
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRationalization of the formation and stability of bosutinib solvated forms†
Eszter
Tieger
*ab,
Violetta
Kiss‡
bc,
György
Pokol
a,
Zoltán
Finta
d,
Jan
Rohlíček
e,
Eliška
Skořepová
bf and
Michal
Dušek
e
aDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Szt. Gellért tér 4, 1111 Budapest, Hungary. E-mail: tiegereszter@gmail.com
bZentiva k.s, U kabelovny 130, 102 37, Prague, Czech Republic
cJanssen Pharmaceuticals, Inc. - Pharmaceutical Companies of Johnson & Johnson, Turnhoutseweg 30, B-2340 Beerse, Belgium
dChinoin Zrt, Tó utca 1-5, 1045 Budapest, Hungary
eInstitute of Physics of the Czech Academy of Sciences, Na Slovance 2, 182 21 Prague, Czech Republic
fDepartment of Solid State Chemistry, University of Chemistry and Technology, Technicka 5, 166 28 Prague, Czech Republic
First published on 22nd November 2016
Abstract
Crystallization of bosutinib from a wide variety of solvents resulted in many distinct structures, and displayed difficulties to crystallize without solvent incorporation. We have prepared 23 solvated/hydrated and one anhydrous solid form and for 11 of them solved the crystal structures. With the goal of rationalizing the high propensity to solvate formation and exploring the stability relationships between the phases, the solid forms were characterized by different experimental and computational methods. Their stability was compared theoretically by calculating the packing efficiency in the crystal structures and the binding energy of the solvent in the crystal lattice using differential scanning calorimetry. Experimental studies were completed by analysing the forms' physical stability in solid state and in suspension and their intrinsic dissolution rate. The survey conducted on the inclusion compounds has resulted in basic understanding of the underlying factors affecting discriminative solvate formation: the solvents are utilised to satisfy previously unused hydrogen bonding capabilities in the host molecule.
Introduction
Polymorphism has been defined as crystal systems for which a substance of the same elemental composition can exist in different crystal structures. The widely disputed term pseudopolymorphism or solvatomorphism refers to those systems where phases differ in their elemental composition through the inclusion of various solvents.1 The differences between anhydrates and hydrates (solvates) are significant. An anhydrous crystalline form is a one component system, and its free energy is specified by temperature and pressure. A crystalline hydrate (solvate) is a two-component system and is specified by temperature, pressure and water (solvent) activity.2The understanding of the physical and chemical properties of different solid forms enables the identification of the most appropriate modification for use in drug development by assessing the risk of form changes under normal and accelerated conditions. The utilization of the most physically stable crystalline form is typically desired as any change in solid form may affect the bioavailability associated with a drug product.3
Pharmaceutical scientists usually search for a solid form with the best properties for therapeutic use and manufacturability. Anhydrous forms are typically preferred over hydrates because they are generally expected to have superior thermal stability and higher aqueous solubility. But often a hydrate is the most stable phase under ambient conditions and therefore this is the selected form for development. The selection process between anhydrous and hydrated/solvated forms is complex, as various factors must be considered including solubility, dissolution profile, processability, hydration–dehydration behaviour, and solid-state stability.4
As generic companies are seeking for new, non-infringing solid forms, solvates are an option to circumvent innovators' patents. Despite their potential to improve the in vitro dissolution kinetics,5–7 solvates are rarely selected for development due to toxicity and regulatory concerns. Up to the present, there are only a few solvates on the market: trametinib,8 dapagliflozin,9 cabazitaxel,10 darunavir,11 warfarin sodium,12 indinavir sulfate,13 and atorvastatin calcium.14
Processing, stability testing and storage can all induce changes in the solvation state of the drug, as they are submitted to various temperatures and solvent activities during different unit operations, giving rise to unexpected solvation/desolvation processes. Therefore during the development of a dosage form, it is essential to determine the conditions of temperature and water/solvent activity under which different solid forms of the drug are stable.15
Bosutinib is a second-generation tyrosine kinase inhibitor used in the treatment of chronic myelogenous leukemia with resistance or intolerance to prior therapy. The API is marketed under the trade name Bosulif®, (Pfizer Inc., New York, NY, USA) as a monohydrate.
Bosutinib has a rich solid form landscape composed of one neat form, five hydrates, many pure and mixed solvates and an amorphous phase. It is known to be a promiscuous solvate former.16
Four solid forms investigated in this study are claimed by Pfizer:17 form I and II monohydrates, denoted here as 1H2O-I and 1H2O-II; form III: a mixed solvate with isopropanol and water (IPA–2H2O) and form V anhydrate (AH). The crystal structure of 1H2O-I was reported recently.18
The mixed solvate with methanol and water and the dihydrate forms are claimed by Zentiva, referred to as MeOH(H2O) and 2H2O.19
Vaidyanathan and coworkers described the higher order hydrate phase of bosutinib,16 denoted as a hexahydrate. As a result of our investigations and single crystal data, we designated this phase as a heptahydrate, 7H2O. The hexahydrate form, 6H2O, also exists and is examined in this study, but it is probably not identical to the form described in that paper.
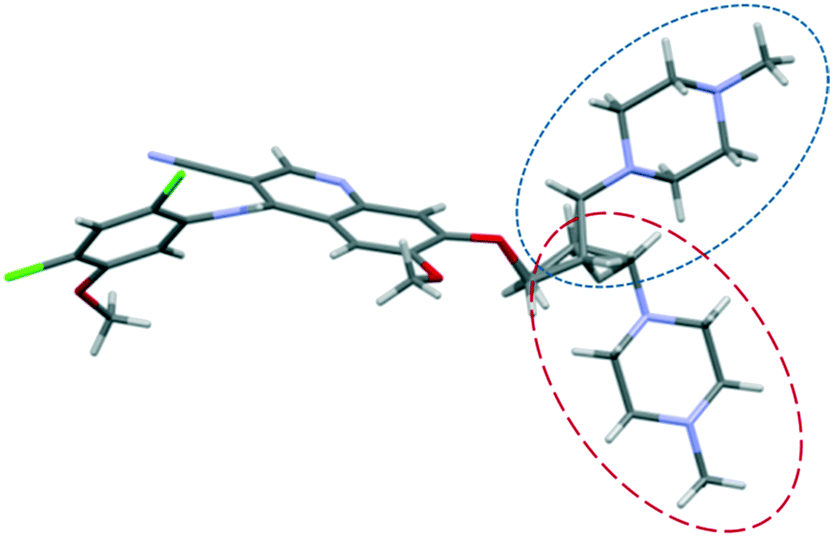
The structure of bosutinib (Fig. 1) shows that one potential hydrogen bond donor and 8 potential acceptor groups exist in this molecule, suggesting it might favor solvate formation.20 To compensate such misbalance between donors and acceptors, the API includes solvent molecules in its crystal lattice.21a,b
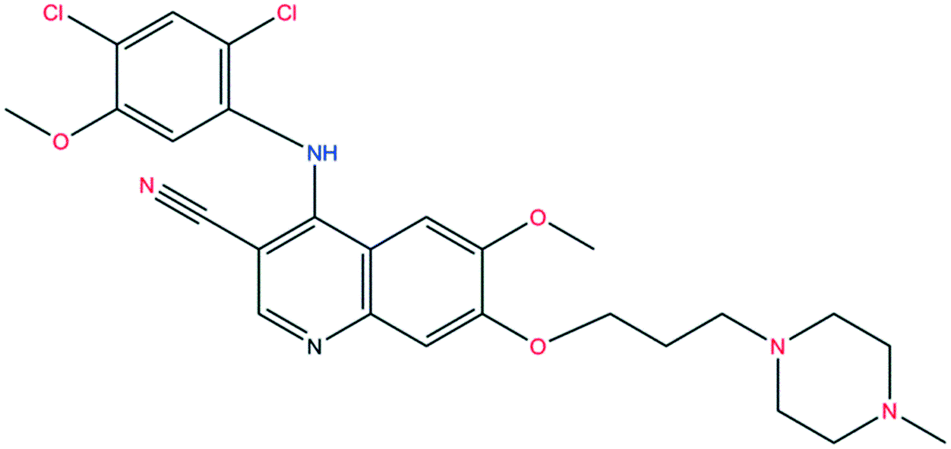 | ||
| Fig. 1 Molecular structure of bosutinib. Hydrogen bond donor sites are marked in blue, acceptor sites in red. | ||
Bosutinib incorporates only specific solvent molecules as essential components of the crystal structure, with certain host–solvent interactions. This kind of discriminative solvate formation often occurs for drugs where the number and disposition of the hydrogen bond donors or acceptors mean that they cannot all be involved in hydrogen bonding.22
Bosutinib's propensity to form solvates in the majority of solvents to which it is exposed presented a problem in discovering a new anhydrous phase. Our aim was to reveal the reason of the high propensity to solvate formation and to rationalize the stability of the solid forms by different approaches.
