DOI:
10.1039/C6CE02157C
(Paper)
CrystEngComm, 2016,
18, 8933-8943
Solid state supramolecular structure of diketopyrrolopyrrole chromophores: correlating stacking geometry with visible light absorption†
Received
10th October 2016
, Accepted 3rd November 2016
First published on 3rd November 2016
Abstract
Mono- and di-alkylated 1,4-diketo-3,6-dithiophenylpyrrolo[3-4-c]pyrrole derivatives (TDPPs) have been synthesised and their solid state packing and absorption properties have been correlated. In this library of compounds the bulkier substituents distort the geometry of the chromophores and shift the lowest energy absorption band as a consequence of reduced π–π stacking and inter-chromophore overlap. Longitudinal displacement of the conjugated core is affected by donor–acceptor intermolecular interactions and twisting of the thiophene ring out of the plane of the DPP core, whereas lateral displacement was correlated to distortion of the NLactam–C(R) bond out of the plane of the DPP core. The di-substituted TDPP with hexyl units exhibit high molecular planarity, strong close packing of the conjugated core and significant red shift of the maximum of absorption in the solid, whereas the mono-substituted compounds with hexyl and ethyl acetate units are the least distorted of the series because of strong intermolecular hydrogen bonding that increases the molecular overlap and planarity of the chromophores. Therefore the family of mono-substituted TDPPs and more specifically the ones with ethyl acetate substituents show good potential for modulating the molecular geometry and optimizing the charge transport in materials for organic electronic applications.
Introduction
The development of new materials for organic solar cells (OSCs) lead to continually improving device efficiencies, in great measure through control of the supramolecular organization of the material in thin films of the active layer incorporating the donors and acceptors.1 The molecular approach makes use of various functionalities – either at the end of the conjugated chain and/or sideways to the core – in attempts to control the supramolecular organization through π–π stacking, hydrogen bonding2 and other supramolecular interactions.3 Both covalent and multicomponent supramolecular structures (primary and secondary)4 as well as hierarchical superstructures determine the properties of the materials.5 In organic semiconductors in general the electron mobility is sensitive to intermolecular electronic interactions that depend on the shape of the frontier orbitals and on the relative orientation and position of the aromatic cores of the adjacent molecules. Thus the fundamental properties of materials for OSCs are controlled by modulating the molecular structure, thin film ordering and optimizing the device architecture. In highly crystalline small molecule thin films absorption and charge carrier transport are determined by packing and orbital overlap but limited by the crystalline domain sizes and their interconnection.6 Derivatives of small molecules such as 1,4-diketo-pyrrolo[3-4-c]pyrroles (DPPs, Scheme 1) have become an attractive alternative to polymers for photovoltaic applications due to their facile synthesis and purification, solution processability,7 as well as efficient charge transport and high light absorption.8 DPPs are thus a class of intensively studied organic building blocks being used as high-performance pigments9 and light harvesting chromophore in OSCs.10 DPP chromophores consist of two condensed lactam units that are habitually decorated at the “end” of the conjugated core by aromatic rings and at the lateral amide-nitrogen atoms by alkyl chains.11 The two electron withdrawing amide units on the core make DPPs good electron acceptor moieties that can be associated to different electron donating or withdrawing aromatic units resulting in an increase of π-conjugation and control over the frontier orbitals energies. Of the wide variety of chromophores derived from DPP, the series of 1,4-diketo-3,6-dithienylpyrrolo[3-4-c]pyrroles (TDPP) derivatives is one of the most investigated for a number of reasons, including synthetic accessibility, and high planarity (small dihedral angle between the DPP core and the thiophene rings) which results in high π-delocalization and favourable π–π intermolecular interactions. The importance of the donor group as well as the presence of one or two alkyl side units on the solid state packing, photophysical and charge transport properties has been recently highlighted for a series of di-hexyl-phenyl, -furyl, -thiophenyl and -selenophenyl,12 and di- and mono-hexyl-phenyl and -thiophenyl DPPs,13 respectively. Based on single crystal X-ray diffraction, a higher torsion angle of the aromatic unit and the DPP core of about 33° was found for phenyl DPP because of steric repulsion between the proton of the phenyl unit and the alkyl side chain attached to the DPP core, whereas five-membered rings remain almost coplanar with the DPP unit. On the other hand, mono-hexyl TDPP has been found to have superior charge carrier mobility to its di-alkylated analogue apparently due to improved close packing driven by intermolecular hydrogen bonding between the free amide groups. Similar studies on TDPPs in the solid state revealed the importance of the alkyl units (branched and linear hydrocarbons, and hydrophilic ethyleneoxy units) in preserving high planarity and therefore close packing of the molecules.14 Hydrophilic ethyleneoxy side chains reduce the twisting out of plane of the thiophene unit, but the observed intermolecular π–π distance (4.021 Å compared to 3.614 Å and 3.785 Å for hexyl and ethylhexyl, respectively) suggests reduced π–π interactions as a result of random orientation of the chain possibly to maximize favourable intermolecular interactions between the ethyleneoxy and thiophene units. In phenyl DPPs possessing benzyl substituents favourable benzyl based intermolecular interactions have been shown to improve the overlap of the π–π co-facial dimers and the charge transfer properties.15 Similarly, higher co-planarity and intramolecular interaction in TDPPs compare to phenyl DPPs favour close packing of chromophores allowing singlet exciton fission to occur suggesting the potential of TDPPs of enhancing the efficiency of solar cells devices.16 These observations highlight the importance of molecular design in controlling the alignment and the π–π overlap of DPPs as ways of optimizing the charge transport in organic conducting materials. We present herein the synthesis, spectroscopic properties and thorough solid state structural analysis of two new series of mono- and di-alkylated TDPP derivatives, with a focus on the differences in the solid state packing caused by the nature of the side-appended alkyl units.
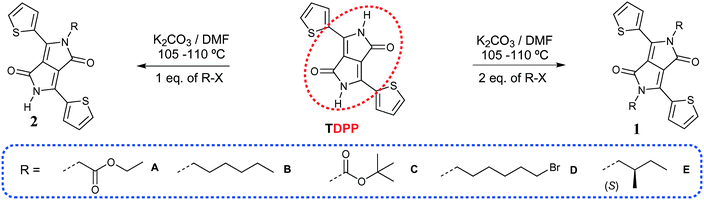 |
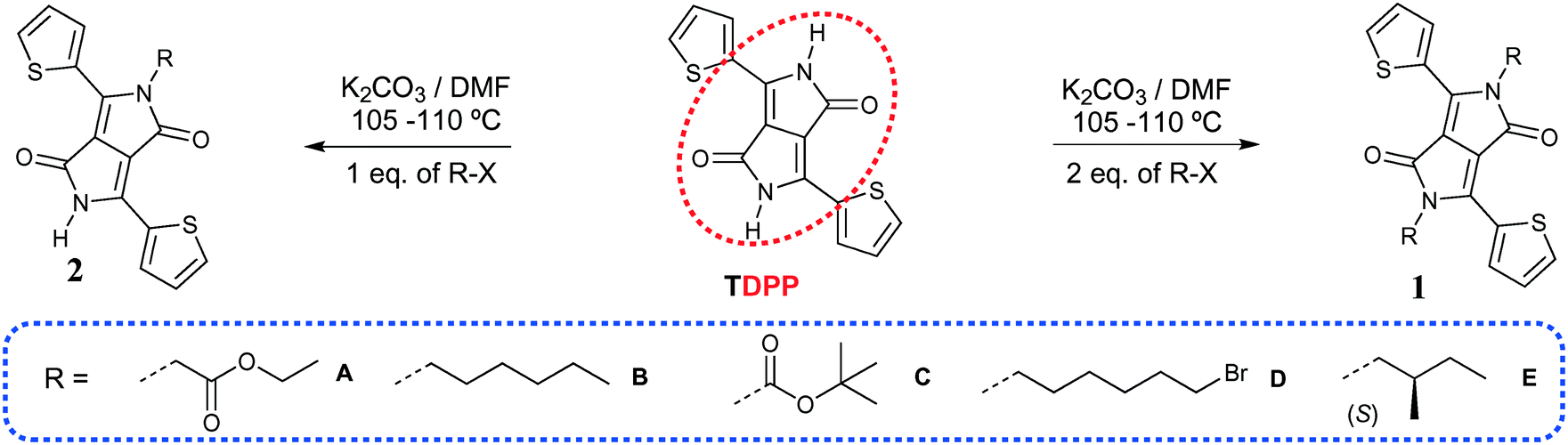
| | Scheme 1 Synthesis of TDPPs 1A–E and 2A–E. | |
Results and discussions
The TDPPs discussed here were synthesized according to similar procedures described previously17 by using either one or two equivalents of the alkyl halide derivatives as shown in Scheme 1. When two equivalents of alkylating agent were added TDPPs 1 were obtained exclusively whereas using fewer equivalents both derivatives 1 and 2 were obtained. They were separated by flash column chromatography (see experimental section in the ESI† for detailed conditions). Synthesis of unsymmetrical TDPP 1CD was performed in two successive steps, reacting TDPP in a first step with di-tert-butyl-dicarbonate to give 2C, followed by subsequent reaction with 1,6-dibromo hexane (Scheme S1, ESI†).
