 1).
| Component 1 | Component 2 | Free MTSLa | |||||||
|---|---|---|---|---|---|---|---|---|---|
| τ radicalr (ns) | Fraction (%) | A ZZ (MHz) | τ radicalr (ns) | Fraction (%) | A ZZ (MHz) | τ radicalr (ns) | Fraction (%) | A ZZ (MHz) | |
| a Residual free spin-label fraction, contribution shows almost complete removal of free spin label from all samples. | |||||||||
| C38 MTSL | 0.89 | 100.0 | 110.0 | n/a | n/a | n/a | n/a | n/a | n/a |
| C38 pyMTSL | 0.63 | 20.0 | 108.5 | 2.20 | 79.5 | 102 | 0.05 | 0.5 | 108.0 |
| C200 MTSL | 0.89 | 99.7 | 115.0 | n/a | n/a | n/a | 0.02 | 0.3 | 109.8 |
| C200 pyMTSL | 0.81 | 17.0 | 109.8 | 8.70 | 82.7 | 94 | 0.05 | 0.3 | 108.0 |
| C288 MTSL | 2.23 | 42.0 | 100.0 | 10.00 | 57.7 | 97 | 0.02 | 0.3 | 109.8 |
| C288 pyMTSL | 3.20 | 25.0 | 101.0 | 10.00 | 74.9 | 101 | 0.05 | 0.1 | 108.0 |
For CcP positions 38 and 200, MTSL has a rotation correlation time (τradicalr) of 0.9 ns, which is the single component contributing to these spectra. When using the pyMTSL tag, the radical has a majority contribution with a 2.5 fold (position 38C) and a more than ten-fold increased rotation correlation time (position 200C) (see Table 1). For the 288 position the MTSL label is rotating more slowly than at the other positions: two contributions are observed with almost equal weight and correlation times of 2 ns respectively 10 ns, which is significantly more immobilized than the MTSL at the other positions. Replacing MTSL by pyMTSL increases the rotation correlation time of the faster fraction to 3 ns and shifts the population to 75% of slower fraction, showing that also at this position, the pyridine substituent leads to a slowing down of the rotation.
The rotation correlation times of pyMTSL vary per position, showing that local interactions contribute to the mobility of pyMTSL and that the mobility of pyMTSL is not exclusively determined by its side-chain structure. Furthermore, none of the rotation correlation times reach the value of the protein, showing that the spin label is not completely anchored to the protein, and demonstrating that the linker connecting the nitroxide-ring to the protein backbone possesses local degrees of freedom. Note that EPR reports on the correlation time of the radical, whereas PREs reflect the rotational correlation time of the radical-nuclear vector (τr).
Experimental optimization of SL conformers
We determined the conformer orientations for both MTSL and pyMTSL using intra-molecular PREs to see if a difference could be observed between these tags.2 In order to account for potential differences in the most favourable SL orientations on free CcP compared to CcP in complex with Cc, all intramolecular PRE measurements were done in the presence of 300 μM non-isotopically labelled Cc (1:1 molar eq.), which was the same concentration as used for the intermolecular PRE measurements. The PREs were measured for the SLs at three positions of CcP and were then converted into distances between the affected nuclei and the paramagnetic centre. These distance restraints were used to determine the favourable orientations of a single conformer or an ensemble of four conformers at each position for both MTSL and pyMTSL (Fig. 4).
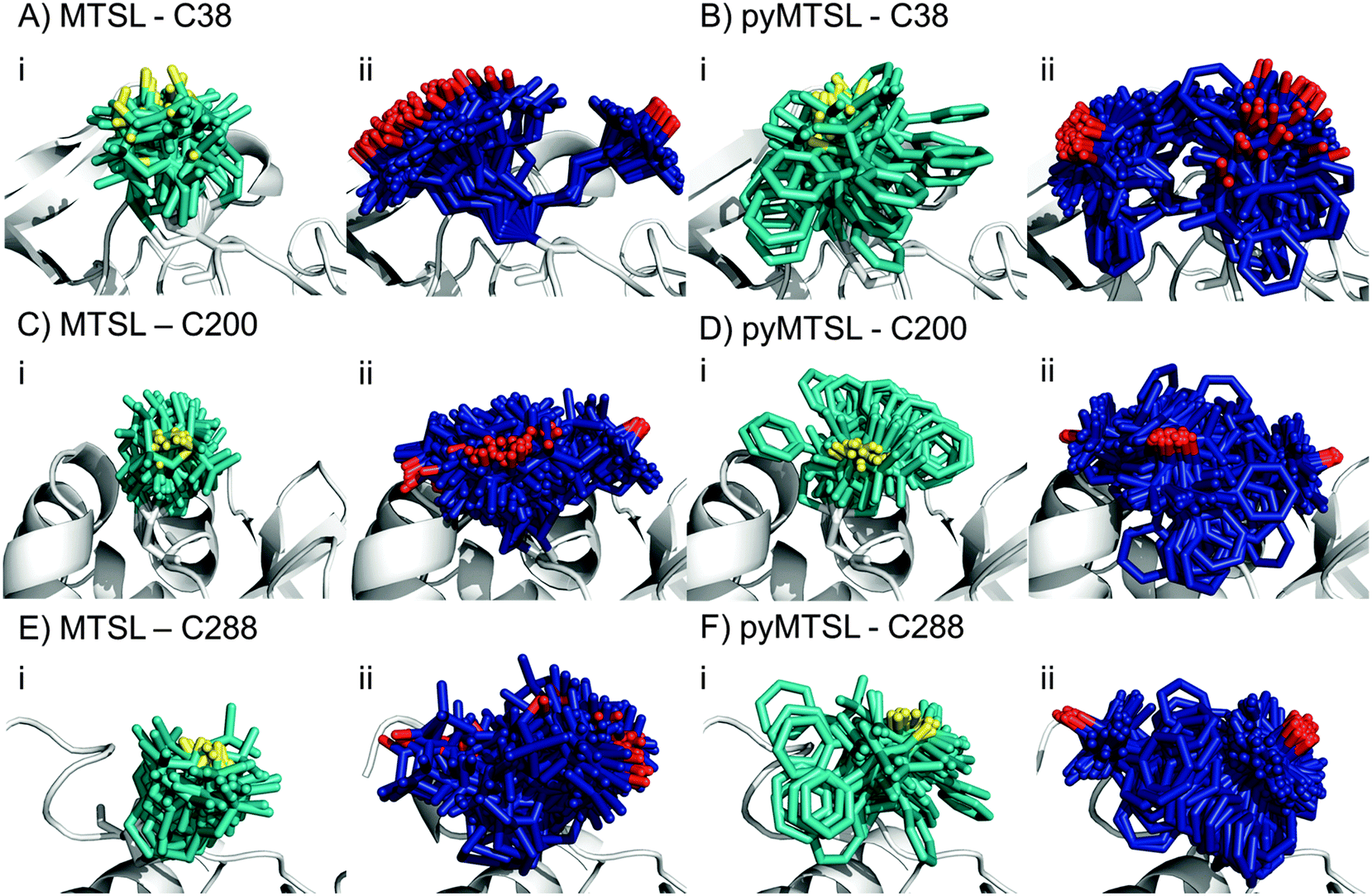 | ||
| Fig. 4 The twenty lowest energy conformer orientations found using intra-molecular PRE based distance restraints for MTSL attached at C38 (A), C200 (C) or C288 (E) or pyMTSL attached at C38 (B), C200 (D) or C288 (F) on the surface of CcP (grey ribbon). The PRE data were fit using a single conformer (i) or an ensemble of four conformers (ii). For a single conformer, the SLs are shown in cyan sticks and the nitroxide oxygen atom is shown in yellow. For the conformer ensemble, the SLs are shown in blue sticks and the nitroxide oxygen atom is shown in red. | ||
When the SL orientations were fit to the data using only a single conformer, the position of the nitroxide radical for the twenty lowest energy structures converged to a fairly precise position at all three attachment sites (Fig. 4-i). Also, little difference was seen between MTSL and pyMTSL. When using an ensemble of four conformers, however, the most favourable orientations were more dispersed and two or three distinct populations appeared (Fig. 4-ii). Table 2 reports the Q-values and average violations (see Materials and methods for the definition of these measures) of the various fits. Interestingly, the ensemble fit yields results that are only marginally better than those obtained with the single conformers. Thus, on the basis of these data it cannot be decided which is the better description of SL conformations.