Their stability was compared theoretically by calculating the packing efficiency in the structures and the binding energy of the solvent in the crystal lattice using differential scanning calorimetry. Experimental studies were completed by analysing the forms' physical stability in the solid state and in suspension and their intrinsic dissolution rate. Numerous solid forms were discovered, but in this paper, we confine our study to the forms, for which the crystal structure solution was successful.
Experimental section
Materials and methods
Preparation method of 7H2O, 6H2O, IPA–2H2O, MeOH(H2O), Diox–2H2O, DMSO–3H2O, 2-BuOH. Concentrated solutions of bosutinib were prepared in different solvents (see the ESI†) close to the boiling point of the solvents. The clear solutions were filtered through a filter paper to get rid of the insoluble particles and were slowly cooled down to 0 °C. The obtained products were collected by filtration and were air-dried. Bosutinib 1H2O-II was obtained by dehydrating 7H2O in a vacuum oven at 40 °C and 150 mbar pressure. Bosutinib·2H2O was crystallized by quickly cooling the saturated solution in dichloromethane.
Preparation method of AH. 1 g of bosutinib free base was dissolved in 90 mL of dichloromethane at room temperature and was filtered through a filter paper into a round bottom flask. The solution was evaporated in a vacuum rotary evaporator to complete dryness using a 40 °C pre-heated water bath and 150 mbar pressure. The dry residue was removed from the flask by rinsing it with a few mL of diethyl ether and the obtained suspension was stirred for 4 h at ambient temperature. The obtained products were collected by filtration and were air-dried.
Differential scanning calorimetry. DSC measurements were performed using a Mettler-Toledo 822e DSC. Samples were placed into standard aluminum pans (40 μL) sealed with a pierced lid. The sample cell was heated under a nitrogen purge at a rate of 10 °C min−1, from 25 °C up to a final temperature of 300 °C with a 50 mL min−1 nitrogen purge.
Thermogravimetric analysis. TGA analyses were performed using a NETZSCH TG 209 thermogravimetric analyser (NETZSCH-Gerätebau GmbH, Germany). Each sample was placed in an aluminum sample pan and inserted into the TG furnace. The furnace was heated under a nitrogen purge at a rate of 10 °C min−1, from 25 °C up to the final temperature of 300 °C.
Critical water activity. A series of acetone/water, acetonitrile/water, 2-propanol/water and methanol/water binary mixtures of varying water contents was prepared and saturated with the compound. A small amount of all obtained forms was added to each. The suspensions were shaken at room temperatures for 14 days in a thermoshaker, then the solids were isolated and characterized by XRPD and Raman spectroscopy. The water activity of the binary mixtures was calculated using an NRTL (non random two-liquid) model of VLE (vapor liquid equilibrium) data.
Rotating disk dissolution rates. Disks were prepared by compressing ∼25 mg of the drug in a die with a 100 kg load force (in the case of 7H2O, 50 kg was used to avoid dehydration) for 120 s using a screw press (Sirius Analytical Instr. Ltd., Forest Row., UK). The exposed surface area for the resulting disk was 0.125 cm2. In the case of AH, both 0.125 cm2 and 0.07 cm2 surface areas were applied in an attempt to avoid/decrease disintegration. A USP dissolution set-up maintained at 37 °C was used for the study. Dissolution measurements were performed using the Sirius inForm platform (Sirius Analytical Instr. Ltd., Forest Row., UK) with built in pH measurement and UV fibre optic spectroscopy. Each dissolution vessel contained 60 mL of an aqueous dissolution medium maintained at a constant pH (0.1 M, acetate buffer pH 4.5). The disk holder (die) was immersed into the dissolution medium and rotated at 100 rpm. The amount of dissolved API was determined from multi wavelength UV absorption measured using an in situ dip probe. Spectra were recorded every 30 s.
Data collection – single crystal. All bosutinib crystals were prepared by slow cooling of saturated solutions. The single crystal X-ray diffraction analysis was carried out at a temperature of 120 K using a diffractometer Xcalibur, Atlas, Gemini ultra with a mirror–monochromator and a CCD detector. Cu Kα radiation with a wavelength of 1.5418 Å was also used. The collection and data reduction program employed was CrysAlisPro (Agilent Technologies, version 1.171.36.28). An empirical correction for absorption was done by the scaling algorithm SCALE3 ABSPACK. The structure was solved by direct methods (program SIR92 (ref. 23)) and refined in the program CRYSTALS 14.40b.24 All non-hydrogen atoms were refined anisotropically. The simulated XRPD patterns were calculated using MERCURY software25 (version 3.3).
Structure determination from powder data. The samples were ground and placed into a 0.3 mm borosilicate glass capillary and were measured at room temperature, 120 K and 150 K in transmission mode on a PANalytical Empyrean powder diffractometer from 4° to 80° 2theta with CuKα1,2 radiation (λ = 1.54184 Å, focusing mirror; the step size was 0.013° 2theta).
Refinement of the crystal structures of AH and 1H2O-II. The procedure was almost identical for both crystal structures. Powder diffraction data of both samples were indexed by the program Conograph.26 After confirmation of the unit cell by Le Bail refinement in the program JANA2006,27 the structure solution was performed by using a direct space approach as it is implemented in the program FOX.28
The molecular model of bosutinib was taken from, at that time, the already refined crystal structure of 1H2O-I (the model was allowed to be flexible). In the case of 1H2O-II, one oxygen atom was added to the asymmetric part of the unit cell to simulate the presence of the water molecule. In both cases, the best solution was used for the Rietveld refinement.
In the Rietveld refinement, the initial model was kept as a rigid body and only one overall isotropic ADP parameter was refined at the beginning to fit the scale parameter and to see the correctness of the model. In the final Rietveld refinement, we removed the rigid body constraint and we refined positions and one isotropic ADP parameter for all non-hydrogen atoms. Hydrogen atoms bonded to parent atoms were kept in their theoretical positions (Uiso of each hydrogen atom was kept as 1.2 × Ueq of the parent atom). The molecular model was restrained with 38 bonds and 53 bond-angles restraints to get a result with reasonable geometry. Each restrained bond and bond-angle value was set individually according to the crystal structure of 1H2O-I.
Both AH and 1H2O-II were originally measured at different temperatures, so the phases were remeasured at 120 K in order to compare the density and packing efficiency to the other forms. The unit cells at 120 K of these two phases were obtained by refining the unit cell parameters. Details of the original refinement can be found in the ESI.†
Packing coefficient calculations. The packing coefficient is calculated by using the following equation:
C k = ZVmolVcell−1, where Vmol is the molecular volume (Å3), Vcell is the volume of the unit cell (Å3), and Z is the number of molecules in the unit cell.
The molecular volume was calculated with Hyperchem (Hypercube Inc.), which employs van der Waals radii in the calculation of the molecular volume of 1.75 Å for carbon, 1.78 Å for chlorine, 1.30 Å for fluorine, 1.20 Å for hydrogen, 1.55 Å for nitrogen, 1.52 Å for oxygen, and 1.82 Å for sulfur atoms.20
Void space calculation. To display and analyze the solvent/water channels, first the solvent/water molecules were deleted from the cif file and then the void map was calculated using Mercury (contact surface, probe radius 0.8 Å and approx. Grid spacing 0.7 Å).
Results
Crystallization experiments in different organic solvents confirmed that bosutinib is a promiscuous solvate former. Up to the present, 5§ hydrates, 9 mixed solvates, 9 pure solvates and 1 anhydrous form were isolated (see Table 1).| Anhydrate | AH |
| Hydrate | 1H2O-I; 1H2O-II; 2H2O; tetrahydrate;§ 6H2O; 7H2O |
| Mixed solvate | IPA–2H2O; MeOH(H2O); Diox–2H2O; DMSO–3H2O; ethanol solvate monohydrate; ethanol solvate dihydrate; chloroform solvate dihydrate; 1-propanol water mixed solvate; tetrahydrofuran water mixed solvate |
| Solvate | MeOH, 2-BuOH; 1-butanol solvate; 1-propanol solvate; acetonitrile solvate; dimethylformamide solvate; acetic acid solvate; N-methyl-2-pyrrolidone solvate |
Bosutinib was found to be unlikely to crystallize without solvent incorporation. Only one anhydrous form, AH, was prepared, which was described as a disordered structure. AH is obtained by the crystallization of the dried amorphous form in anhydrous media. The disorder is manifested in the splitting of the nitrile peak both in the vibrational and ssNMR spectra (see the ESI†) which is not present in any solvated form. This phenomenon clearly shows that two conformations are present in the structure, which possess different energetic states, therefore appear at different frequencies. Every AH batch showed this splitting, but all solvated forms crystallize without disorder, hence we can assume that the inclusion of solvents stabilizes the structure and eliminates this disorder.