Crystal structure analysis
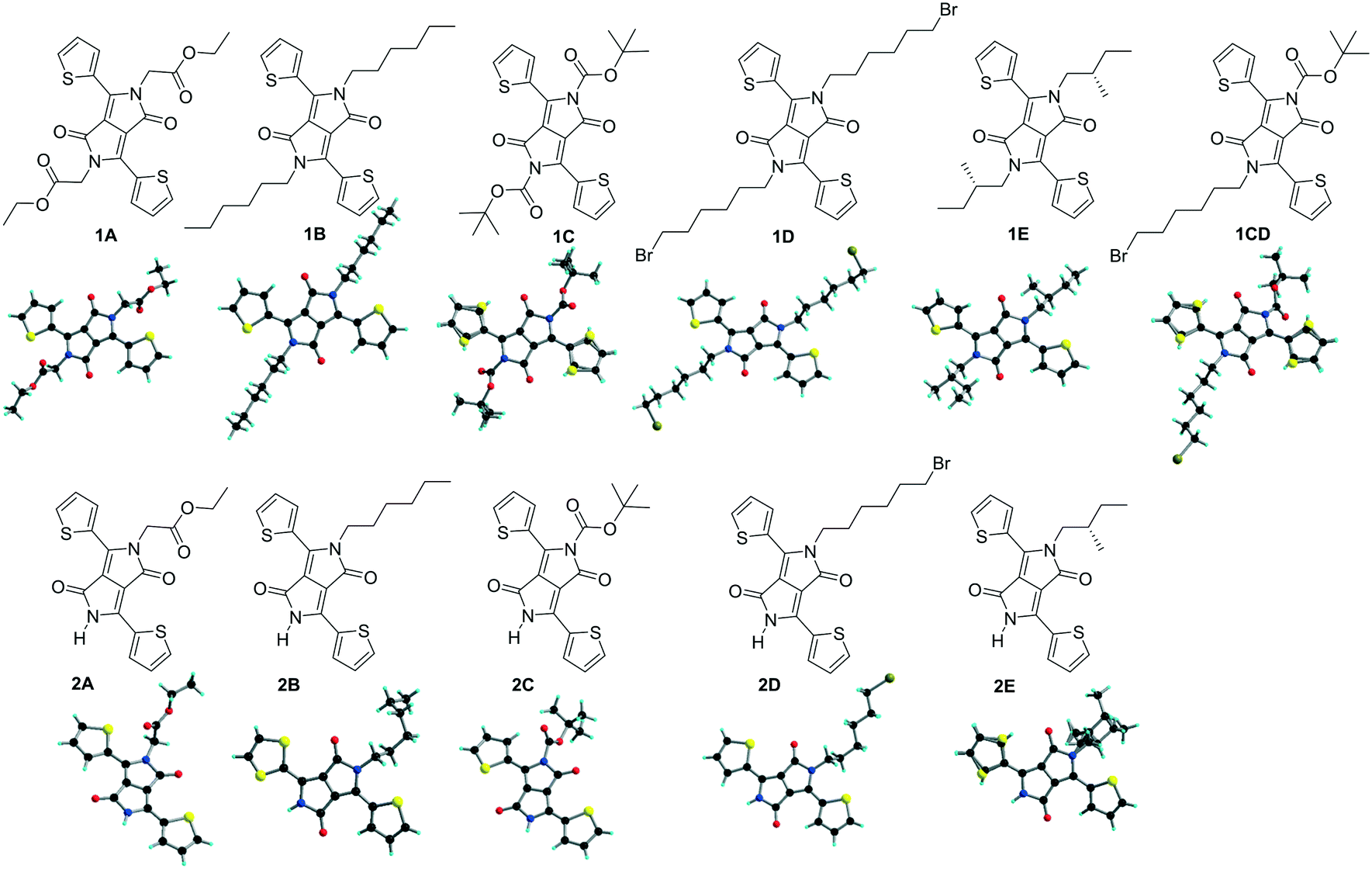
Single crystals with needle and plate shapes were grown by evaporation from dichloromethane/ethyl acetate for most compounds, and dichloromethane (1C) and a mixture of dichloromethane and tetrahydrofuran (2A). Single crystals of 1B14 and 2B13 have been described elsewhere and the crystallographic data for the comparative discussion were taken from the available cif files. The homogeneity of the crystalline forms was checked by comparing the powder X-ray diffraction of the solid samples with theoretical patterns simulated from their single-crystal structures (Fig. S1, ESI†). The experimental peaks in the region of 5−30° match well with simulated ones for the tested compounds. 2E shows very weak diffraction peaks indicating a lower degree of crystallinity in the powder compared to other derivatives. In general, the molecular packing in the powders is very similar to that in single crystals, and there is no clear evidence of polymorphism. The molecular and single crystals structures of all TDPP derivatives discussed are shown in Fig. 1.
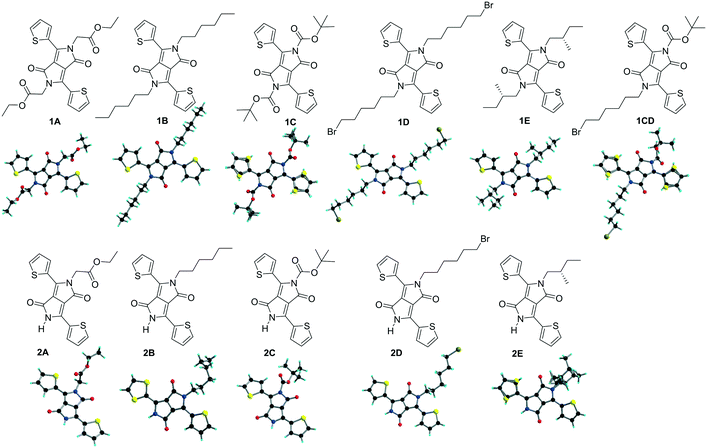 |
| | Fig. 1 Structure of the discussed TDPPs. The structures of 1B and 2B have been taken from previously reported files.13,14 | |
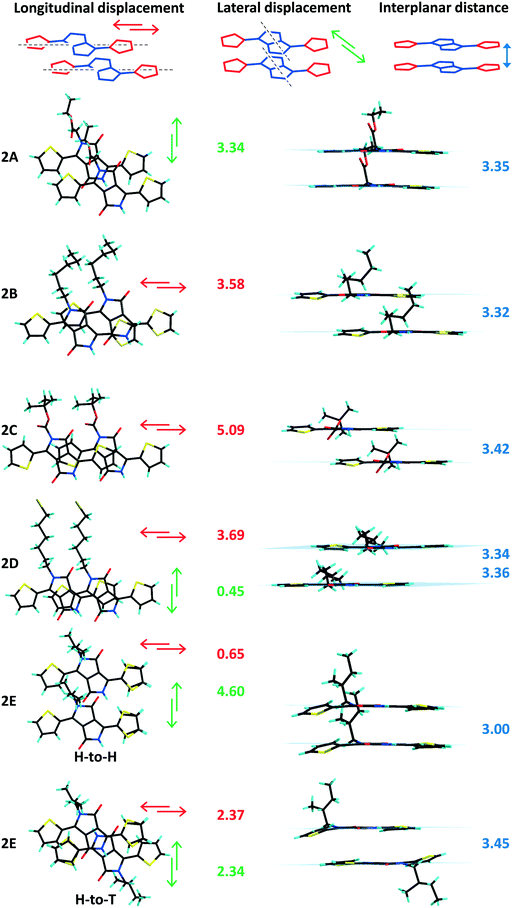
TDPPs 1A–D and 2A–D crystallize in centrosymmetric space groups of the monoclinic and orthorhombic systems whereas chiral compounds 1E and 2E in the non-centrosymmetric space groups P21 and R3 of the monoclinic and trigonal system, respectively (Tables S1 and S2, ESI†). At the molecular level in the crystals, the thiophene rings are arranged in an anti orientation in most of the di-substituted compounds 1 and in 2C, and a syn orientation in the mono-alkylated 2A and 2D, in both cases the sulphur atoms of the thiophene rings face the alkyl chain. In 2C the sulphur atoms of the thiophene units are pointing towards the DPP carbonyl moiety whereas in 1C, 1CD and 2E the thiophenyl units shows structural disorder of the sulphur atom (Fig. 1). The planarity of the molecules has been found to be related to the nature of the appended alkyl substituents and will be addressed later in this work as the degree of twisting of the thiophenyl units and the –N–C(R) bonds out of the plane of the DPP core (Tables 1 and 2).