| Position | MTSL | pyMTSL | |||
|---|---|---|---|---|---|
| 1 conformer | 4 conformers | 1 conformer | 4 conformers | ||
| C38 | AV (Å) | 2.94 | 2.44 | 2.83 | 2.57 |
| Q-factor | 0.20 | 0.17 | 0.18 | 0.17 | |
| C200 | AV (Å) | 1.65 | 1.61 | 1.82 | 1.80 |
| Q-factor | 0.15 | 0.14 | 0.15 | 0.14 | |
| C288 | AV (Å) | 2.35 | 2.28 | 2.16 | 2.13 |
| Q-factor | 0.18 | 0.17 | 0.16 | 0.15 | |
Essentially the same position was found for the nitroxide radical when using either MTSL or pyMTSL. For C38-MTSL (Fig. 4A and B-ii), the ensemble showed two distinct populations, one with a very well-defined location of the nitroxide radical and one more dispersed, with ratio of approximately 25%:75%. For pyMTSL, the positions are similar but the populations have a ratio closer to 50%:50%. Furthermore, the well-defined and disperse populations are swapped when compared to those for MTSL. The EPR data (Fig. 3A) showed a marked decrease in mobility for pyMTSL compared to MTSL at position C38 but this difference was not reflected in the number of calculated SL orientations. Furthermore, the populations found by NMR were not reflected in the EPR simulations (Table 1). For example, for C38 MTSL the EPR simulations found only one highly mobile population while for C38 pyMTSL the EPR simulations found two populations but in a ratio of 20.0%:79.5% as compared to roughly 50%:50% found by NMR. Differences between the ratios of populations found by EPR and NMR were also seen for both SLs at C200 and for MTSL at C288 (see discussion below).
For position C200, the EPR data showed a similar decrease in mobility for pyMTSL compared to MTSL, which is reflected in a much more defined positioning of the nitroxide radical for pyMTSL compared to MTSL in the calculated SL ensemble orientations, although the general location of the radicals was the same in both cases (Fig. 4C and D). When using an ensemble of four conformers at this position, three distinct populations were found with a ratio of about 25%:50%:25% for both SLs. However, the EPR simulations found only one population for C200 MTSL and only two populations for C200 pyMTSL with a ratio of 17.0%:82.7% (Table 1).
The NMR spectra for C288-(py)MTSL showed chemical shift perturbations for peaks of the residues near the spin label attachment site (small shifts for residues 284 and 285; larger shifts for residues 286–294) indicating that the presence of the spin label affects the local backbone structure of the C-terminal loop. Therefore, the position of the nitroxide radical was also determined by docking pseudoatom(s) to CcP using the intra-PRE data, representing the paramagnetic centre(s) unrestrained by the covalent linkage to the CcP backbone. The resulting positions and fit to the data were very similar to those found when the SLs were attached to CcP, indicating that linking the SL to the backbone of CcP seen in the crystal structure did not interfere with determining the experimentally most favourable orientations. As observed for the other SL positions, the ensemble of conformers at C288 showed more than one population for both SLs, with a ratio of about 25%:75%. Interestingly, the EPR simulations also showed two populations for both SLs at C288 with a ratio of 42.0%:57.7% for MTSL and a ratio of 25.0%:74.9% of pyMTSL.
Overall, there seems to be little correlation between the populations of SL conformers found using NMR and those found during the EPR simulations; C288 pyMTSL is the only position for which ratio between the populations found with NMR matched those found with EPR. Furthermore, the τradicalr values found during the EPR simulations (0.63–3.20 ns for the fast components; 2.20–10.00 ns for the slow components) were much lower than the estimated τc of the protein (16 ns).14,18 For spin labels, the value of τs is large so τc is dominated by τr in the formula τc−1 = τr−1 + τs−1.4–6 The value of τr is affected by any motions that change the vector between the paramagnetic centre and the nucleus such as protein tumbling, SL mobility, and local protein dynamics6 but of these usually only protein tumbling is taken into account35 as it is generally assumed to dominate the relaxation measurements.36 Despite shorter τradicalr values for the SLs, the assumption that protein tumbling dominates τc appears to hold true here as previous tests on the same system using lower τc values of 4 or 12 ns did not improve the docking results.14 This discrepancy between τr and τradicalr can be understood if it is assumed that the short correlation of the radical is caused mainly by small movements of the SL but have little influence on the length and orientation of the radical-nuclear vector in the external field. Rearrangement of this vector is dominated by rotation of the protein.
The EPR spectra for position C288 showed that both MTSL and pyMTSL were highly immobilized (Fig. 3C), so highly defined locations for the most favourable conformer orientations may be expected. When using a single conformer, the nitroxide radical positions were indeed precisely defined (Fig. 4E and F-i) and also the radical positions for the pyMTSL ensemble are well defined (Fig. 4E-ii). However, it should be noted that the calculations give no evidence for stronger steric restrictions. The restraints used in the calculations are to the oxygen of the SL. Other atoms can sample the conformational space as far as allowed by the restrained position of the oxygen atom. This is illustrated by the pyridyl ring that occupies a wide range of orientations for the pyMTSL in each of three positions. It is not obvious that this range is more limited for C288.
To see how well the calculated structures of the most favourable orientations fit the PRE data, the distances from the paramagnetic centre to the amide protons were back-predicted using r−6 averaging over all conformer orientations present in the best twenty solutions (Fig. 4) and then compared to the experimentally observed distances (Fig. S2, ESI†). The quality of fit parameters for all data sets are given in Table 2.
Very little difference is seen between the calculated distances, and thus the quality parameters, for the single conformer and ensemble solutions. Therefore, it cannot be established whether the single conformer or the ensemble is the better description for the SL, so both have been used in the docking calculations to allow for a comparison. Also little difference is present between solutions for MTSL and pyMTSL, in accord with similarity between the input PRE data sets for both SL types. There are, however, significant differences in the quality of fit between the SL positions. The best fits are observed for C200. The reason is not evident.
Docking of Cc to CcP
During the docking, first a single SL conformer was used that was either free to rotate or fixed in the experimentally determined most favourable orientation. Docking was repeated 10000 times from random starting positions of Cc and an ensemble was generated from the twenty lowest energy solutions for MTSL (Fig. 5A–D) and pyMTSL (Fig. 5E–H).