As in the pharmaceutical industry a stable anhydrous form is the first choice of a solid form, experiments were executed with the intention of obtaining another (disorderless) anhydrate. For this purpose the following methods were applied:
1. Desolvation – drying
Desolvation of the solvates/hydrates resulted in reduction of the crystallinity (except for 7H2O which provided 1H2O-II by dehydration). Removal of the essential solvent stabilizing the structures caused them to collapse to an amorphous phase. However the disruption of the crystalline lattice is often followed by a rearrangement into an anhydrous phase, but no subsequent recrystallization of the amorphous structures occurred.2. Elevated temperature slurry
Close to the boiling temperature of the guest, its thermal mobility increases to such a degree that entrapment fails and a guest-free compound is crystallized instead of an inclusion complex.29–31Since bosutinib has the propensity to (at least partially) convert to the thermodynamically stable 1H2O-I if even traces of water are present, furthermore it is capable to absorb water from the atmosphere, so a high temperature slurry could only result in an anhydrous form if water is eliminated. In general, every phase contains more or less amount of water except for the 1- and 2-butanol solvates. These ones were used as starting materials in the elevated temperature slurry experiments. 1-Butanol solvate and 2-BuOH were suspended in various high boiling point solvents. The suspensions were stirred at 90 °C in the Crystal16TM (Technobis group, The Netherlands) for 3 days, then slowly cooled back to room temperature and filtered on glass filters. Both solvates converted to the disordered AH in cyclopentyl methyl ether, n-heptane, methyl isobutyl ketone, n-propyl-acetate and toluene. Interestingly, the slurries in 1-pentanol resulted in 1H2O-I particles with a 5% yield as the limited water content present in the system only enabled this degree of conversion.
3. Desolvation at high relative humidity
High relative humidity can sometimes trigger the desolvation of a solvate to a stable anhydrous form,32 but storing solvated bosutinib forms in the presence of water vapour yields 1H2O-I or 7H2O in the case of 1H2O-II (see the Solid state stability section).Accepting that no new anhydrous form was discovered, we decided to investigate the structure and stability of the solvated forms. The comparison of solvate stability with different amounts and types of solvents is complex due to the lack of an unambiguous comparison parameter. The stability of solvates can be assessed by comparing the thermal (kinetic) stability, thermodynamic stability in slurry experiments, behaviour under stress conditions, intrinsic dissolution rate and packing efficiency.
X-ray powder diffraction
The XRPD patterns (Fig. 2 and 3) show clear distinction among all the forms. The patterns of 1H2O-II and 7H2O are similar, but 7H2O has significantly wider interplanar distances. Also, the patterns of Diox–2H2O, DMSO–3H2O and IPA–2H2O are similar.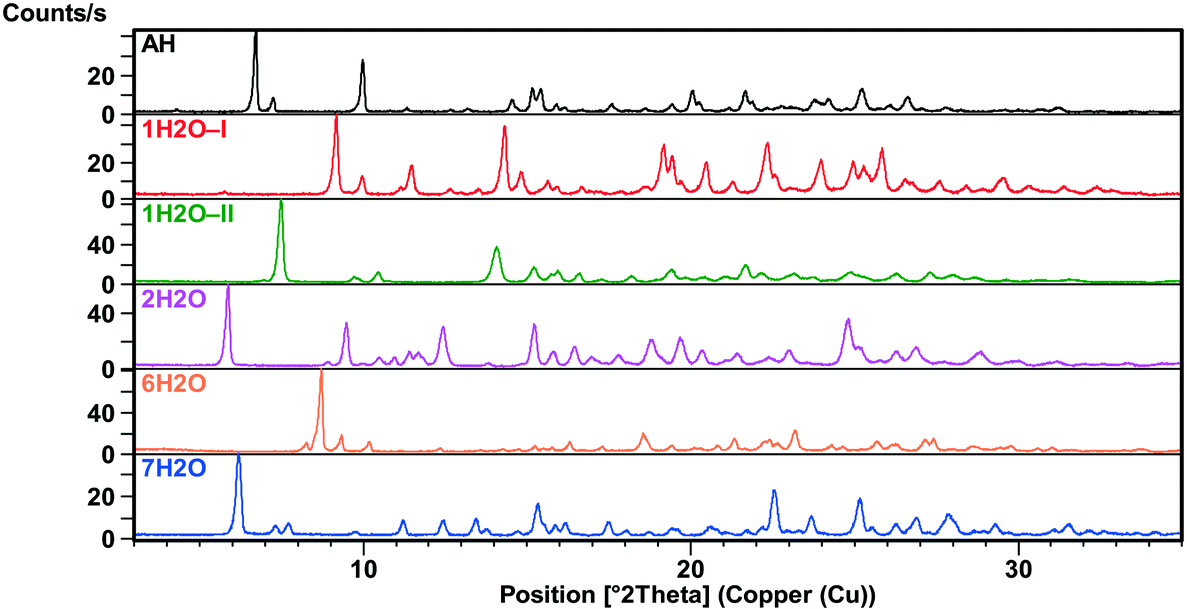 | ||
| Fig. 2 XRPD patterns of the bosutinib hydrates and anhydrate. | ||
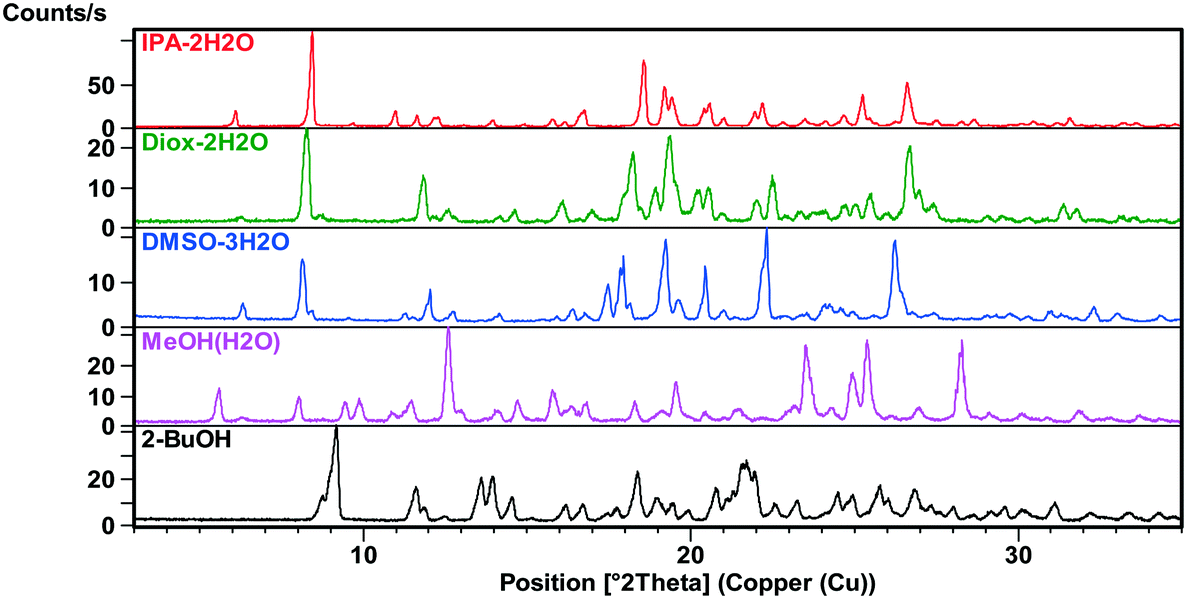 | ||
| Fig. 3 XRPD patterns of the bosutinib solvates. | ||
All investigated forms were stable under ambient storage conditions (in the laboratory cupboard where the conditions are close to the long term stability conditions (25 °C ± 2 °C/60% RH ± 5% RH)) for 2 years. The only exceptions are MeOH(H2O) and ethanol water mixed solvates where slow transformation to the thermodynamically more stable 1H2O-I was observed.
However, the structure of the pure methanol solvate, MeOH, is elucidated (see the Crystal structure section), but in practice, only the methanol–water mixed solvate (as bulk powder), MeOH(H2O), was obtained with the same structural characteristics. Probably due to the sufficient diameter of the channels, one part of methanol was substituted by water. It can be considered a host–guest complex33 in which the amount of the solvent guest is variable. This solvent exchange with water was observed in the past.34,35 Like water, methanol can act both as a proton donor or acceptor, hence their exchange is unhindered. The pattern predicted from the crystal structure of MeOH matched the experimental pattern of MeOH(H2O) (see the ESI†), indicating that the crystal structure is representative of bulk powders. Therefore when describing the crystal structures or packing efficiency, MeOH is considered, while in the investigation of the bulk powder's properties such as thermal characteristics, stability and intrinsic dissolution rate, MeOH(H2O) is studied.