Table 1 Measured values (Å) of the thiophene⋯lactam centroids, inter-planar distances and molecular displacement (along the long and short axes) together with dihedral angles (°) between the planes of DPP and the adjacent thiophene groups (DPP-Th) and the planes of lactam unit and CLactam–N–C(R) of the alkyl substituent, for TDPP derivatives 1
| TDPP |
Th⋯lactam centroids (Å) |
Inter-planar distance (Å) |
Displacement (Å) |
DPP-Th (°) |
Lactam-[CLactam–N–C(R)] (°) |
| Long |
Short |
|
1A
|
3.40; 3.57 |
3.35 |
4.59 |
|
2.85 and 4.66 |
5.48 and 7.64 |
|
1B
14
|
3.61 |
3.50 |
4.09 |
|
9.67 |
4.96 |
|
1C
|
4.13 |
3.40 |
|
4.77 |
14.18 |
24.96 |
|
1D
|
3.56; 3.60 |
3.37 |
4.18 |
|
8.83 |
2.61 |
|
1E
|
a: 3.69; 3.74 |
a: 3.58 |
a: 4.32 |
|
a: 18.24 and 14.57 |
a: 3.43 and 4.35 |
|
|
b: 3.69; 3.72 |
b: 3.35 |
b: 4.49 |
|
b: 20.70 and 16.84 |
b: 3.98 and 4.39 |
|
1CD
|
3.82 |
3.54 |
4.99 |
2.33 |
17.90 (C) |
31.76 (C) |
| 2.66 (D) |
0.71 (D) |
Table 2 Measured values (Å) of the thiophene⋯lactam centroids, inter-planar distances and molecular displacement (along the long and short axes) together with dihedral angles (°) between the planes of DPP and the adjacent thiophene groups (DPP-Th) and the planes of lactam unit and CLactam–N–C(R) of the alkyl substituent, for TDPP derivatives 2
| TDPP |
Th⋯lactam centroids (Å) |
Inter-planar distance (Å) |
Displacement (Å) |
DPP-Th (°) |
Lactam-[CLactam–N–C(R)] (°) |
| Long |
Short |
|
2A
|
4.81 |
3.35 |
|
3.34 |
0.79; 3.89 (R) |
9.77 |
|
2B
13
|
3.67; 3.71 |
3.32 |
3.58 |
|
3.46; 11.62 (R) |
1.30 |
|
2C
|
3.61; 3.64 |
3.42 |
5.09 |
|
4.20; 13.7 (R) |
a: 4.51 |
| b: 5.97 |
|
2D
|
3.57; 3.60 |
3.34; 3.36 |
3.66 |
0.45 |
a: 3.99; 6.49 (R) |
a: 9.11 |
| b: 1.97; 2.59 (R) |
b: 2.66 |
|
2E, H-to-H |
4.73 |
3.00 |
0.65 |
4.60 |
6.11; 19.82 (R) |
a: 14.13 |
| b: 17.49 |
|
2E, H-to-T |
4.04 |
3.45 |
2.37 |
2.34 |
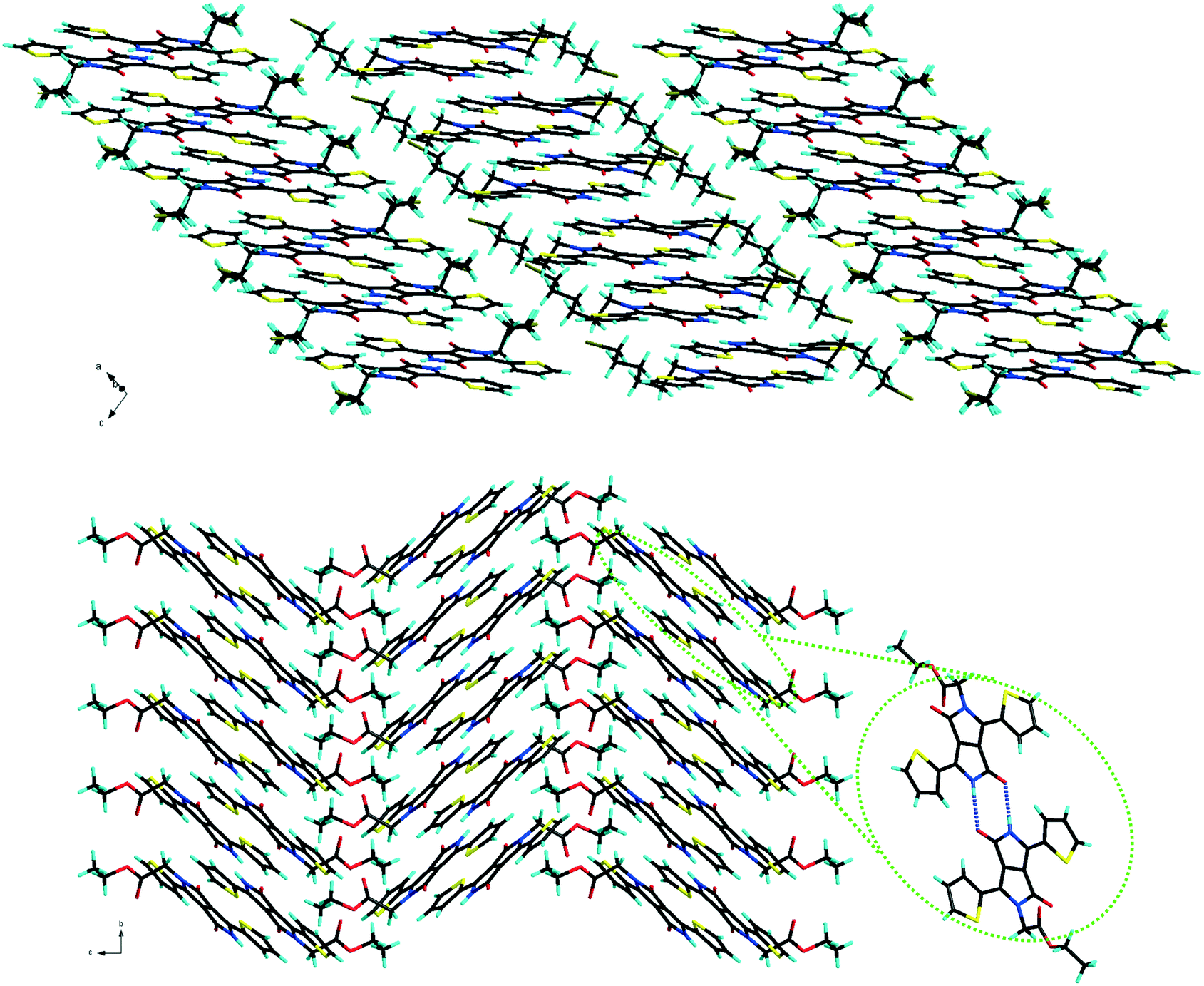
At the supramolecular level, in the crystal packing, the TDPPs bearing two alkyl units (compound family 1) show slipped stacking in a classical edge-to-face herringbone fashion similar to the packing of previously described dihexyl-phenyl DPP and thiophene DPP 1B (Fig. S2, ESI†).12 The mono-alkylated derivatives form strong hydrogen bonds between two molecules through the free amide functionalities and the dimers thus formed further adopt slipped packing as a result of π–π stacking. Most of the mono-alkylated TDPPs (compound family 2) prefer to pack in a slipped co-facial herringbone arrangement similar to the packing observed in previously reported 2B13 except for 2D (which has a slipped one dimensional π stacking of the dimers) and 2E, that shows the classical herringbone type seen in di-alkylated DPPs (Fig. 2 and Fig. S3, ESI†).