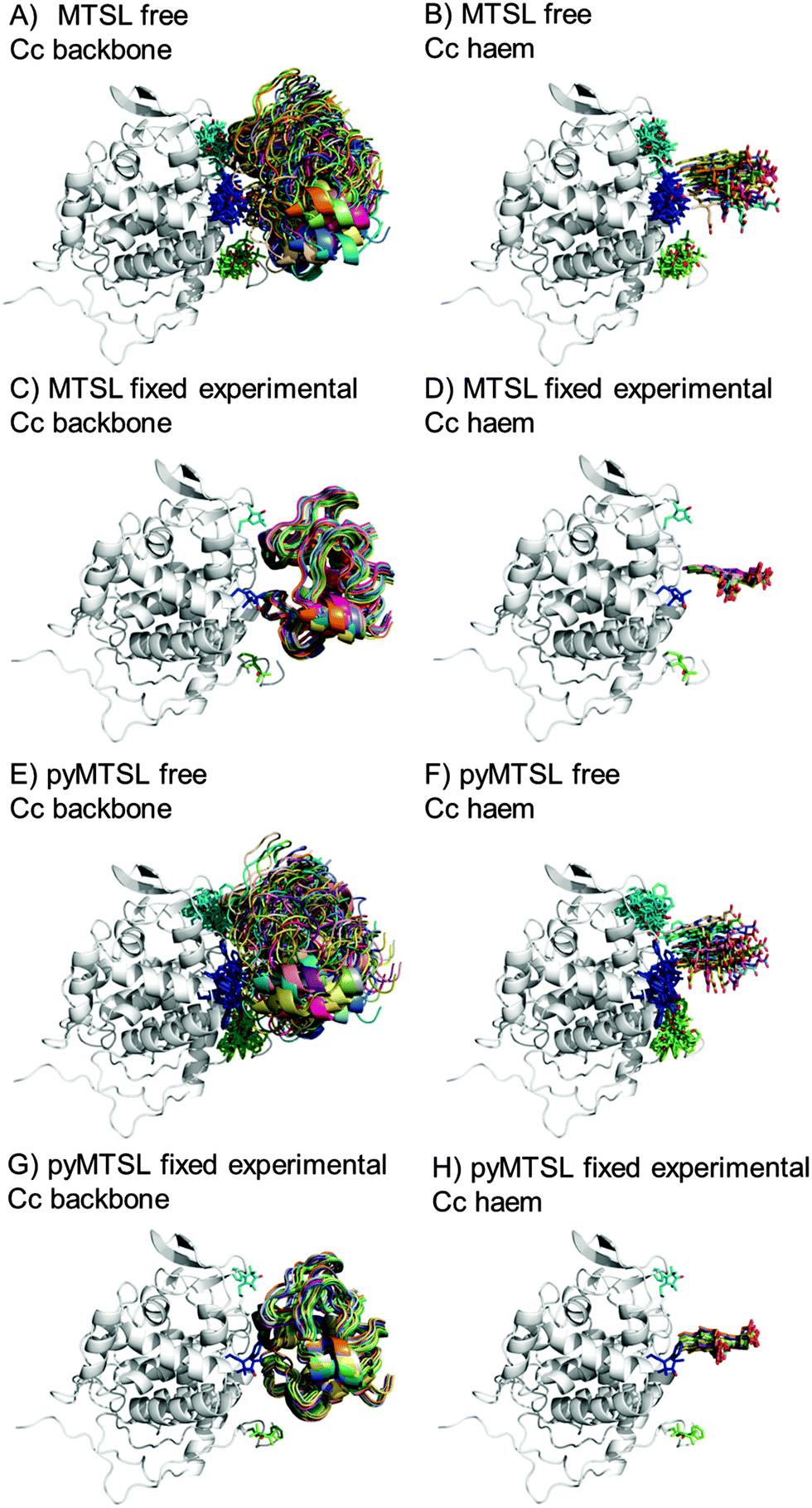 | ||
| Fig. 5 Twenty lowest energy solutions for docking Cc to CcP driven by intermolecular PRE data using a single SL conformer that was free to move during docking (MTSL A, B; pyMTSL E, F) or that was fixed in the experimentally determined most favourable orientation (MTSL C, D; pyMTSL G, H). CcP is shown in grey ribbons and Cc is shown in multi-coloured ribbons. The SLs are shown in sticks at positions 38 (teal), 200 (blue) and 288 (green), with the nitroxide oxygen atom in red and the Cc haem group is shown in multi-coloured sticks. The docking was done using the Cc and CcP structures taken from PDB entry 2PCC.13 | ||
When the SL is free to move during docking, the position of Cc as well as the SL orientations in the resulting ensemble of the twenty lowest energy solutions were much more dispersed than when the SL orientations were fixed; this can been seen most clearly when the positions of the Cc haem groups are compared. Similar results were observed in a previous study.15 Fixing the SL orientations provides a more precise description of the Cc position, indicating that the docking is reproducible.14,18 Note that the higher precision does not imply a result that is closer to the benchmark, in this case the position of Cc observed in the crystal structure, as is discussed below.
The distances between the Cc amide protons and the SL nitroxide radicals in the ensemble were compared to the experimental distances. Some discrepancies in the fit are to be expected since the data were obtained for the Cc–CcP complex in solution, which is known to consist of 30% encounter complex and 70% stereo-specific complex, while the structure comparison was done with the crystal structure, which consists only of the stereo-specific complex.14 Nevertheless, there was a good overall fit between the back-calculated and experimental distances (Fig. S3, ESI†). The fit was evaluated using the AV and Q-factors (Table 3).
| Position | MTSL | pyMTSL | |||
|---|---|---|---|---|---|
| 1 conformer free | 1 conformer fixed exp. | 1 conformer free | 1 conformer fixed exp. | ||
| C38 | AV (Å) | 1.98 | 2.55 | 2.01 | 2.53 |
| Q-factor | 0.10 | 0.15 | 0.10 | 0.14 | |
| C200 | AV (Å) | 2.13 | 2.51 | 1.88 | 2.19 |
| Q-factor | 0.10 | 0.12 | 0.09 | 0.12 | |
| C288 | AV (Å) | 1.21 | 2.17 | 1.44 | 2.04 |
| Q-factor | 0.07 | 0.12 | 0.08 | 0.11 | |
From the AV and Q-factors, it is clear that a freely rotating conformer much better describes the inter-molecular PRE data than does a single fixed conformer, despite the fixed position being determined experimentally with intra-molecular PRE data. This is not surprising since there are more degrees of freedom during the docking in this case; both the SL orientation and the position of Cc are fit to the inter-molecular PRE data when the SL is free to move.
The results were also compared to the stereo-specific orientation found in the crystal structure (Fig. 6). Overall, the Cc positions were similar to that of the X-ray crystal structure, although the calculated Cc ensemble was rotated slightly around the stereo-specific binding interface in all data sets. The RMSD of the Cα atoms was calculated by first generating an average structure from all the Cc orientations, done by taking the linear average of the individual structures, and then comparing that to the stereo-specific orientation in the X-ray crystal structure. The RMSD is sensitive to differences caused by both rotation and translation, while the DRMS is mainly sensitive to translation. The DRMS is calculated by determining the Cα–Cα distance matrix of for all Cα pairs from the two structures and then taking the root-mean-square deviation (eqn (3)). The DRMS is always smaller than the RMSD and a large RMSD in combination with a small DRMS indicates that the two structures are mostly rotated relative to each other.37