Crystal structure
Analysis of the crystal structures of solvated forms of a compound can provide good insights into the nature of the interaction between the solvent molecules and the functional groups of the molecule. The crystal data are summarized in Tables 2 and 3.| AH | 1H2O-I | 1H2O-II | 2H2O | 6H2O | 7H2O | |
|---|---|---|---|---|---|---|
| Formula | C26H29Cl2N5O3 | C26H31Cl2N5O4 | C26H31Cl2N5O4 | C26H33Cl2N5O5 | C26H41Cl2N5O9 | C26H43Cl2N5O10 |
| Cell parameters | a = 24.212(5) | a = 12.323(10) | a = 9.459(6) | a = 9.785(10) | a = 12.321(2) | a = 9.155(10) |
| b = 8.122(2) | b = 13.972(10) | b = 12.421(12) | b = 10.474(10) | b = 12.793(2) | b = 13.043(10) | |
| c = 13.040(3) | c = 30.268(10) | c = 12.820(6) | c = 14.805(10) | c = 20.792(3) | c = 15.086(10) | |
| α = 90 | α = 90 | α = 80.040(4) | α = 80.999(10) | α = 90 | α = 69.094(10) | |
| β = 91.864(12) | β = 90 | β = 76.990(5) | β = 89.131(10) | β = 102.051(12) | β = 88.905(10) | |
| γ = 90 | γ = 90 | γ = 72.040(3) | γ = 70.023(10) | γ = 90 | γ = 77.835(10) | |
| Volume | 2562.96 Å3 | 5211.5 Å3 | 1387.31 Å3 | 1407.27 Å3 | 3205.24 Å3 | 1641.83 Å3 |
| Crystal system | Monoclinic | Orthorhombic | Triclinic | Triclinic | Monoclinic | Triclinic |
| Space group | P21/a | Pbca |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/n |
P |
| R-factor | 0.0832 | 0.0410 | 0.0874 | 0.0469 | 0.0313 | 0.0495 |
| Density | 1.375 g cm−3 | 1.398 g cm−3 | 1.311 g cm−3 | 1.337 g cm−3 | 1.323 g cm−3 | 1.328 g cm−3 |
| Temperature | 120 K | 120 K | 120 K | 120 K | 120 K | 120 K |
| Z | 4 | 8 | 2 | 2 | 4 | 2 |
| IPA–2H2O | Diox–2H2O | DMSO–3H2O | MeOH | 2-BuOH | |
|---|---|---|---|---|---|
| Formula | C29H41Cl2N5O6 | C30H41Cl2N5O7 | C56H82Cl4N10O14S2 | C54H66Cl4N10O8 | C30H39Cl2N5O4 |
| Cell parameters | a = 10.387(4) | a = 10.294(10) | a = 10.634(2) | a = 15.042(10) | a = 14.674(6) |
| b = 10.651(4) | b = 11.194(10) | b = 14.836(3) | b = 11.684(10) | b = 14.845(5) | |
| c = 14.865(5) | c = 14.896(10) | c = 22.083(4) | c = 31.449(10) | c = 15.273(6) | |
| α = 104.472(3) | α = 107.974(10) | α = 72.096(17) | α = 90 | α = 90 | |
| β = 97.659(3) | β = 98.064(10) | β = 81.948(17) | β = 92.372(10) | β = 114.517(5) | |
| γ = 91.287(3) | γ = 90.706(10) | γ = 81.289(17) | γ = 90 | γ = 90 | |
| Volume | 1575.54 Å3 | 1613.70 Å3 | 3260.49 Å3 | 5522.34 Å3 | 3026.95 Å3 |
| Crystal system | Triclinic | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group |
P |
P |
P |
P21/n | P21/c |
| R-factor | 0.0539 | 0.0423 | 0.0561 | 0.0592 | 0.0490 |
| Density | 1.321 g cm−3 | 1.347 g cm−3 | 1.350 g cm−3 | 1.353 g cm−3 | 1.327 g cm−3 |
| Temperature | 120 K | 120 K | 120 K | 120 K | 120 K |
| Z | 2 | 2 | 2 | 4 | 4 |
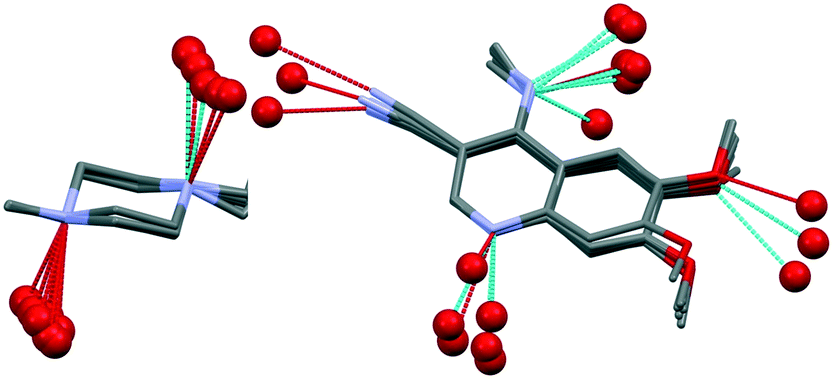
Bosutinib is a flexible molecule which can adopt different conformations in its crystals. If the quinolinone moiety is superposed for each form, the rest of the molecule is not aligned (Fig. 4).
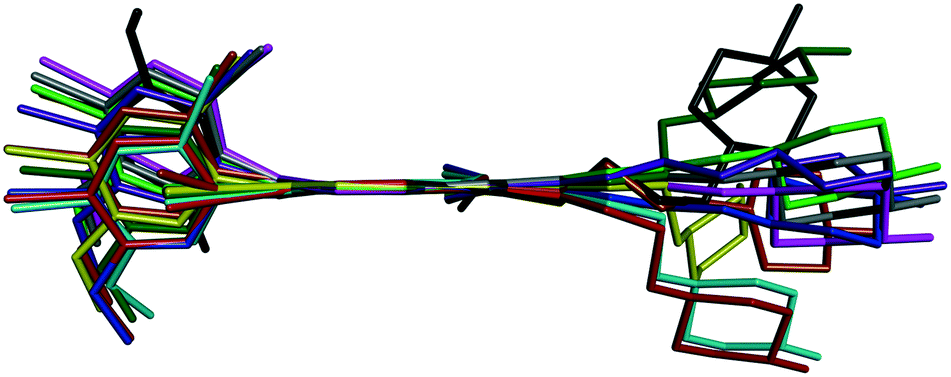 | ||
Fig. 4 Overlay of bosutinib in all solved structures viewed perpendicular to the quinoline ring. 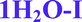 , ,  , , 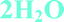 , , 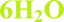 , , 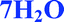 , , 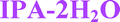 , , 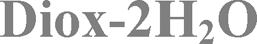 , , 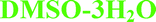 , ,  , , 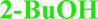 , , 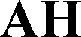 . . | ||
The conformations differ in the spatial orientation of the methylpiperazine and the substituted aniline ring moiety. In the case of AH, the rotation of the C–N sigma bond causes the substituted aniline ring to be mirrored, while the solvated forms differ only in the tilt angle of this fragment. The orientation of the methylpiperazine moiety varies in all conformations, dictated by the structural arrangement in each solid form.
Small adjustments in conformation, such as changes in torsion angles by a few degrees and rotation of different groups, can change the lattice energy significantly. With these differences in the conformation, the API adapts to the distinct type and size of guests, different hydrogen bonds are available and different packings are derived from these conformational changes. Discriminative solvate formation occurs, with the solvate selectivity based on the solvents' functionality to provide strong intermolecular interactions. The molecule possesses eight H-bond acceptor groups and one H-bond donor group (Fig. 1), which enable a number of possible intermolecular hydrogen bonding arrangements. The high degree of torsional freedom and large variety of hydrogen bonding possibilities enable versatile solvate formation.
Half of the solvates are mixed solvates. In those structures, mainly only water is directly bonded to adjacent bosutinib molecules due to its small size as it can fit into voids inaccessible to the larger organic solvent molecules and versatile hydrogen-bonding capabilities. The only exception is IPA–2H2O, where the 2-propanol OH is engaged in a H-bond with the nitrile group of the API.
However distinct solid forms show diverse bonding behaviour, allowing the API to form a variety of structures, there are common features in the structural arrangements. There is no direct connection between bosutinib molecules; they are only connected through the guest molecules. This supports our hypothesis that the main contributing factor for the solvent inclusion is the effect of the extensive H-bond network established by the solvent molecules.