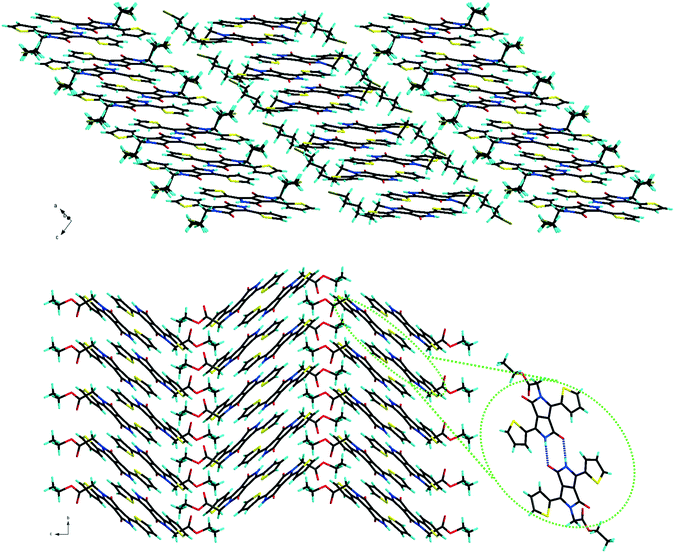 |
| | Fig. 2 Molecular packing of 2D showing anisotropy of the π stacking along a axis (top) and 2A showing co-facial herringbone arrangement with a highlight of the dimer formed by hydrogen bonding (bottom). | |
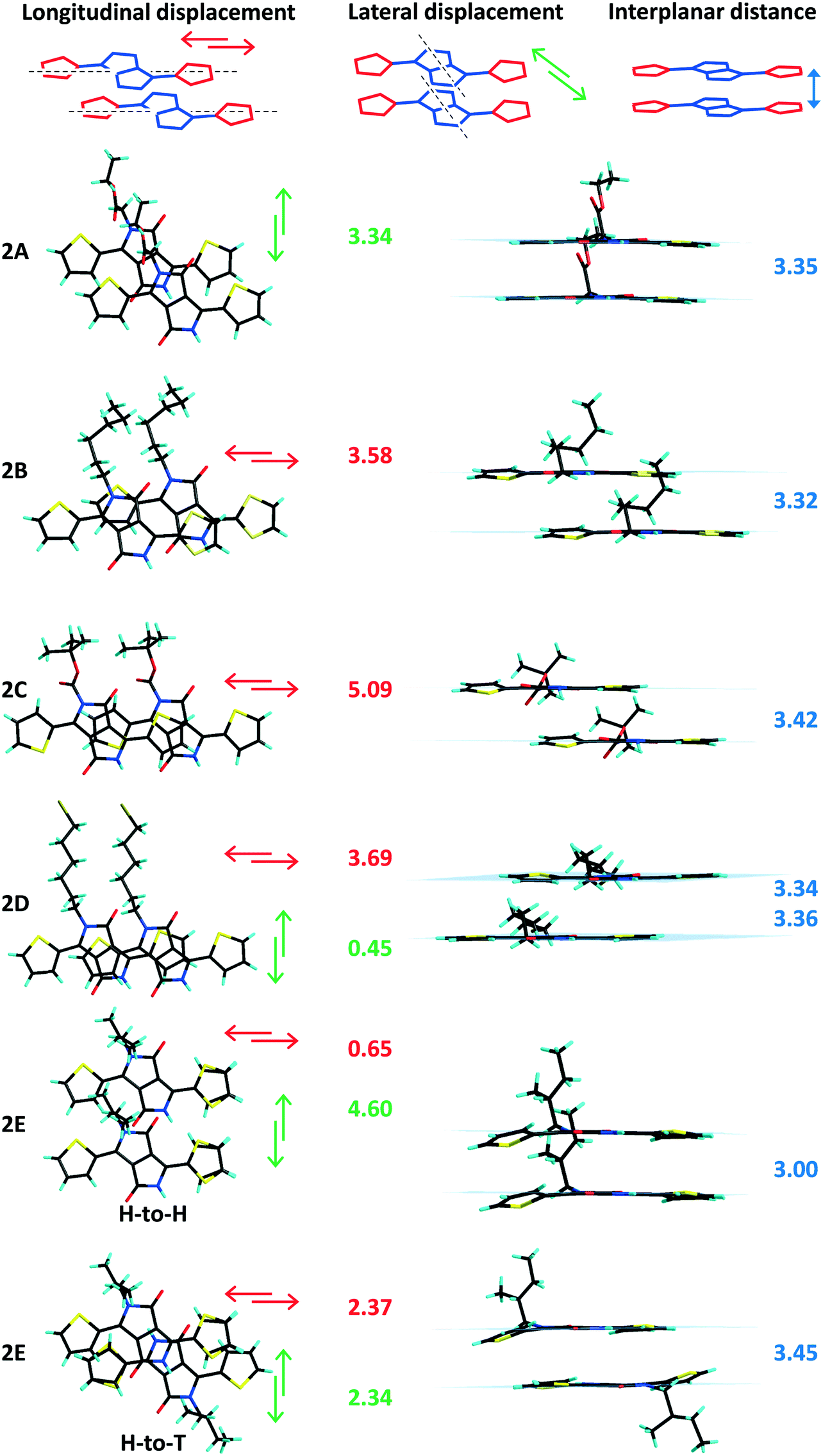
The longitudinal (long axis) slipped stacking is probably a consequence of the thiophenyl units overlapping with the lactam rings of neighbouring molecules, resulting in donor–acceptor reinforced π–π interactions as suggested by the short intermolecular distances observed between the centroids of the electron donor thiophene ring (D) and the electron deficient lactam unit (A) (Fig. S4, ESI†). Significant short D–A intermolecular π–π distances of as little as 3.40 Å and up to 3.82 Å were observed for 1 (A, B, D, E and CD) and 2B–D supported as well by the short plane to plane distances (Fig. 3 and 4). In contrast, molecules 1C, 2A and 2E show high D–A centroid–centroid distances of 4.13, 4.81 and 4.04; 4.73 Å, respectively, as a consequence of both longitudinal and lateral (short axis) slipping (Tables 1 and 2). Furthermore as a result of high lateral slipping 2E shows inter-planar distance between head-to-head molecules of about 3.00 Å and between head-to-tail molecules of 3.45 Å. The latter has a short intermolecular D–A distance (4.04 Å) suggesting stronger π–π stacking in a head-to-tail fashion rather than head-to-head observed in all the other derivatives (Fig. 4 and Fig. S5, ESI†).
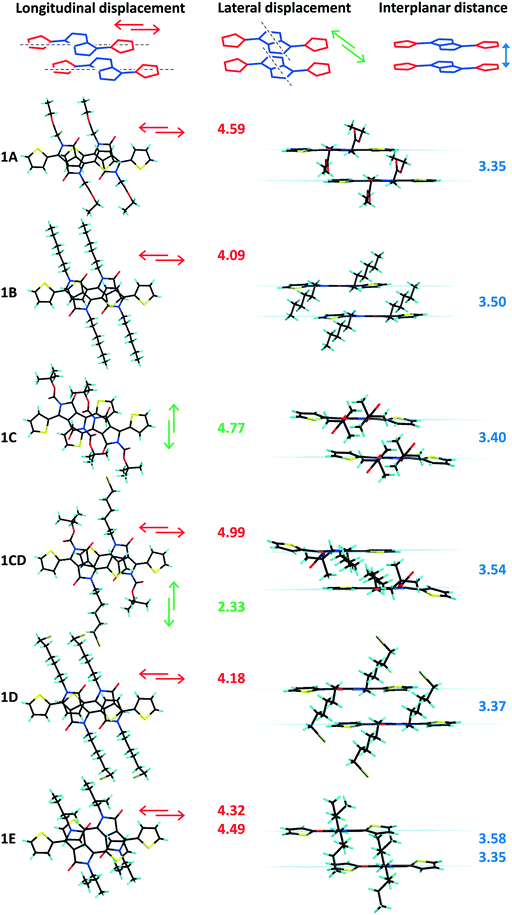 |
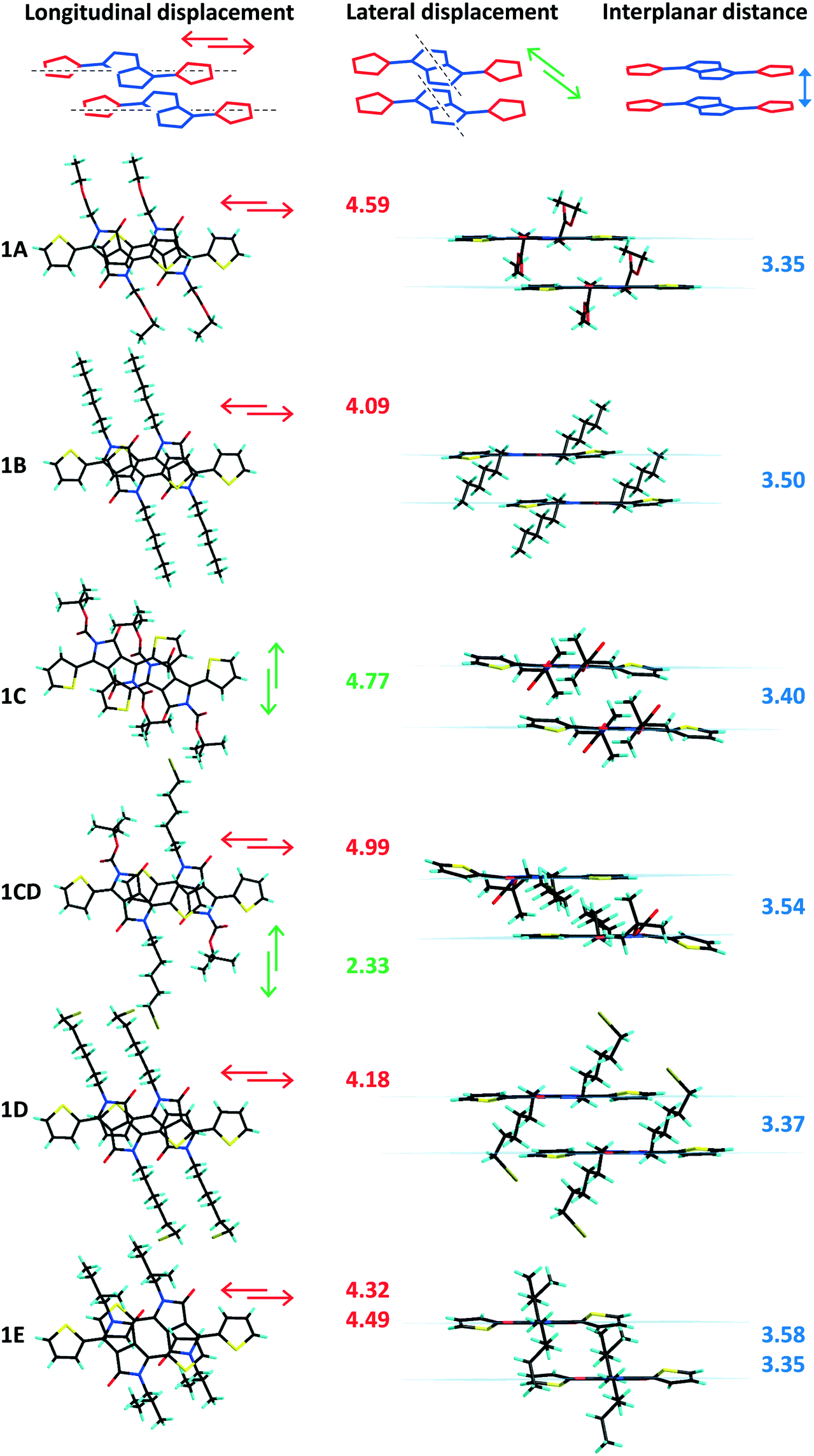
| | Fig. 3 Side and top views of the crystal packing of 1A–E and 1CD, plane to plane distances (blue) together with molecular plane displacement values (longitudinal in red and lateral in green) given in Å. | |
 |
| | Fig. 4 Side and top views of the crystal packing of 2A–E, plane to plane distances (blue) and molecular displacement plane values (longitudinal in red and lateral in green) given in Å. | |
In both series of DPPs inter-planar distances, displacement between parallel molecules (“slipping”) and planarity have been correlated with the nature of the substituent (linear or branched) attached to the nitrogen atom of the lactam ring. Any distortion of planarity of the molecule is expected to have an influence on the charge delocalization along the molecular backbone and is believed to influence their packing in the solid, limiting the contact of the nearby molecules. For analogous phenyl-DPPs the large dihedral angle between the phenyl and lactam rings of 34.13° was associated with steric interactions between the proton of the phenyl ring and the alkyl substituents.12 In thiophenyl-DPP these steric repulsions are significantly reduced, so that the out of plane twisting of the thiophene ring has a relatively small influence on packing. Nevertheless the impact of the bulkiness of the chain is significant and worthy of further discussion. Large dihedral angles between the thiophene and the DPP core originate from steric repulsions between the thiophene rings and the nearby substituents on the nitrogen atoms of the lactam rings irrespective of the orientation of the thiophenyl units (Tables 1 and 2). Beside the thiophene ring twisting out of the molecular plane, distortion of the NLactam–C(R) bond was also found to be highly dependent on the substituent's nature and its bulkiness. Thus the highest inter-planar distances, molecular slipping and planarity distortion have been observed for branched alkyl units C and E (Fig. 3 and 4 and Fig. S7, ESI†). The structures of 1C and 1E show twisting of the thiophene rings out of the plane of the DPP unit by 14.2° and 14.6–20.7°, respectively, suggesting this is mainly caused by the bulkiness of the substituent and not the disposition of the thiophenyl units with respect