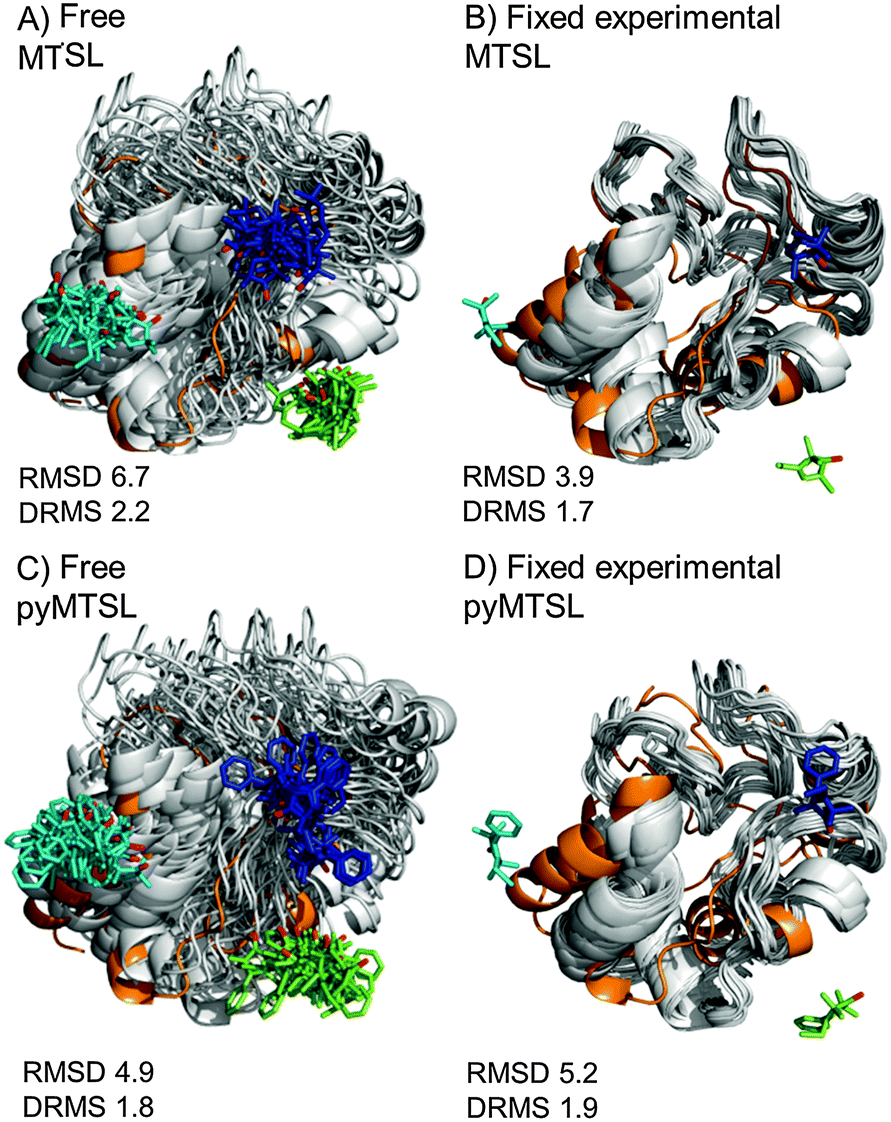 | ||
| Fig. 6 Comparison of the Cc positions as viewed from spin labelled CcP. The Cc position in the crystal structure (orange; PDB entry 2PCC)13 is compared to the twenty lowest energy solutions for docking Cc to CcP (grey ribbons), driven by intermolecular PRE data using a single conformer that was free to move during docking (A and C) or that was fixed in the experimentally determined most favourable orientation (B and D). The SLs (MTSL A, B; pyMTSL C, D) are shown in sticks at positions 38 (teal), 200 (blue) and 288 (green), with the nitroxide oxygen atoms in red. | ||
For both fixed and free MTSL and pyMTSL, the RMSD values were much higher than the DRMS values, indicating that a large proportion of the difference between the calculated Cc orientations compared to the crystal structure is due to rotation of Cc, while the binding interface is similar. For MTSL, fixing the SLs in the experimentally determined most favourable orientations improved the fit to the crystal structure and decreased the DRMS from 2.2 Å for the freely rotating SLs to 1.7 Å with the SLs fixed (Fig. 6A and B). Also, the RMSD is significantly reduced from 6.6 Å to 3.9 Å. This indicates that for MTSL, experimentally determining the most favourable SL orientations prior to protein docking improved the accuracy of the final structure. For pyMTSL, the DMRS values were similar in both cases, with a DRMS of 1.8 Å and 1.9 Å when using freely rotating and fixed SLs, respectively.
SL conformer ensemble
The protein docking calculations were repeated using the same inter-molecular PRE data and an ensemble of four SL conformers. Previous PRE studies on the Cc–CcP complex used an ensemble of four MTSL tags that were either fixed in selected positions during rigid body docking14 or were free to move during a subsequent dynamic docking step.15 Both approaches were repeated here along with a rigid body docking in which the SLs were fixed in the experimentally determined most favourable conformer orientations, using the orientations found in the lowest energy solution obtained with the intra-molecular PRE data. Cc was docked to CcP using inter-molecular PRE data and an ensemble was generated from the twenty lowest energy solutions for MTSL (Fig. 7) and pyMTSL (Fig. 8).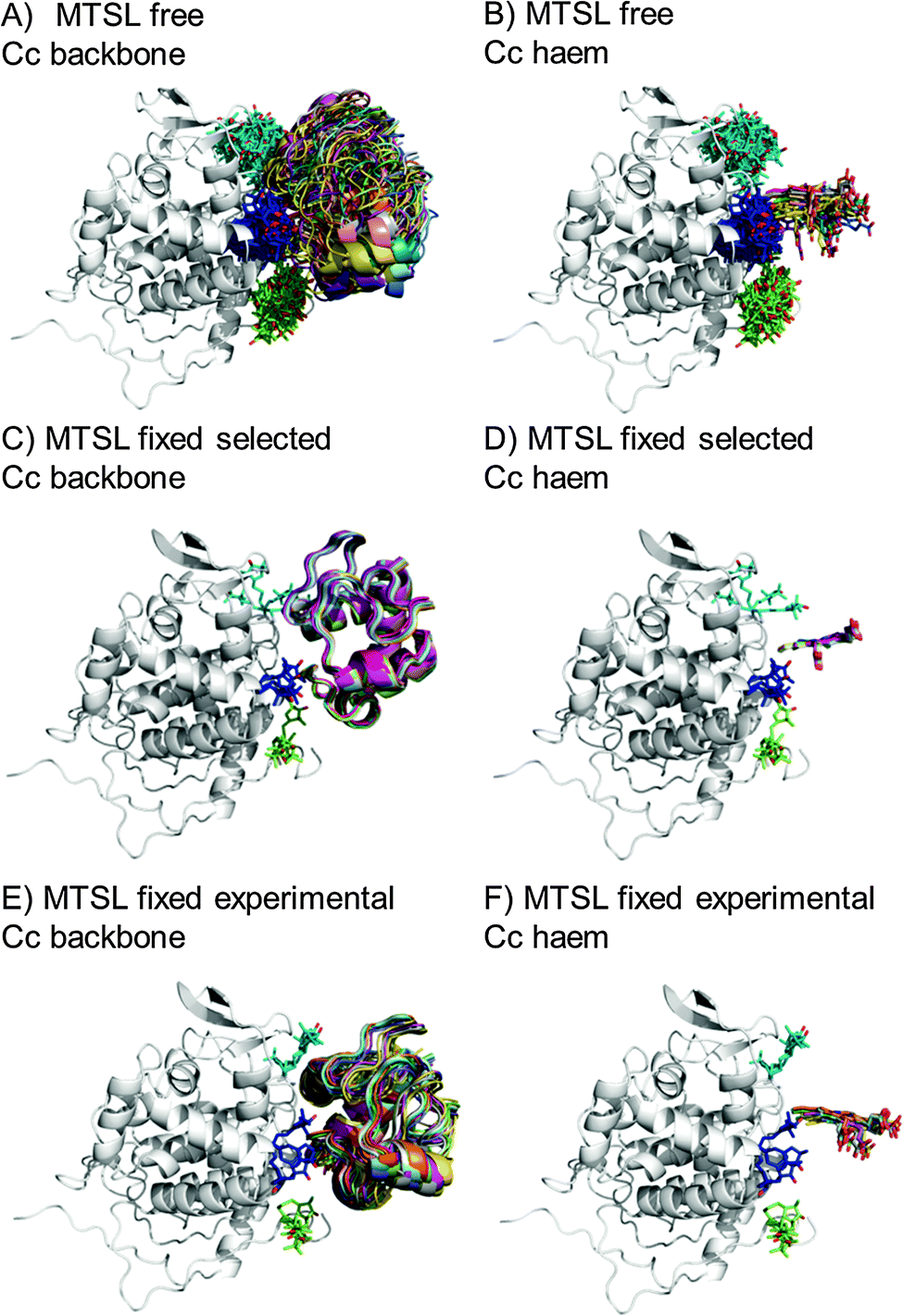 | ||
| Fig. 7 Twenty lowest energy solutions for docking Cc to CcP driven by intermolecular PRE data using an ensemble of four MTSL conformers that were free to move during docking (A and B) or that were fixed in selected positions (C and D) or experimentally determined most favourable orientations (E and F). CcP is shown in grey ribbons and Cc is shown in multi-coloured ribbons. The SLs are shown in sticks at positions 38 (teal), 200 (blue) and 288 (green), with the nitroxide oxygen atom in red and the Cc haem group is shown in multi-coloured sticks. The docking was done using structures from PDB entry 2PCC.13 | ||
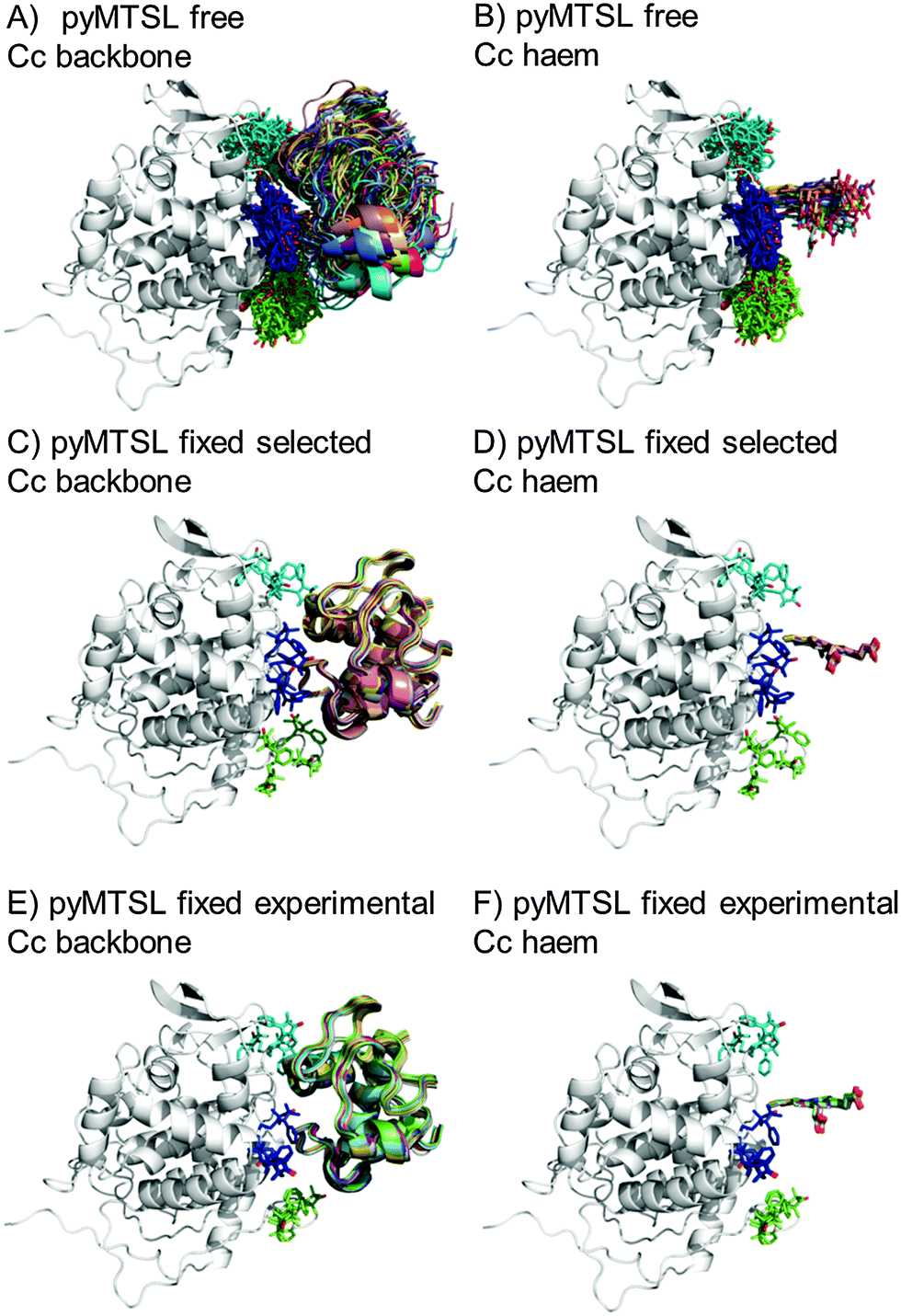 | ||
| Fig. 8 Twenty lowest energy solutions for docking Cc to CcP driven by intermolecular PRE data using an ensemble of four pyMTSL conformers that were free to move during docking (A and B) or that were fixed in selected positions (C and D) or experimentally determined most favourable orientations (E and F). CcP is shown in grey ribbons and Cc is shown in multi-coloured ribbons. The SLs are shown in sticks at positions 38 (teal), 200 (blue) and 288 (green), with the nitroxide oxygen atom in red and the Cc haem group is shown in multi-coloured sticks. The docking was done using structures from PDB entry 2PCC.13 | ||