Intermolecular hydrogen bonds between one nitrogen atom (1H2O-I, 1H2O-II and 2H2O) or both nitrogen atoms (6H2O, 7H2O, Diox–2H2O, DMSO–3H2O and IPA–2H2O) of the methylpiperazine ring and water molecule were observed in all hydrated forms (Fig. 5, left). In the case of the pure solvates, MeOH and 2-BuOH, this hydrogen bond is created by the alcoholic hydroxyl group. An analogous situation occurs in the case of the nitrogen atom of the secondary amine situated between the quinoline and the 2,4-dichloro-5-methoxy aniline moieties, which is always engaged in hydrogen bonding with water or with alcohol molecules when water is not present in the crystal structure.
 | ||
| Fig. 5 Overlay of the methylpiperazine ring in all solved structures of bosutinib (except for AH) with displayed hydrogen bonds (left) and overlay of the substituted quinoline ring with displayed hydrogen bonds (right). The rest of the molecule was omitted for clarity. | ||
On the other hand, the nitrogen atom of the quinoline group is only bonded to water molecules and in the case of MeOH and 2-BuOH this nitrogen atom does not participate in the hydrogen bonding network. In the case of the higher order hydrates (2H2O, 6H2O and 7H2O), the oxygen atom of the methoxy group is also hydrogen bonded to water molecules and in three cases (6H2O, 7H2O and IPA–2H2O), the nitrogen atom of the nitrile group is also involved in hydrogen bonding (Fig. 5, right).
The similarities in the crystal packing were investigated by xPac36 and CrystalCMP37 softwares. The packing of bosutinib molecules in the crystal structures of Diox–2H2O, IPA–2H2O and DMSO–3H2O is identical as it is shown in Fig. 6. These three crystal structures represent identical 3D packing of the API. 2D packing similarities can be found between crystal structures of 1H2O-II and 7H2O and between MeOH and 2H2O. In other crystal structures (1H2O-II, 2H2O, 6H2O, 7H2O, MeOH and BuOH), bosutinib dimers are connected by π–π interactions of the quinoline rings. AH forms similar dimers to 2H2O and MeOH.
 | ||
Fig. 6 Overlay of bosutinib crystal structures of  , , 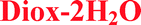 and and  showing their identical packing (left) and overlay of the asymmetric part of the unit cells showing the identical position of host molecules (right). showing their identical packing (left) and overlay of the asymmetric part of the unit cells showing the identical position of host molecules (right). | ||
1H2O-II is a high energy polymorph of 1H2O-I, obtained exclusively by dehydrating 7H2O. The similarity in the packing motif of the 2D layers of 7H2O and 1H2O-II is depicted in Fig. 7.
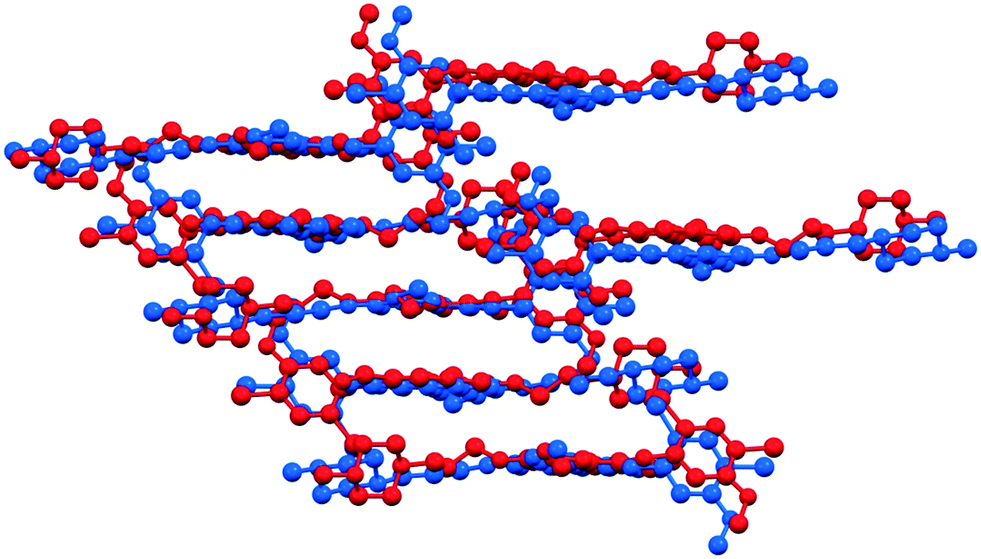 | ||
Fig. 7 Comparison of the packing of  and and  . . | ||
7H2O and 1H2O-II are closely related, but with different orientations of the methylpiperazine and the substituted aniline ring. During dehydration, six water molecules leave the structure involving changes in hydrogen bonding and one water molecule remains to stabilize the structure. Due to the high stoichiometry and wide channels of 7H2O (Fig. 8), the dehydration is relatively facile. While the desolvation of other hydrates and solvates results in an amorphous form, in this case it can occur through moderate structural rearrangements in the crystal lattice leading to a kinetically stabilized metastable form.
 | ||
| Fig. 8 Void space in the structure of 7H2O viewed along the crystallographic a and c axes. | ||
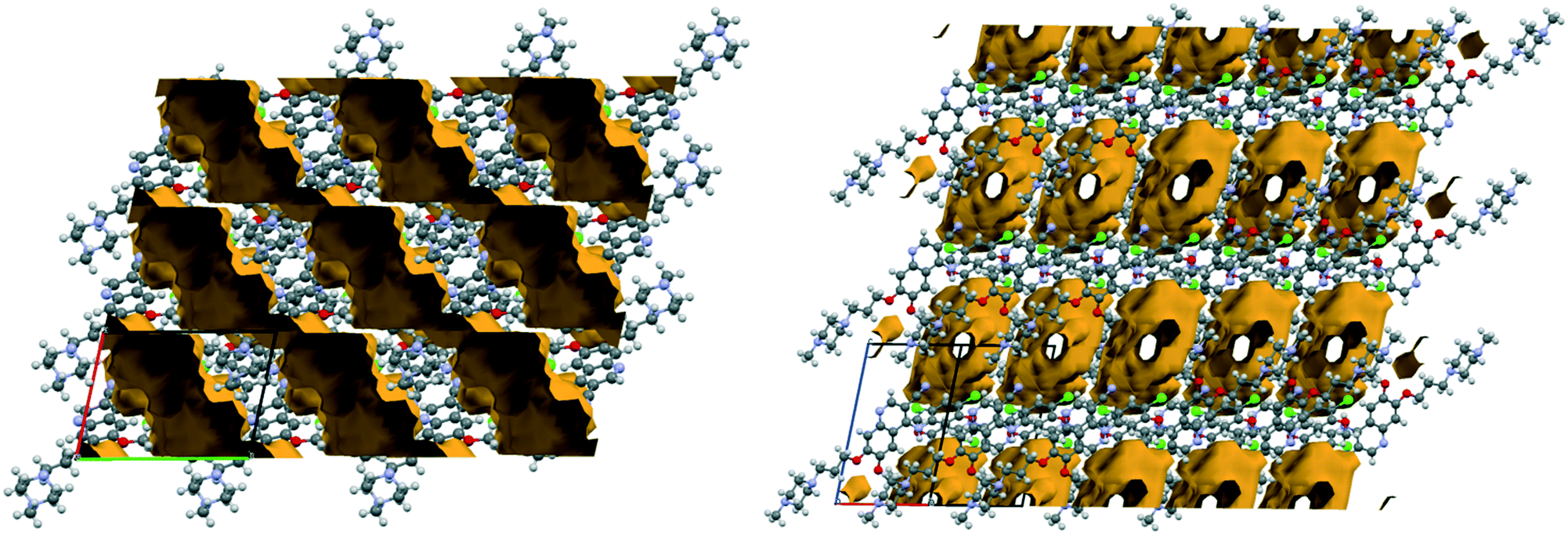
In the structure of AH, according to the best description of the disorder (see the details in the ESI†), the methylpiperazine moiety adopts two orientations with the site occupancy factors of 0.631![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.369; a so called conformational site disorder38 is observed (Fig. 9).
:0.369; a so called conformational site disorder38 is observed (Fig. 9).
 | ||
| Fig. 9 Description of the disorder in the crystal structure of AH. | ||
There is no strong H-bond in the structure of AH, however it would be expectable between the secondary amine and the nitrile group, or between the secondary amine and methoxy oxygen of the phenyl ring from the neighboring molecule, but their orientations and distances are unfavourable. The nitrile group establishes C–H⋯N interactions with the quinoline ring and with the methoxy group.
Despite the disordered structure and the lack of strong H-bonds, AH showed stability in the solid state. It seems that the collective interactions in the crystal structure have more significant influence in determining the overall lattice energy, thus stability of a crystal form than the presence or absence of specific types of intermolecular interactions. This solvent-free form can achieve lattice energy minimization through other weak intermolecular interactions (van der Waals forces), which can be just as effective as strong H-bonding in other, solvated crystal structures.39
With the inclusion of the solvent molecules into the lattice, the API molecules arrange differently to accommodate more energetically favourable structures. The API molecules adapt to new conformations that allows them to participate in more H-bond interactions resulting in the formation of new crystal structures. Multipoint hydrogen bonding is clearly a dominant factor that governs the inclusion of a large number of solvent molecules. A wide range of groups are capable of providing the required characteristics. Probably steric effects play equally important role in the solvent selectivity.