to the alkyl chains (anti disposition of the sulphur atoms facing the lactam carbonyl group in 1C and the alkyl chain in 1E). This effect can be easily observed in 1CD bearing both linear and bulky chains. The dihedral angle with the DPP core of the thiophenyl unit interacting with the tert-butoxycarbonyl (BOC) substituent (C) is 17.9° whereas the one in the vicinity of the hexyl-bromide (D) remains almost coplanar with the DPP unit (2.66°). In contrast, mono-alkylated TDPPs (family 2) have different dihedral angles of the planes of DPP moiety and the two thiophenyl groups ranging from 0.79° to 6.11° for the thiophene ring adjacent to the free lactam unit and from 2.59° to 19.8° for the thiophene ring interacting with the alkyl chain, demonstrating the influence of the substituent on the overall molecular conformation (Table 2, Fig. S8 and S9, ESI†).
Secondly, compared to other members of their series, 1C, 1CD and 2E also exhibit higher dihedral angles between the plane of the lactam ring and the corresponding CLactam–N–C(R) plane (Tables 1 and 2, Fig. S6, ESI†). The twisting of the BOC unit out of the DPP plane is surely associated with electrostatic repulsion between the carboxylate directly attached to the nitrogen atom and the carbonyl of the lactam ring, whereas in 1A and 2A, with the carboxylate one methylene carbon removed from the lactam ring, the NLactam–C(R) bond is less distorted; the values of the corresponding dihedral angles are similar to the ones of the linear branched derivatives. Therefore, in some cases, flexibility of the molecule overcomes the impact of the alkyl unit bulkiness ensuring its planarity. Additionally the crystallization energy and intermolecular interactions could either overcome or impose strains in the packing of the substituents. Similar observations were made for polymorphs of other thiophenyl-DPPs bearing two BOC units where the deviation of planarity and conformation of the BOC along the NLactam–C(R) bond were found to be directly related.18 In that case one phase contains almost planar molecules with a very small twist of the NLactam–C(R) bond (5.28°) whereas the second phase has a higher distortion from planarity and dihedral angle of the CLactam–N–C(R) plane and the lactam unit (23.4°). The distortion out of the molecular plane of the CLactam–N–C(R) bond could also result from the large lateral displacement of the molecules and thus increased steric impediments between the substituents and the aromatic rings of the nearby molecules. Thus, molecules 1C, 1CD and 2E that have the largest dihedral angles of the CLactam–N–C(R) plane and the lactam unit (25.0°, 31.8° and 17.5°) have the most significant lateral displacement of adjacent molecules in the stack (Tables 1 and 2). Despite the high distortion of planarity 1C, 1E and 2E (head-to-head) are showing short inter-planar distances very likely because of the large molecular displacement (Fig. 3). Shorter inter-planar distances in 1A and 2A could be associated to increased planarity resulting from significant intramolecular carbonyl-thiophene CHTh⋯OLactam interactions reinforced by alkyl-CH⋯OLactam intrastack intermolecular interactions arising from superior flexibility of the substituent compared to BOC unit (Tables S4–S6, Fig. S7, ESI†). In the two families of compounds discussed in here the longitudinal displacement has been associated to either strong D–A interactions or distortion of the thiophenyl ring out of the plane of the DPP whereas in derivatives with lateral displacement different degrees of NLactam–C(R) bond distortion have been observed. In order to further correlate the π–π packing with molecular planarity and intermolecular electronic coupling solution and solid absorption investigations have been performed on both families of compounds and are discussed below.