As observed when using a single SL, allowing the SLs to rotate during docking resulted in a large, dispersed ensemble of the twenty lowest energy solutions for both MTSL and pyMTSL, while fixing the SL conformer orientations produced a well-defined position for Cc. The back-calculated distances between the Cc amide protons and the SL nitroxide radical oxygens were similar when using either free or fixed SL conformers and there was a good overall agreement with the experimental distances (Fig. S4, ESI†). The fits were evaluated using the AV and Q-factors (Table 4).
| Position | MTSL | pyMTSL | |||||
|---|---|---|---|---|---|---|---|
| 4 conformers free | 4 conformers fixed sele. | 4 conformers fixed exp. | 4 conformers free | 4 conformers fixed sele. | 4 conformers fixed exp. | ||
| C38 | AV (Å) | 2.35 | 2.53 | 2.66 | 2.00 | 2.18 | 2.40 |
| Q-factor | 0.12 | 0.14 | 0.15 | 0.10 | 0.11 | 0.13 | |
| C200 | AV (Å) | 1.93 | 2.96 | 2.17 | 2.00 | 2.10 | 2.03 |
| Q-factor | 0.10 | 0.14 | 0.12 | 0.08 | 0.11 | 0.11 | |
| C288 | AV (Å) | 1.32 | 1.51 | 2.24 | 1.60 | 1.80 | 2.32 |
| Q-factor | 0.08 | 0.09 | 0.12 | 0.09 | 0.10 | 0.12 | |
Again, the best fits were found when the SL orientations were free to move during dynamic docking; i.e., the SL conformer orientations were fit to the PRE data along with the position of Cc. This indicates that, although fixing the SL orientations results in a very precise determination of the Cc position, using four fixed positions for the nitroxide radical and, thereby, limiting the degrees of freedom, cannot describe the observed PRE data completely.
Surprisingly, optimizing the SL positions using experimental data did not yield a better fit to the inter-molecular PRE data than simply selecting four orientations from the sterically allowed possibilities (see Material and methods for details of how the conformers were selected). The back-calculated distances are determined using the orientation of the complex found in the X-ray crystal structure, assuming that the orientation of the complex in solution is very similar to the one observed in the crystalline state. However, it is known that the complex is in fast exchange on the NMR timescale between this stereo-specific orientation, accounting for 70% of the complex in solution, and a more dynamic the encounter state, accounting for the remaining 30%.14,18,19 The experimental PREs are a (non-linear) average of all orientations in solution, so comparing them to only the stereo-specific orientation will limit the quality of the fit. Nevertheless, the overall fit to the PRE data was reasonably good for all conditions.
The results for docking Cc to CcP were also compared to the crystal structure of the stereospecific complex (Fig. 9). As shown previously,8,9 the best results were obtained for MTSL when using multiple conformers. Furthermore, as seen when using a single SL conformer, many of the resulting Cc orientations are rotated with respect to the stereospecific binding site resulting in high RMSD values but small DRMS values for the ensembles. For MTSL, fixing the conformers in the selected orientations produced the worst fit the with crystal structure (RMSD = 6.7 Å; DRMS = 2.2 Å). The fit improved when the SLs could freely rotate (RMSD = 4.8 Å; DRMS = 1.8 Å) but the best fit was obtained when using the experimentally determined conformer orientations (RMSD = 2.5 Å; DRMS = 1.5 Å). This indicated that the most accurate description of the protein complex was achieved by predetermining the conformer orientations using intra-molecular PREs, even though the quality of fit to the experimental data was not the best for this solution.