Thermal analysis
Typically solvate stability is evaluated by characterizing the dehydration conditions and observed phase changes. The stability of isolated site solvates where the solvent molecules are strongly bound depends on the solvent–host interactions and the spatial characteristics.40In the case of the bosutinib solvates, the desolvation events are usually broad endotherms due to the broad range in energy to evaporate the solvent(s) from the samples (Fig. 10 and 12). Nonetheless, desolvation endotherms can become sharp for very stable solvates (2-BuOH) which release the solvent at a temperature where its vapour pressure is sufficiently high to evaporate the solvent immediately. In the case of 2-BuOH, the onset of desolvation (T = 101.3 °C) is close to the boiling point of the pure solvent (99.5 °C). The relative sharpness of the DSC endotherm may be attributed to the location of these guest molecules in the crystals as they are firmly held by bosutinib molecules. Also, the bigger size of the solvent can result in a hindered exit from the closely packed structure.
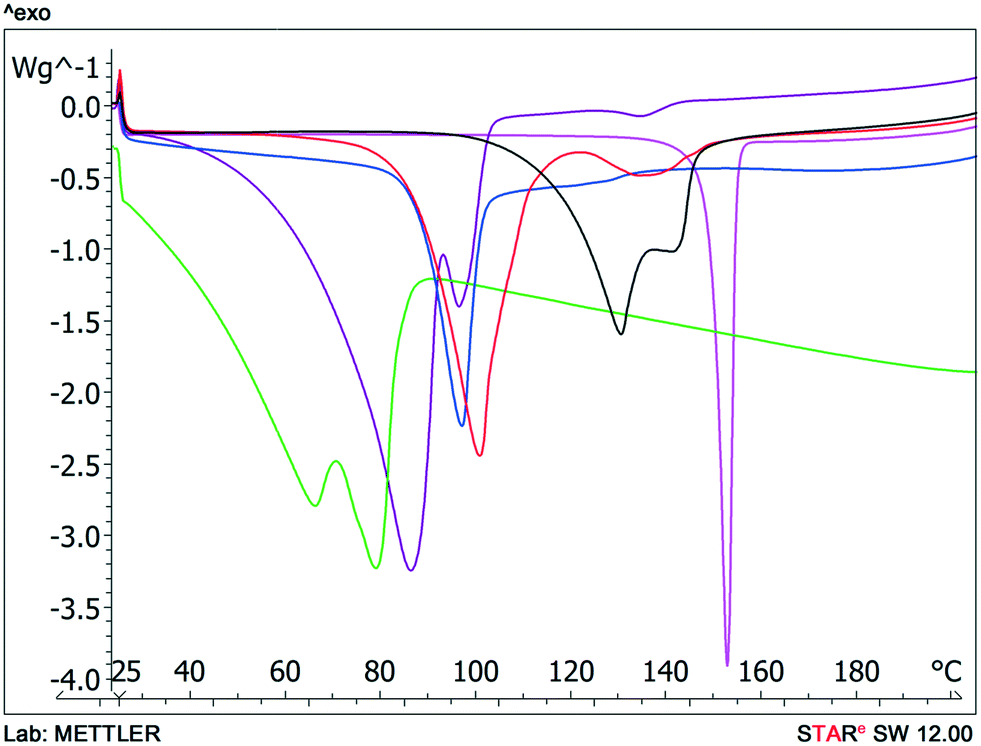 | ||
Fig. 10 DSC scans of bosutinib hydrates and anhydrate  , ,  , ,  , ,  , ,  , ,  . . | ||
We could rank the strengths of host–guest interactions in the solvates based on the desolvation onset temperature. Desolvation is a kinetically driven event and therefore dehydration kinetic parameters can be strongly affected by various sample and experimental factors like particle size distribution, relative humidity and heating rate. Still, the crystal structure is usually the dominant factor.41–44
Solvent molecules in a tunnel-like arrangement, like in 6H2O or 7H2O, exit in the direction of channels with anisotropic dehydration resulting in a smooth desolvation process (Fig. 11). The mixed solvates release their solvents at lower temperatures than the pure solvate 2-BuOH. The DSC thermograms show incompletely resolved endotherms corresponding to the departure of different solvents. The desolvation temperatures differ, indicating different binding strengths of the various solvents in the crystals (Fig. 13).
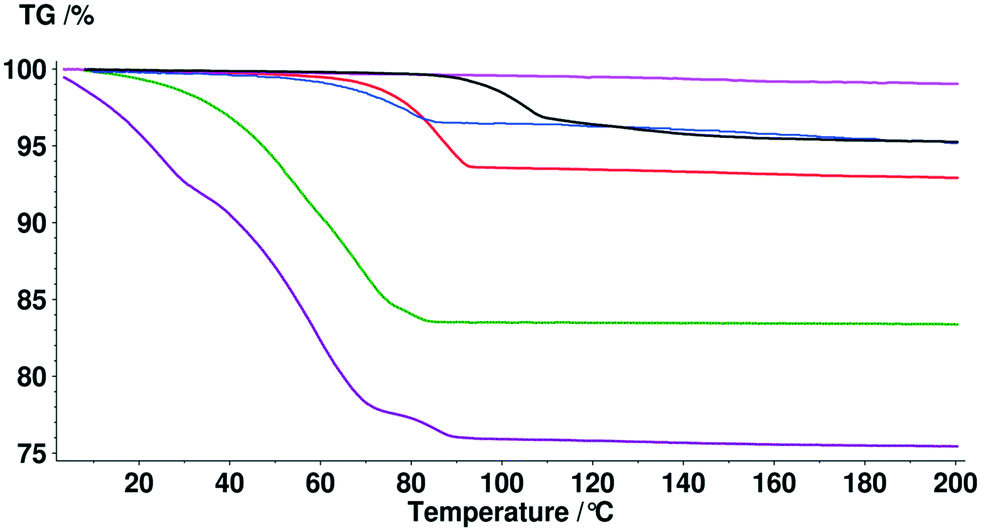 | ||
Fig. 11 TG curves of bosutinib hydrates and anhydrate  , ,  , ,  , ,  , , 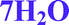 , , 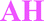 . . | ||
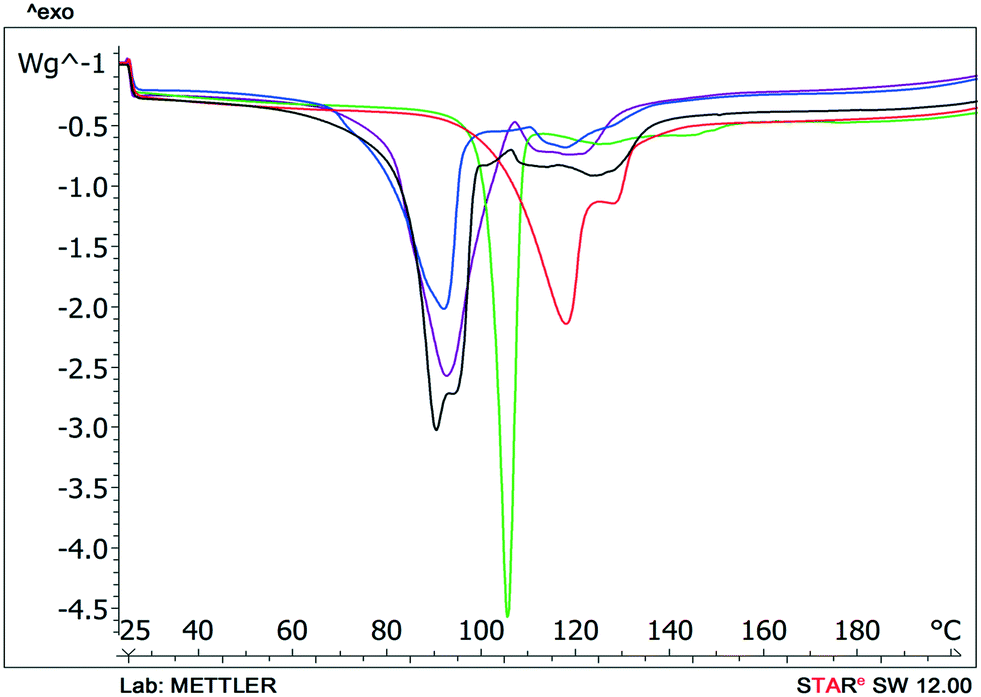 | ||
Fig. 12 DSC scans of bosutinib solvates 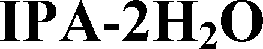 , ,  , ,  , ,  , ,  . . | ||
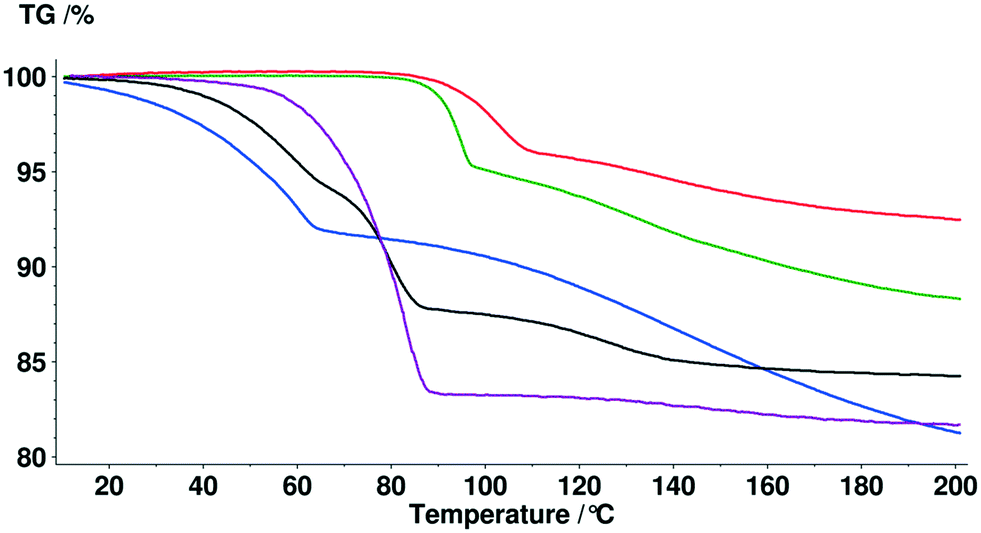 | ||
Fig. 13 TG curves of bosutinib solvates  , ,  , ,  , ,  , ,  . . | ||
The quantitative estimate of the stability of these solvates can be established by determining the binding energies.45–49
| ΔHS = [(ΔHSexp × 100)/ΔmS%] × MS |
The binding energy for the solvents (ΔHS) can be calculated from the enthalpy of desolvation (ΔHSexp, defined by the DSC curve), the percentage mass loss (ΔmS%, defined from TG data) and the molecular mass of the solvent (MS). We compared the calculated values to the corresponding enthalpy of vaporization of the pure solvents to their interactions in the liquid state in order to determine whether the solvent molecules are strongly/loosely bound in the host lattice. We confine the calculations to pure solvates/hydrates as the release of water and organic solvents from the mixed solvates is not resolved, the endotherms overlap and therefore the computations would not be precise.