Spectroscopic properties
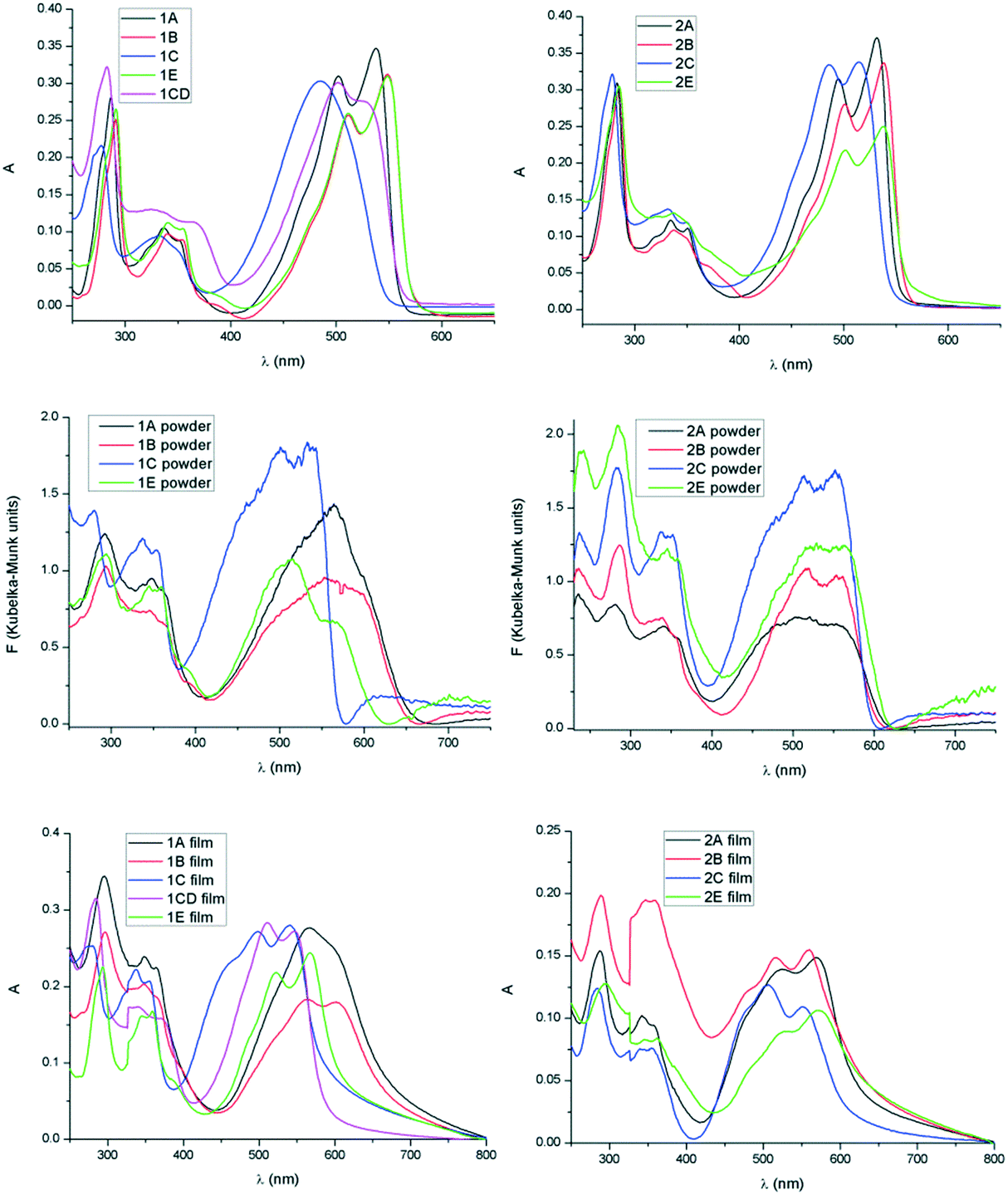
The UV-visible absorption spectra of all compounds have been examined in solution and using diffuse reflectance spectroscopy in the solid state. All TDPP derivatives exhibit dual band absorption profiles common for compounds with a donor–acceptor primary structure.19 The absorption profiles show intense low energy bands at approximately 500 nm and weaker high energy bands at around 340 nm attributed to strong intramolecular interactions from the thiophenyl donor to the DPP acceptor and π–π transitions localized on either the donor or the acceptor, respectively (Fig. 5, Tables 3 and 4).20 Along the series of alkyl units a shift of the absorption maximum correlates with the electron withdrawing effect of the substituent. Therefore in both mono- and di-alkyl DPPs a blue shift occurs in the order B (or E), A, C when moving from simple hydrocarbons to carboxylate. The electron withdrawing effect of the substituent can be further seen in non-symmetrical 1CD that is blue shifted compared to analogous symmetrical 1B and red shifted compared to 1C (Fig. 5, top left).
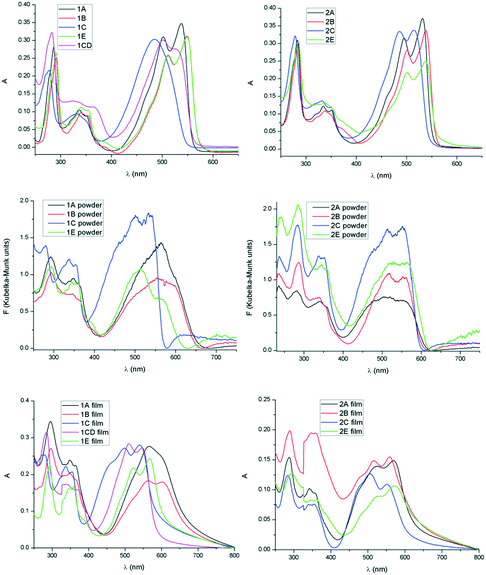 |
| | Fig. 5 Absorption spectra of compounds 1A–C, E and 2A–C, E in solution (CH2Cl2 10−5 M, top), as powder (by diffuse reflectance mode and transformed to Kubelka–Munk units,21 middle) and as film (bottom). | |
Table 3 Absorption data in dichloromethane solution, conc. 10−5 M, as powder and film for TDPP derivatives 1
| TDPP |
λ
max (nm) solution |
ε M−1 cm−1 |
λ
max (nm) powder |
λ
max (nm) film |
|
1A
|
538 |
34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
578 |
567 |
|
1B
|
548 |
31055 |
603 |
601 |
|
1C
|
485 |
30270 |
544 |
540 |
|
1E
|
548 |
31055 |
575 |
568 |
|
1CD
|
528 |
27435 |
|
544 |
Table 4 Absorption data in dichloromethane solution, conc. 10−5 M, as powder and film for TDPP derivatives 2
| TDPP |
λ
max (nm) solution |
ε M−1 cm−1 |
λ
max (nm) powder |
λ
max (nm) film |
|
2A
|
531 |
37067 |
572 |
569 |
|
2B
|
538 |
33705 |
563 |
559 |
|
2C
|
514 |
33791 |
560 |
552 |
|
2E
|
538 |
24505 |
573 |
571 |
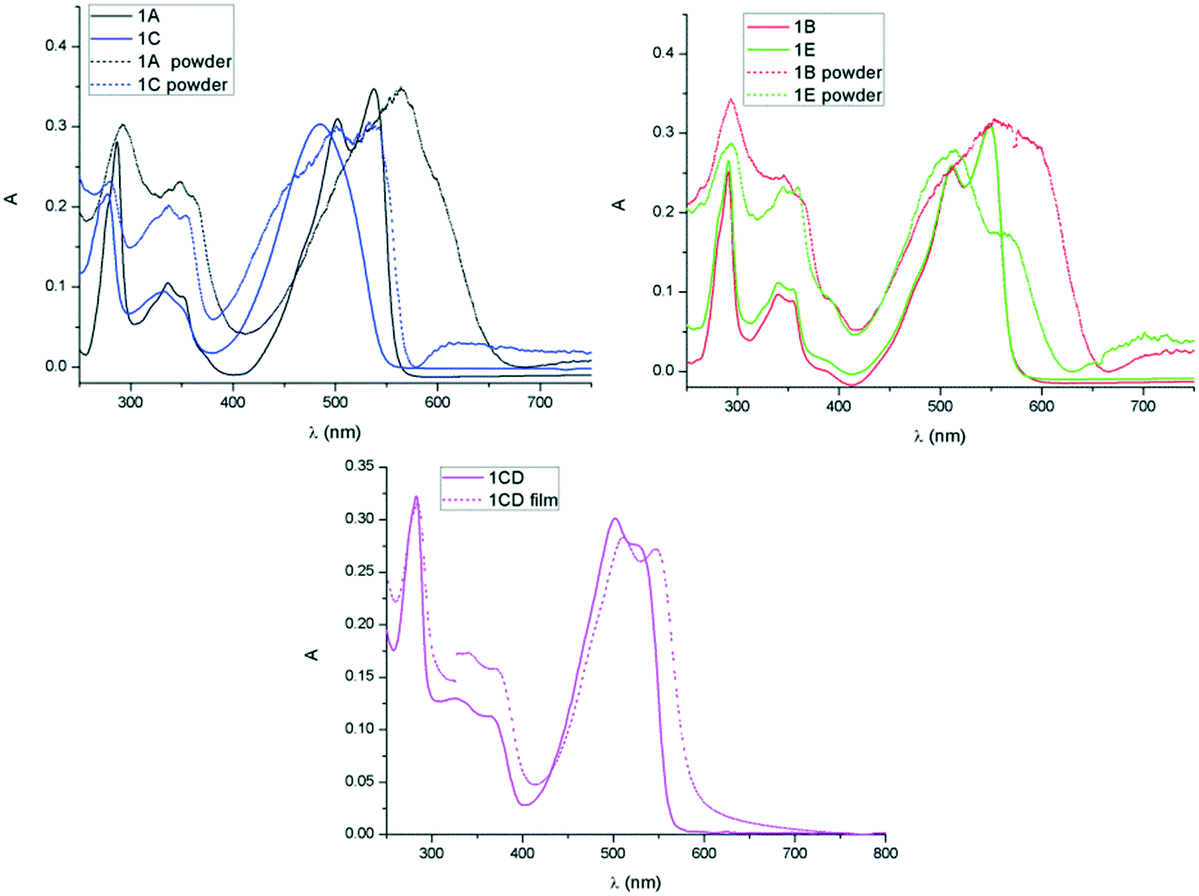
In all derivatives the absorption peaks of the solids (powder and film) are broader and red shifted by about 50 nm compared to the solution state bands, which is indicative of increased intermolecular interactions in the solid state. In the absorption spectra of the films the low energy regions show shoulder peaks that result from vibronic coupling of adjacent molecules.22 The emerging shoulder of the lower energy band has been associated to the formation of H-type aggregates and the red shift of the lower energy band to the formation of J-type aggregation.23 The maximum of absorption of 1E is only slightly red shifted whereas 1B possessing linear chains shows higher red shift which is consistent with J-type aggregation and increased conjugation24 because of the highly planar conformation in the solid state (Fig. 6 and Table 3). A similar effect was observed in the solid state spectra of 1A, a higher red shift suggesting a more dominant J type stacking compared to 1C, probably as consequence of reduced steric impediments of the A substituent and thus increased molecular planarity. These observations are in line with previous results on simple di-alkylated TDPP derivatives that show complex J and H type aggregation depending on the nature of the hydrocarbon chain, linear versus branched.25
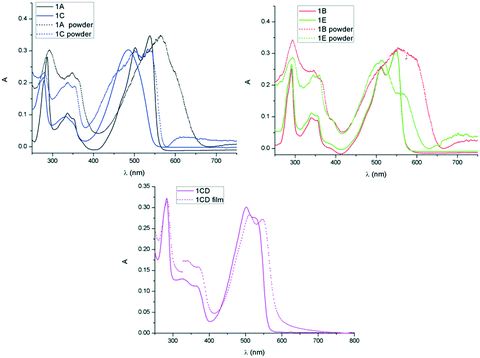 |
| | Fig. 6 Comparison of absorption spectra in solution and solid of 1A and 1C (top left), 1B and 1E (top right) and 1CD (bottom). | |
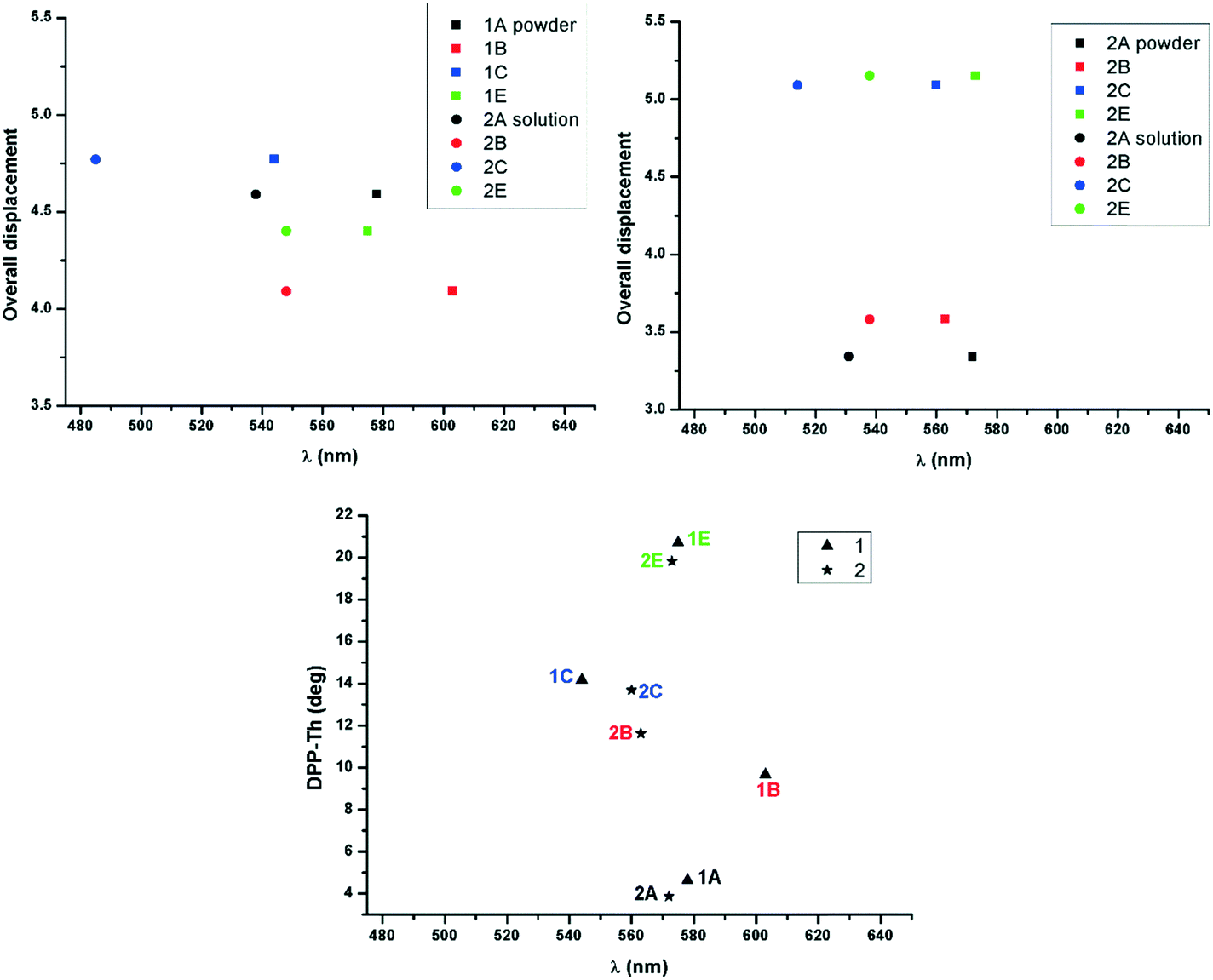