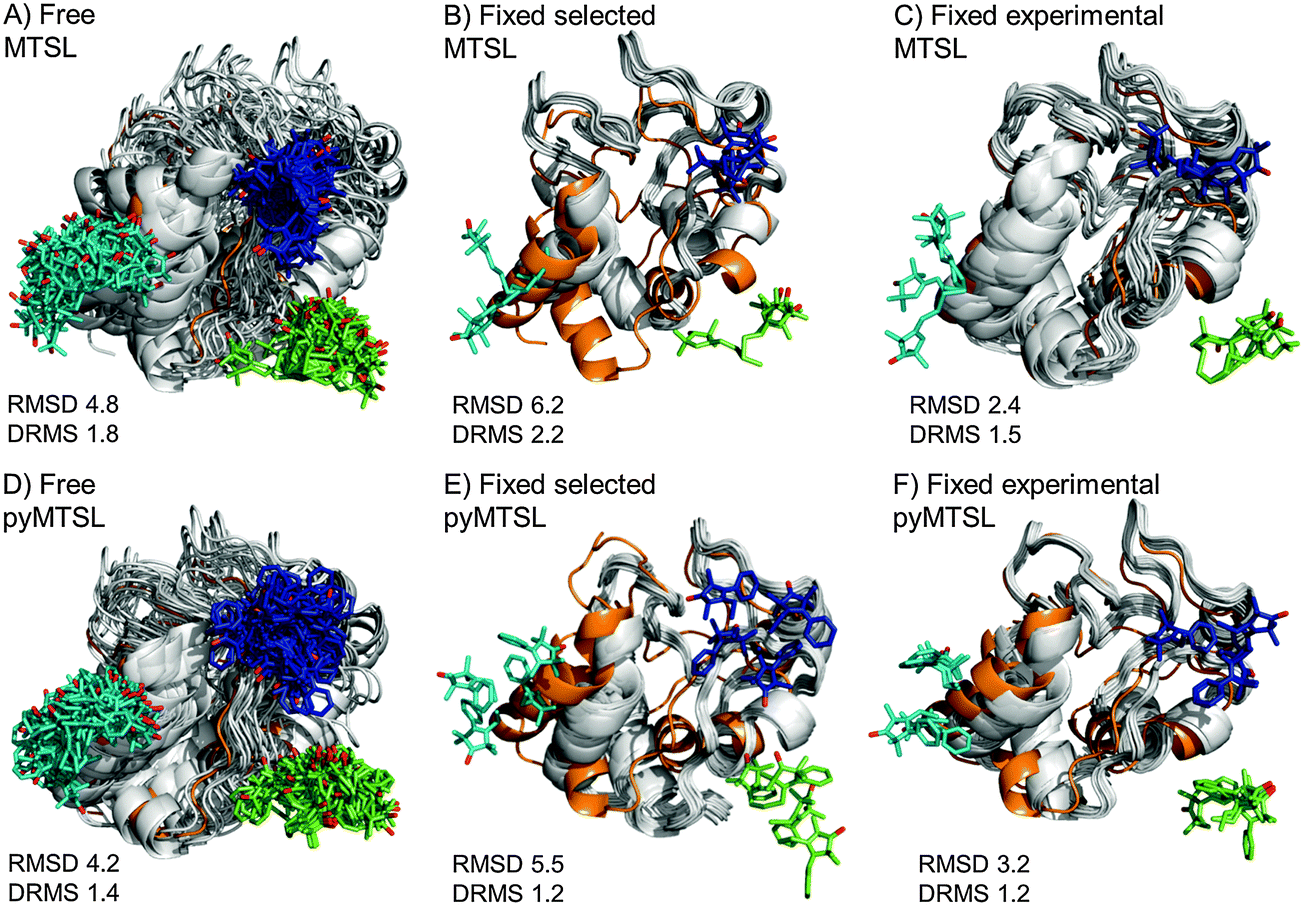 | ||
| Fig. 9 Comparison of the Cc positions as viewed from spin labelled CcP. The Cc position in the crystal structure (orange; PDB entry 2PCC)13 is compared to the twenty lowest energy solutions for docking Cc to CcP (grey ribbons), driven by intermolecular PRE data using an ensemble of four conformers that were free to move during dynamic docking (A and B) or that were fixed in selected positions (C and D) or experimentally determined most favourable orientations (E and F). The SLs are shown in sticks at positions 38 (teal), 200 (blue) and 288 (green), with the nitroxide oxygen atoms in red and the Cc haem group shown in multi-coloured sticks. | ||
In a previous study, PREs from MTSL at positions 38, 200 and 288 were fixed in selected positions while Cc was docked to CcP.14 The same conformers were used in this study. An RMSD with the crystal structure of 2.2 Å was found in that study, which is much smaller than the RMSD values that we observed, of 6.7 Å. This large discrepancy is likely due to small differences in the experimental PREs and the fact that normalization of the Ipara/Idia data (as described in the Materials and methods) was not done in the previous study. Although the Ipara/Idia ratio for residues unaffected by PRE is expected to be 1.0, we frequently observe values that are on average slightly higher or slightly lower. The reason for this is unclear but is likely due to slight differences between the diamagnetic and paramagnetic samples. The transverse relaxation rate is very sensitive to the exact fraction bound, because the rotational correlation time of the CcP–Cc complex is much larger than that of free Cc. If the concentrations of the proteins vary slightly, for example, due to slight aggregation, the fraction bound Cc can differ between the paramagnetic and diamagnetic samples. Since it is a global effect on all residues, and the deviation in the average Ipara/Idia ratio can be both larger and smaller than 1.0, non-specific contact between Cc and the paramagnetic protein cannot be the cause of this effect. This normalization was not done in the previous study14 resulting in shorter distance restraints than were obtained in this study, particularly for position C38, which allowed Cc to find a position much closer to the SLs than was allowed in this study. These findings demonstrate that the outcome of the docking is very sensitive to relatively small differences in the experimental data set.
For pyMTSL, the DRMS values showed that using pyMTSL resulted in a very good fit to the stereo-specific state but the RMSD values were quite high. Therefore, the differences between the final Cc positions found after docking and the stereo-specific state were mainly due to rotation and not translation of Cc. When the SL was free to rotate, the resulting fit had an RMSD of 4.2 Å and DRMS 1.4 Å. Fixing the conformers in the selected orientations resulted in greater rotation of Cc relative to the stereo-specific state (RMSD = 5.5 Å; DRMS = 1.2 Å). Fixing the SLs in the experimentally determined orientations resulted in a lower RMSD of 3.2 Å and DRMS 1.2 Å, in line with the results for MTSL. Therefore, in this case, it was necessary to represent pyMTSL using multiple conformers to get the best results.
As was shown previously,14 fixing the SL orientations during docking produces a highly defined ensemble of solutions. However, precision is of no importance in the absence of accuracy, which can be difficult to achieve when the fixed SL orientations must be selected without experimental data. The use of intra-molecular PRE to predetermine the most favourable SL orientations improved the accuracy of structure determination despite the fact that this did not necessarily improve the fit between the experimental and back-calculated data. In this case, we used the stereo-specific orientation of Cc observed in the X-ray crystal structure as the benchmark for accuracy under the assumption that this is also the main state of the complex in solution. However, as mentioned above, this stereo-specific state only accounts for 70% of the complex in solution, with the remaining 30% in the encounter state,14,16–19 resulting in discrepancies in the fit. Furthermore, under most conditions, a significant rotation of Cc around the stereo-specific binding interface was observed. This is likely due to the fact that PREs are highly sensitive to minor states38 giving them a disproportionally large influence on the final orientation of Cc determined by the docking calculations. Although fixing the SLs in the experimentally determined most favourable orientations helped to reduce this effect, we conclude that PREs do not yield very reliable restraints for determining protein orientation within a complex.