In the case of the hydrates, the calculated binding energy is inversely proportional to the stoichiometry (Table 4). In the structures of the monohydrates and 2H2O, the water molecules are strongly held and the calculated binding energies exceed the enthalpy of vaporization of pure water. The calculations are consistent with the superior stability of 1H2O-I as the calculated binding energy is significantly higher. In the higher order hydrates, the water molecules are located along channels and this arrangement promotes dehydration. Therefore the calculated energies do not exceed the enthalpy of vaporization of pure water. The 2-butanol molecules in 2-BuOH are also very strongly bound to the API molecules indicated by the high calculated binding energy value.
| Solid form | Desolvation enthalpy, kJ mol−1 | Weight loss by TGA% | Calculated binding energy, kJ mol−1 | Enthalpy of vaporization of pure solvent at 25 °C, kJ mol−1 | Boiling point of pure solvent, °C |
|---|---|---|---|---|---|
| 1H2O-I | 96.46 | 3.52 | 89.94 | 43.99 | 100 |
| 1H2O-II | 64.04 | 3.56 | 59.04 | 43.99 | 100 |
| 2H2O | 101.98 | 5.99 | 54.10 | 43.99 | 100 |
| 6H2O | 229.60 | 16.47 | 39.30 | 43.99 | 100 |
| 7H2O | 279.67 | 24.03 | 31.91 | 43.99 | 100 |
| 2-BuOH | 97.44 | 10.88 | 109.81 | 49.72 | 99.5 |
Solid state stability
Phase transformations can occur in the solid state as a response to variations in humidity and temperature. Stability testing evaluates the effect of environmental factors on the quality of the drug substance. In this case, accelerated stability testing was applied in order to determine the phase boundaries, understand the solid-state stability and identify appropriate storage conditions. Investigation of the physical stability of the drug substance is an integral part of the systematic approach of solid form selection as it contributes to determining whether the solid form is suitable for use in pharmaceutical manufacturing.For this purpose, the samples were exposed to different atmospheres for 10 days. Unfortunately, we could not prepare DMSO–3H2O in a larger scale, so its behaviour was not investigated. Desiccators containing various saturated solutions were applied to provide a range of relative humidities. The following salts were selected: phosphorus pentoxide (0% RH), magnesium chloride (33–26% RH¶) and sodium chloride (75% RH). The desiccators were stored at 25, 50 and 80 °C during the studies. The results are depicted in Fig. 14.
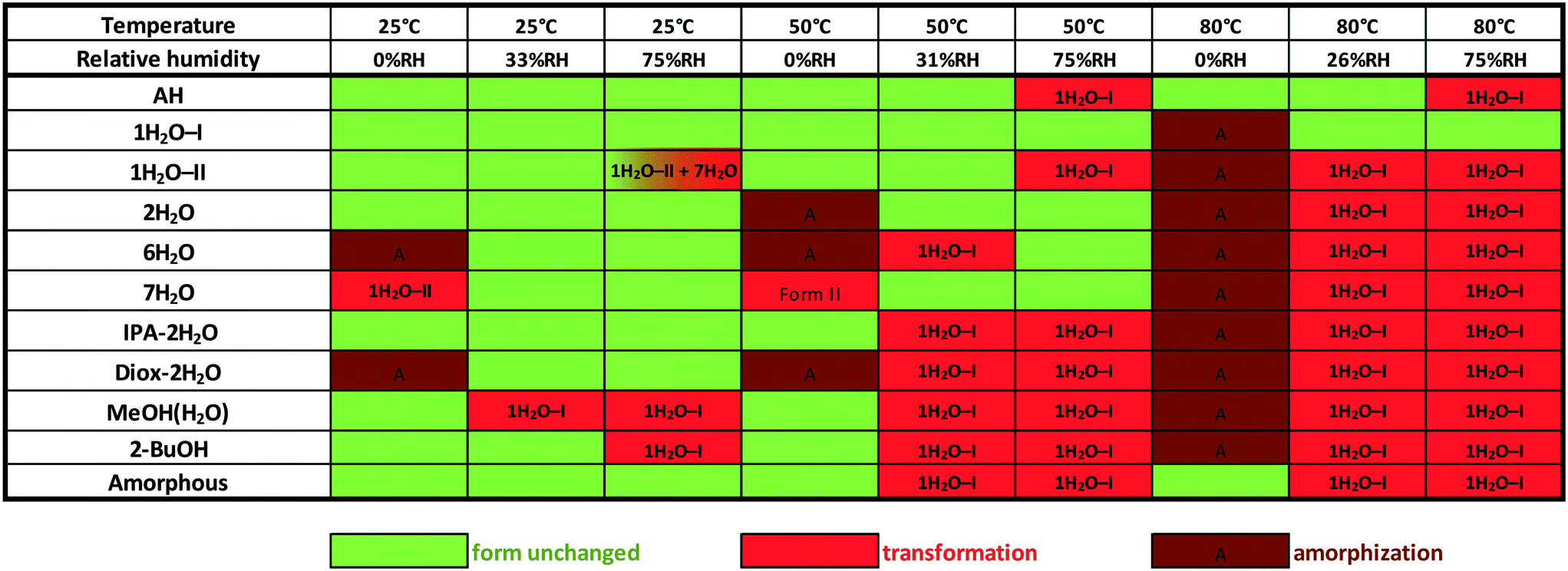 | ||
| Fig. 14 Solid state stability of bosutinib forms at various temperatures and relative humidity levels. | ||
1H2O-I showed the highest physical stability among the investigated forms. Amorphization occurred at 80 °C and 0% RH, but besides this conditions, the structure remained unchanged. The solvates slowly exchange their solvent against water and the other solid forms also restructures to 1H2O-I at elevated temperature and relative humidity.
The second most resistant form is AH which only converts to 1H2O-I at high RH levels (≥75%) at elevated temperatures. The amorphous form showed comparable results, but it is more sensitive to humidity at higher temperatures. 2H2O was found to be stable enough for pharmaceutical use as its structure did not change at 50 °C, 75% RH. Amorphization occurred under anhydrous conditions and it transformed to 1H2O-I at high RH levels at 80 °C. 1H2O-II is less stable than 1H2O-I, which is consistent with this polymorph being produced only by dehydration. Due to the slower kinetics of the solid–solid phase transition, 1H2O-II only partially converted to 7H2O at high relative humidity at 25 °C (the conversion was complete during the DVS measurement, see the ESI†). 7H2O dehydrates to 1H2O-II under anhydrous conditions, but its structure remained unchanged up to 50 °C when moisture was present. The most resistant mixed hydrate is IPA–2H2O, where there is a direct connection between 2-propanol and API molecules, but it should be protected against moisture. 2-BuOH is also sensitive to high relative humidity levels.