The absorption maximum in the solid has been correlated with the molecular geometry and relative displacement in the stacks for the two series of derivatives (Fig. 7). In the di-alkylated series the highest red shift of absorption maximum of the lowest energy band has been observed for molecule 1B whose geometry proved to be least distorted in the crystal (small dihedral angle between the DPP and the adjacent thiophenyl unit). Therefore the molecular overlap and red shift is increasing in the order 1C, 1E, 1A and 1B in the di-alkylated series and 2E, 2C, 2B and 2A in the mono-alkylated series. In the mono-alkylated series differences of the maximum of absorption with the nature of the alkyl unit are less significant (Fig. 5, right and Table 4). Thus a slight or no red shift of the maximum of absorption of the lowest energy transition was observed for 2A and 2B compared to 2E and 2C possibly because of small molecular displacement and therefore higher contribution to the absorption band of the H-type aggregation. This feature might be because intermolecular hydrogen bonding allows the alignment of the transition dipoles,26 and therefore strong close packing and π–π stacking. In both series the molecular planarity is increasing in the order E, C, B and A (Fig. 7, bottom). The molecular overlap in 2A is highest of the series as a consequence of planarity and intermolecular hydrogen bonding. Therefore mono-alkyl TDPP based on A alkyl units are expected to have superior charge carrier mobility reported for example on 2B, because of improved close packing.13
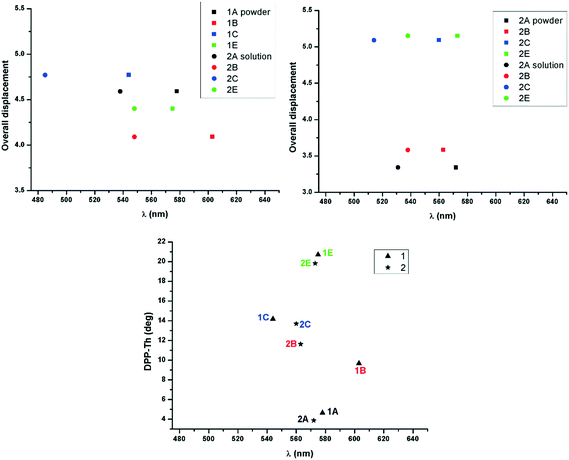 |
| | Fig. 7 Maximum of absorption in the solid and solution plotted against the molecular displacement for 1 (top left) and 2 (top right), and in the solid against the dihedral angles between the planes of DPP and the adjacent thiophene rings (bottom). | |
Conclusions
The intermolecular packing of TDPP derivatives in single crystals has been found to be highly dependent on the nature of the alkyl units at the lateral amide-nitrogen atoms. The spectroscopic properties of the two series of TDPPs were directly correlated to the geometry of the aromatic core that was found to be strongly influenced by the side substituent. As previously shown linear chains allow close packing of the molecules due to reduce steric impediments whereas branched chains will increase their solubility. Nevertheless branched solubilising chains have to be carefully chosen as they not only influence the close packing of the molecules through their bulkiness but have as well a huge impact on the planarity of the molecule and thus affecting both the close packing and the conjugation. In both series of compounds, bulkier substituents, methyl-butyl (E) and BOC (C), distort the planarity of the conjugated system showing high dihedral angles between the DPP core and the adjacent thiophene or the CLactam–N–C(R) plane. Moreover 1C, 1CD and 2E that have the largest dihedral angles of the CLactam–N–C(R) plane and the corresponding lactam rings show the most significant lateral displacement of neighbouring molecules. These packing effects influence directly the visible light absorption properties of the materials. The maximum of absorption of solid 1E is only slightly red shifted with respect to solution whereas 1B possessing linear chains shows higher red shift which is consistent with J type aggregation and increased conjugation because of the higher planar conformation. All mono-alkylated TDPPs show similar absorption maxima in the solid (differences of about 10 nm) because of strong close packing as a consequence of both increased planarity because of reduced steric impediments and intermolecular hydrogen bonding of the free lactam units. Molecular displacement correlated with the red shift of the lowest energy band suggests linear alkyl chains (type B) as ideal substituents for close packing of the molecules in the solid, while favourable π-core geometry is also achieved with the ethyl acetate substituent (type A). Beside the effect on the molecular planarity in the solid, ethyl acetate substituents could be useful in switching the solubility of the TDPPs towards polar solvents potentially giving a control over the packing in the bulk materials, a possibility we are exploring presently. These results are particularly pertinent given the very recent report on the geometry dependence of singlet fission in TDPP derivatives,27 an effect which is promising for the application of these materials in optoelectronic devices. We believe that the identification of these multifarious aggregation modes will also aid in the interpretation of the supramolecular arrangements in devices in which precise structural characterisation is not possible.