Much better results can be obtained when combining PCS or RDC with the PRE data.39–48 This was demonstrated recently by Hiruma et al. for the cytochrome P450cam–putidaredoxin complex. Paramagnetic tags were placed at two locations on cytochrome P450cam as well as on one location on putidaredoxin and intermolecular PCS, RDC and PRE were obtained. The PCS and RDC back-calculated from the final, well-defined structure matched the experimental data very well, as did the PRE data for the one of the tag positions on cytochrome P450cam. For the second position, however, the experimental PREs were stronger than expected for many residues and the authors concluded that this was due to a minor state influencing the PRE results. Nevertheless, the use of PCS and RDC in combination with PRE data allowed for the structure of the major state to be successfully resolved, as judged by the subsequently determined crystal structure of the complex, which showed a 1.7 Å RMSD with the mean of the NMR ensemble of structures.49 This work was done using different paramagnetic lanthanoid ions, which can be attached to the protein via double armed caged lanthanide NMR probes (CLaNP), having the additional benefit of being highly immobilized and therefore limiting spatial averaging of the observed paramagnetic effects.50,51
For structural modelling paramagnetic restraints can also be combined with other NMR data, such as RDCs obtained using external alignment media and NOEs.39,42–47 This was done recently by Shi et al. who used PRE data to complement both NOE and RDC data for the integral-membrane protein phospho-lamban (PLN). PLN is a small protein consisting of two helical domains linked by a flexible loop. The NMR structure of the monomeric protein in dodecylphosphocholine micelles had previously been solved by their group using NOE and solvent PRE but the resulting ensemble of conformers in the membrane was very poorly defined.52 Combining the NOE data with RDCs resulted in 100 low energy structures that were highly refined (backbone RMSD = 1.6 Å) but were grouped into four families exhibiting a four-fold degeneracy in the relative orientations of the two helices. By also incorporating PRE data into the structure refinement, the degeneracy of the RDC data was overcome and the correct family of structures was obtained with high resolution (backbone RMSD = 1.2 Å).48 This highlights the benefit of combining multiple data sets when resolving the structure of dynamic and/or multi-domain proteins, especially when together they can provide both distance and orientation information.
Conclusions
This work has combined both intra- and inter-molecular PRE data to investigate the role of SL mobility on complex structure determination. While little difference was found between MTSL and pyMTSL, the accuracy of final results, as judged by similarity to the crystal structure of the complex, was highly dependent on the number and choice of SL conformer orientations used during the docking. It was also found that fixing the SL orientations during docking resulted in highly precise ensembles for the Cc position but that this level of precision was not correlated with a better match to the stereo-specific orientation of Cc. Although pre-determination of the favourable SL orientations using intra-molecular PRE data did help to improve the accuracy of docking results for the Cc–CcP complex, this did not necessarily improve the fit between the experimental and back-calculated data, so additional cases should be studied to assess the value of this technique for highly dynamic complexes. Overall, it seems that PRE determined distance restraints used in isolation are not ideal for determining the protein orientation within a dynamic complex and much better results can be obtained when combining PCS or RDC with the PRE data.49Acknowledgements
This work was supported by the Netherlands Organization for Scientific Research (NWO), grants 700.58.441 and 700.58.014 and grant number 10SMPA04 of the Foundation for Fundamental Research on Matter (FOM), which is part of NWO.References
- G. M. Clore, Exploring sparsely populated states of macromolecules by diamagnetic and paramagnetic NMR relaxation, Protein Sci., 2011, 20(2), 229–246 CrossRef CAS PubMed.
- J. Schilder, M. A. S. Hass, P. H. Keizers and M. Ubbink, Paramagnetic NMR spectroscopy and lowly populated states, in Recent Developments in Biomolecular NMR, ed. M. G. Clore and J. Potts, Royal Society of Chemistry, 2012, pp. 130–150 Search PubMed.
- O. H. Griffith and H. M. Mcconnel, A nitroxide-maleimide spin label, Proc. Natl. Acad. Sci. U. S. A., 1966, 55(1), 8–11 CrossRef CAS.
- I. Bertini, C. Luchinat and S. Aime, NMR of paramagnetic substances, Coord. Chem. Rev., 1996, 150, 29–110 CrossRef.
- J. L. Battiste and G. Wagner, Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear Overhauser effect data, Biochemistry, 2000, 39(18), 5355–5365 CrossRef CAS PubMed.
- G. Jeschke, Conformational dynamics and distribution of nitroxide spin labels, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 72, 42–60 CrossRef CAS PubMed.
- J. Iwahara, C. D. Schwieters and G. M. Clore, Ensemble approach for NMR structure refinement against 1H paramagnetic relaxation enhancement data arising from a flexible paramagnetic group attached to a macromolecule, J. Am. Chem. Soc., 2004, 126(18), 5879–5896 CrossRef CAS PubMed.
- G. A. Bermejo, M. P. Strub, C. Ho and N. Tjandra, Determination of the solution-bound conformation of an amino acid binding protein by NMR paramagnetic relaxation enhancement: use of a single flexible paramagnetic probe with improved estimation of its sampling space, J. Am. Chem. Soc., 2009, 131(27), 9532–9537 CrossRef CAS PubMed.
- C. Tang, C. D. Schwieters and G. M. Clore, Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR, Nature, 2007, 449(7165), 1078–1082 CrossRef CAS PubMed.
- C. Tang, J. Iwahara and G. M. Clore, Visualization of transient encounter complexes in protein–protein association, Nature, 2006, 444(7117), 383–386 CrossRef CAS PubMed.
- N. L. Fawzi, M. R. Fleissner, N. J. Anthis, T. Kalai, K. Hideg, W. L. Hubbell and G. M. Clore, A rigid disulfide-linked nitroxide side chain simplifies the quantitative analysis of PRE data, J. Biomol. NMR, 2011, 51(1–2), 105–114 CrossRef CAS PubMed.
- T. Yonetani, Studies on cytochrome c peroxidase. II. Stoichiometry between enzyme, H2O2, and ferrocytochrome c and enzymic determination of extinction coefficients of cytochrome c, J. Biol. Chem., 1965, 240(11), 4509–4514 CAS.
- H. Pelletier and J. Kraut, Crystal-structure of a complex between electron-transfer partners, cytochrome c peroxidase and cytochrome c, Science, 1992, 258(5089), 1748–1755 CAS.
- A. N. Volkov, J. A. Worrall, E. Holtzmann and M. Ubbink, Solution structure and dynamics of the complex between cytochrome c and cytochrome c peroxidase determined by paramagnetic NMR, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(50), 18945–18950 CrossRef CAS PubMed.
- Q. Bashir, Dynamics in electron transfer protein complexes, Doctoral thesis, Leiden University, 2010 Search PubMed.
- J. Schilder, F. Lohr, H. Schwalbe and M. Ubbink, The cytochrome c peroxidase and cytochrome c encounter complex: the other side of the story, FEBS Lett., 2014, 588(10), 1873–1878 CrossRef CAS PubMed.
- A. N. Volkov, M. Ubbink and N. A. van Nuland, Mapping the encounter state of a transient protein complex by PRE NMR spectroscopy, J. Biomol. NMR, 2010, 48(4), 225–236 CrossRef CAS PubMed.
- Q. Bashir, A. N. Volkov, G. M. Ullmann and M. Ubbink, Visualization of the encounter ensemble of the transient electron transfer complex of cytochrome c and cytochrome c peroxidase, J. Am. Chem. Soc., 2010, 132(1), 241–247 CrossRef CAS PubMed.
- K. Van de Water, N. A. J. van Nuland and A. N. Volkov, Transient protein encounters characterized by paramagnetic NMR, Chem. Sci., 2014, 5(11), 4227–4236 RSC.
- W. B. Pollock, F. I. Rosell, M. B. Twitchett, M. E. Dumont and A. G. Mauk, Bacterial expression of a mitochondrial cytochrome c. Trimethylation of lys72 in yeast iso-1-cytochrome c and the alkaline conformational transition, Biochemistry, 1998, 37(17), 6124–6131 CrossRef CAS PubMed.
- A. S. Morar, D. Kakouras, G. B. Young, J. Boyd and G. J. Pielak, Expression of 15N-labeled eukaryotic cytochrome c in Escherichia coli, J. Biol. Inorg. Chem., 1999, 4(2), 220–222 CrossRef CAS PubMed.
- L. B. Vitello, M. Huang and J. E. Erman, pH-dependent spectral and kinetic properties of cytochrome c peroxidase: comparison of freshly isolated and stored enzyme, Biochemistry, 1990, 29(18), 4283–4288 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, EasySpin, a comprehensive software package for spectral simulation and analysis in EPR, J. Magn. Reson., 2006, 178(1), 42–55 CrossRef CAS PubMed.
- S. Steigmiller, M. Börsch, P. Gräber and M. Huber, Distances between the b-subunits in the tether domain of F0F1-ATP synthase from E. coli, Biochim. Biophys. Acta, Bioenerg., 2005, 1708(2), 143–153 CrossRef CAS PubMed.
- E. Lescop, P. Schanda and B. Brutscher, A set of BEST triple-resonance experiments for time-optimized protein resonance assignment, J. Magn. Reson., 2007, 187(1), 163–169 CrossRef CAS PubMed.
- W. F. Vranken, W. Boucher, T. J. Stevens, R. H. Fogh, A. Pajon, M. Llinas, E. L. Ulrich, J. L. Markley, J. Ionides and E. D. Laue, The CCPN data model for NMR spectroscopy: development of a software pipeline, Proteins, 2005, 59(4), 687–696 CrossRef CAS PubMed.
- J. A. Worrall, U. Kolczak, G. W. Canters and M. Ubbink, Interaction of yeast iso-1-cytochrome c with cytochrome c peroxidase investigated by [15N, 1H] heteronuclear NMR spectroscopy, Biochemistry, 2001, 40(24), 7069–7076 CrossRef CAS PubMed.
- Y. Gao, J. Boyd, R. J. Williams and G. J. Pielak, Assignment of proton resonances, identification of secondary structural elements, and analysis of backbone chemical shifts for the C102T variant of yeast iso-1-cytochrome c and horse cytochrome c, Biochemistry, 1990, 29(30), 6994–7003 CrossRef CAS PubMed.
- J. S. Fetrow and S. M. Baxter, Assignment of 15N chemical shifts and 15N relaxation measurements for oxidized and reduced iso-1-cytochrome c, Biochemistry, 1999, 38(14), 4480–4492 CrossRef CAS PubMed.
- C. A. Bonagura, B. Bhaskar, H. Shimizu, H. Li, M. Sundaramoorthy, D. E. McRee, D. B. Goodin and T. L. Poulos, High-resolution crystal structures and spectroscopy of native and compound I cytochrome c peroxidase, Biochemistry, 2003, 42(19), 5600–5608 CrossRef CAS PubMed.
- C. D. Schwieters, J. J. Kuszewski, N. Tjandra and G. M. Clore, The Xplor-NIH NMR molecular structure determination package, J. Magn. Reson., 2003, 160(1), 65–73 CrossRef CAS PubMed.
- C. D. Schwieters, J. J. Kuszewski and G. M. Clore, Using Xplor-NIH for NMR molecular structure determination, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48(1), 47–62 CrossRef CAS.
- A. M. Berghuis and G. D. Brayer, Oxidation state-dependent conformational changes in cytochrome c, J. Mol. Biol., 1992, 223(4), 959–976 CrossRef CAS PubMed.
- Y. C. Kim, C. Tang, G. M. Clore and G. Hummer, Replica exchange simulations of transient encounter complexes in protein-protein association, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(35), 12855–12860 CrossRef CAS PubMed.
- G. M. Clore and J. Iwahara, Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes, Chem. Rev., 2009, 109(9), 4108–4139 CrossRef CAS PubMed.
- P. Bernado, J. Garcia de la Torre and M. Pons, Interpretation of 15N NMR relaxation data of globular proteins using hydrodynamic calculations with HYDRONMR, J. Biomol. NMR, 2002, 23(2), 139–150 CrossRef CAS PubMed.
- B. Zagrovic and V. S. Pande, How does averaging affect protein structure comparison on the ensemble level?, Biophys. J., 2004, 87(4), 2240–2246 CrossRef CAS PubMed.
- S. Scanu, J. Forster, M. G. Finiguerra, M. Hashemi Shabestari, M. Huber and M. Ubbink, The complex of cytochrome f and plastocyanin from Nostoc sp PCC 7119 is highly dynamic, ChemBioChem, 2012, 13(9), 1312–1318 CrossRef CAS PubMed.
- B. Simon, T. Madl, C. D. Mackereth, M. Nilges and M. Sattler, An efficient protocol for NMR-spectroscopy-based structure determination of protein complexes in solution, Angew. Chem., Int. Ed., 2010, 49(11), 1967–1970 CrossRef CAS PubMed.
- X. Xu, P. H. Keizers, W. Reinle, F. Hannemann, R. Bernhardt and M. Ubbink, Intermolecular dynamics studied by paramagnetic tagging, J. Biomol. NMR, 2009, 43(4), 247–254 CrossRef CAS PubMed.
- X. Xu, W. Reinle, F. Hannemann, P. V. Konarev, D. I. Svergun, R. Bernhardt and M. Ubbink, Dynamics in a pure encounter complex of two proteins studied by solution scattering and paramagnetic NMR spectroscopy, J. Am. Chem. Soc., 2008, 130(20), 6395–6403 CrossRef CAS PubMed.
- L. Salmon, G. Nodet, V. Ozenne, G. W. Yin, M. R. Jensen, M. Zweckstetter and M. Blackledge, NMR characterization of long-range order in intrinsically disordered proteins, J. Am. Chem. Soc., 2010, 132(24), 8407–8418 CrossRef CAS PubMed.
- C. W. Bertoncini, Y. S. Jung, C. O. Fernandez, W. Hoyer, C. Griesinger, T. M. Jovin and M. Zweckstetter, Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(5), 1430–1435 CrossRef CAS PubMed.
- J. Song, L. W. Guo, H. Muradov, N. O. Artemyev, A. E. Ruoho and J. L. Markley, Intrinsically disordered gamma-subunit of cGMP phosphodiesterase encodes functionally relevant transient secondary and tertiary structure, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(5), 1505–1510 CrossRef CAS PubMed.
- J. R. Huang and S. Grzesiek, Ensemble calculations of unstructured proteins constrained by RDC and PRE data: a case study of urea-denatured ubiquitin, J. Am. Chem. Soc., 2010, 132(2), 694–705 CrossRef CAS PubMed.
- T. C. Leeper, X. Qu, C. Lu, C. Moore and G. Varani, Novel protein-protein contacts facilitate mRNA 3′-processing signal recognition by Rna15 and Hrp1, J. Mol. Biol., 2010, 401(3), 334–349 CrossRef CAS PubMed.
- Y. Takayama and G. M. Clore, Intra- and intermolecular translocation of the bi-domain transcription factor Oct1 characterized by liquid crystal and paramagnetic NMR, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(22), E169–E176 CrossRef PubMed.
- L. Shi, N. J. Traaseth, R. Verardi, M. Gustavsson, J. L. Gao and G. Veglia, Paramagnetic-based NMR restraints lift residual dipolar coupling degeneracy in multidomain detergent-solubilized membrane proteins, J. Am. Chem. Soc., 2011, 133(7), 2232–2241 CrossRef CAS PubMed.
- Y. Hiruma, M. A. Hass, Y. Kikui, W. M. Liu, B. Olmez, S. P. Skinner, A. Blok, A. Kloosterman, H. Koteishi, F. Lohr, H. Schwalbe, M. Nojiri and M. Ubbink, The structure of the cytochrome p450cam-putidaredoxin complex determined by paramagnetic NMR spectroscopy and crystallography, J. Mol. Biol., 2013, 425(22), 4353–4365 CrossRef CAS PubMed.
- P. H. Keizers, J. F. Desreux, M. Overhand and M. Ubbink, Increased paramagnetic effect of a lanthanide protein probe by two-point attachment, J. Am. Chem. Soc., 2007, 129(30), 9292–9293 CrossRef CAS PubMed.
- P. H. Keizers, A. Saragliadis, Y. Hiruma, M. Overhand and M. Ubbink, Design, synthesis, and evaluation of a lanthanide chelating protein probe: CLaNP-5 yields predictable paramagnetic effects independent of environment, J. Am. Chem. Soc., 2008, 130(44), 14802–14812 CrossRef CAS PubMed.
- J. Zamoon, A. Mascioni, D. D. Thomas and G. Veglia, NMR solution structure and topological orientation of monomeric phospholamban in dodecylphosphocholine micelles, Biophys. J., 2003, 85(4), 2589–2598 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp03781f |
| This journal is © the Owner Societies 2016 |