Acknowledgements
We thank the EPSRC, GSK, and the School of Chemistry at the University of Nottingham for funding, and STFC/Diamond for access to Diamond Light Source Beamline I19.28
Notes and references
-
(a) S. S. Babu, S. Prasanthkumar and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 1766 CrossRef CAS PubMed;
(b) A. M. Haruk and J. M. Mativetsky, Int. J. Mol. Sci., 2015, 16, 13381 CrossRef CAS PubMed.
-
(a) T. Aytun, L. Barreda, A. Ruiz-Carretero, J. A. Lehrman and S. I. Stupp, Chem. Mater., 2015, 27, 1201 CrossRef CAS;
(b) J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, J. Am. Chem. Soc., 2016, 138, 173 CrossRef CAS PubMed.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784 CrossRef CAS PubMed.
-
(a) S. Rieth, Z. Li, C. E. Hinkle, C. X. Guzman, J. J. Lee, S. I. Nehme and A. B. Braunschweig, J. Phys. Chem. C, 2013, 117, 11347 CrossRef CAS;
(b) Y. Zhou, C. X. Guzman, L. C. Helguero-Kelley, C. Liu, S. R. Peurifoy, B. Captain and A. B. Braunschweig, J. Phys. Org. Chem., 2016 DOI:10.1002/poc.3548.
- D. Ley, C. X. Guzman, K. H. Adolfsson, A. M. Scott and A. B. Braunschweig, J. Am. Chem. Soc., 2014, 136, 7809 CrossRef CAS PubMed; C. X. Guzman, R. M. K. Calderon, Z. Li, S. Yamazaki, S. R. Peurifoy, C. Guo, S. K. Davidowski, M. M. A. Mazza, X. Han, G. Holland, A. M. Scott and A. B. Braunschweig, J. Phys. Chem. C, 2015, 119, 19584 Search PubMed.
-
(a) P. M. Beaujuge and J. M. J. Frechet, J. Am. Chem. Soc., 2011, 133, 20009 CrossRef CAS PubMed;
(b) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed.
- M. A. Naik and S. Patil, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4241 CrossRef CAS.
- D. Chandran and K.-S. Lee, Macromol. Res., 2013, 21, 272 CrossRef CAS.
- M. Kaur and D. H. Choi, Chem. Soc. Rev., 2015, 44, 58 RSC.
- Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684 CAS.
- C. Kim, J. Liu, J. Lin, A. B. Tamayo, B. Walker, G. Wu and T.-Q. Nguyen, Chem. Mater., 2012, 24, 1699 CrossRef CAS.
-
(a) J. Dhar, N. Venkatramaiah, A. Anitha and S. Patil, J. Mater. Chem. C, 2014, 2, 3457 RSC;
(b) C. Fu, F. Bélanger-Gariépy and D. F. Perepichka, CrystEngComm, 2016, 18, 4285 RSC.
- J. Dhar, D. P. Karothu and S. Patil, Chem. Commun., 2015, 51, 97 RSC.
- M. A. Naik, N. Vnekatramaiah, C. Kanimozhi and S. Patil, J. Phys. Chem. C, 2012, 116, 26128 CAS.
-
(a) J. Calvo-Castro, M. Warzecha, A. R. Kennedy, C. J. McHugh and A. J. McLean, Cryst. Growth Des., 2014, 14, 4849 CrossRef CAS;
(b) J. Calvo-Castro, M. Warzecha, I. D. H. Oswald, A. R. Kennedy, G. Morris, A. J. McLean and C. J. McHugh, Cryst. Growth Des., 2016, 16, 1531 CrossRef CAS.
- P. E. Hartnett, E. A. Margulies, C. M. Mauck, S. A. Miller, Y. Wu, Y.-L. Wu, T. J. Marks and M. R. Wasielewski, J. Phys. Chem. B, 2016, 120, 1357 CrossRef CAS PubMed.
-
(a) A. B. Tamayo, B. Walker and T.-Q. Nguyen, J. Phys. Chem. C, 2008, 112, 11545 CrossRef CAS;
(b) A. B. Tamayo, M. Tantiwiwat, B. Walker and T.-Q. Nguyen, J. Phys. Chem. C, 2008, 112, 15543 CrossRef CAS.
- S. T. Salammal, J.-Y. Balandier, J.-B. Arlin, Y. Olivier, V. Lemaur, L. Wang, D. Beljonne, J. Cornil, A. R. Kennedy, Y. G. Geerts and B. Chattopadhyay, J. Phys. Chem. C, 2014, 118, 657 CAS.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396 CrossRef CAS PubMed.
-
(a) G. L. Gibson, T. M. McCormick and D. S. Seferos, J. Am. Chem. Soc., 2012, 134, 539 CrossRef CAS PubMed;
(b) U. Salzner, J. Chem. Theory Comput., 2014, 10, 4921 CrossRef CAS PubMed.
-
J. Torrent and V. Barron, in Methods of Soil Analysis Part 5—Mineralogical Methods, ed. A. L. Ulery and L. R. Drees, Soil Science Society of America, Inc., Madison, Wisconsin, USA, 2008, vol. 13, pp. 367–385 Search PubMed.
- A. B. Koren, M. D. Curtis, A. H. Francis and J. W. Kampf, J. Am. Chem. Soc., 2003, 125, 5040 CrossRef CAS PubMed.
- W. Shin, T. Yasuda, G. Watanabe, Y. S. Yang and C. Adachi, Chem. Mater., 2013, 25, 2549 CrossRef CAS.
- J. Dhar, T. Mukhopadhay, N. Yaacobi-Gross, T. D. Anthopoulos, U. Salzner, S. Swaraj and S. Patil, J. Phys. Chem. B, 2015, 119, 11307 CrossRef CAS PubMed.
- M. Kirkus, L. Wang, S. Mothy, D. Beljonne, J. Cornil, R. A. J. Janssen and S. C. J. Meskers, J. Phys. Chem. A, 2012, 116, 7927 CrossRef CAS PubMed.
- J. Mizuguchiand and H. Shikamori, J. Phys. Chem. B, 2004, 108, 2154 CrossRef.
- C. M. Mauck, P. E. Hartnett, E. A. Margulies, L. Ma, C. E. Miller, G. C. Schatz, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2016, 138, 11749 CrossRef CAS PubMed.
- H. Nowell, S. A. Barnett, K. E. Christensen, S. J. Teat and D. R. Allan, J. Synchrotron Radiat., 2012, 19, 435 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental section and synthetic procedures, additional solid and solution data and crystallographic data tables. CCDC 1508451–1508459 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce02157c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a and
David B.
Amabilino
*ab
a and
David B.
Amabilino
*ab