The stability order based on the stress test is: 1H2O-I > AH > amorphous > 2H2O > 1H2O-II, IPA–2H2O and 7H2O > 2-BuOH and 6H2O > MeOH(H2O) and Diox–2H2O.
Solvent mediated phase transformation (critical solvent activity investigations)
An effective method to determine the relative physical stability of anhydrous and solvated forms is the solvent mediated solid phase transformation containing mixtures of crystalline forms in various systems with known solvent activities.50 This method can shed light on the solvent activity range under which the solvate is stable. Sometimes solvent mediated phase transformation experiments only yield solvated/hydrated forms, providing no information on the stability or existence of anhydrous forms.Acetone/water, acetonitrile/water, 2-propanol/water and methanol/water systems were chosen for the determination of the critical water/solvent activity between the forms.
The critical water activity measurement in acetone/water (Fig. 15) and acetonitrile/water mixtures (Fig. 16) provided similar results: 1H2O-I is stable up to aw ∼ 0.8 and 7H2O is stable above this limit. The results are in correlation with the DVS measurement (see the ESI†), as 7H2O is formed above 80% RH.
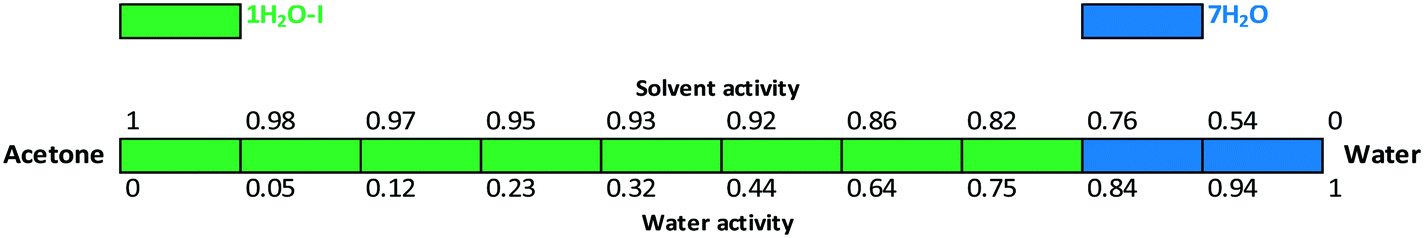 | ||
| Fig. 15 Critical water activity results in acetone/water mixtures. | ||
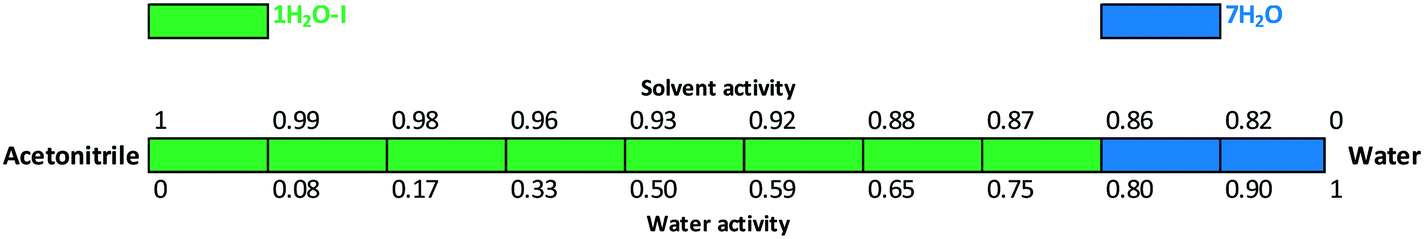 | ||
| Fig. 16 Critical water activity results in acetonitrile/water mixtures. | ||
The water activity investigation performed in the methanol/water system (Fig. 17) indicates that in the case of MeOH(H2O), there is a gradual change in the water and methanol contents as certain portion of the organic solvent can be replaced by water and vice versa. Therefore MeOH(H2O) is stable from 0 up to 0.66 water activity. It is in correspondence with the observed “anomaly” of the crystal structure: the pure methanol solvate structure was solved (whose calculated pattern matches the mixed solvate pattern, see the ESI†), while in practice the mixed solvate was obtained. This implies that there is a continuous transition and the structure can incorporate various ratios of solvents adapting to the surrounding atmosphere/media. The amounts of water and methanol showed significant batch-to-batch variation depending on the crystallization media. Below 0.8 methanol activity, 1H2O-I is stable and 7H2O is only obtained in pure water. The formation of 7H2O is not favoured in methanol/water mixtures.
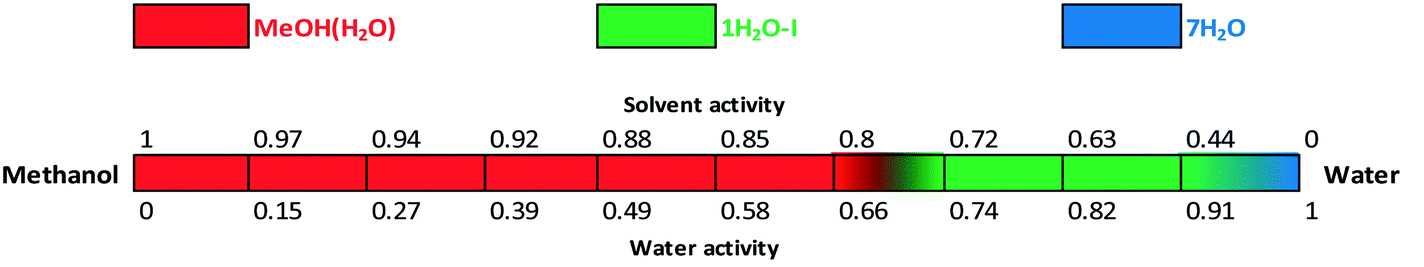 | ||
| Fig. 17 Critical water and solvent activity results in methanol/water mixtures. | ||
The critical water activity measurement in 2-propanol/water shows that the critical water activity of IPA–2H2O is 0.3; pure 2-propanol solvate was not obtained (Fig. 18). IPA–2H2O is stable up to aw ∼ 0.87, above which 7H2O is the favoured solid form. Fig. 15, 16 and 18 clearly show that bosutinib has high affinity to water as 1H2O-I can crystallize even in anhydrous media. The suspended API contains a certain amount of water, but the dissolved drug influences the water activity to a much smaller extent than does the organic solvent.50
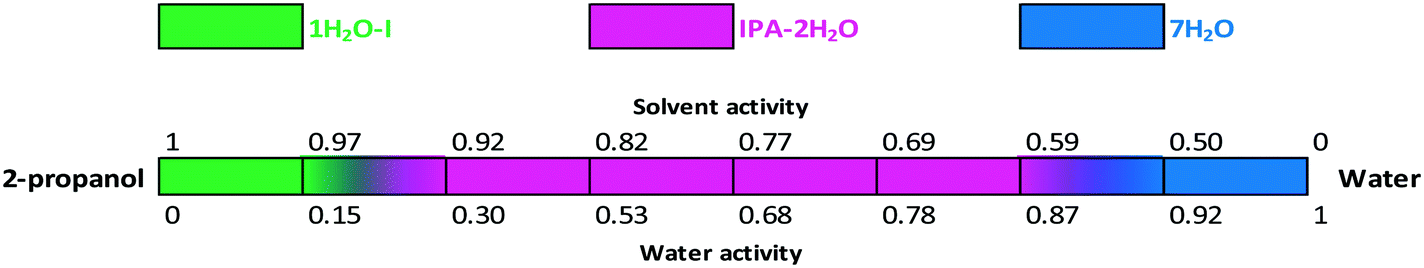 | ||
| Fig. 18 Critical water and solvent activity results in 2-propanol/water mixtures. | ||
The stability boundaries indicate that the stability of mixed solvates is more complex, as both water and solvent activity can play a significant role depending on the structural characteristics. In the case of MeOH(H2O), anhydrous conditions result in a stable solvate and there is a gradual change in the composition of the incorporated solvents. On the other hand, in the case of IPA–2H2O, both solvent and water activity are essential for the maintenance of the structure and the pure solvate structure is not favoured. While 1H2O-1 can crystallize in pure 2-propanol, acetonitrile or acetone, for the crystallization of IPA–2H2O the required amount of both water and 2-propanol must be present.
Intrinsic dissolution rate
The dissolution rate and solubility in a solvent medium are one of the most important characteristics of a drug substance, as these quantities determine the bioavailability of the drug for its intended therapeutic use.1 In this paper, the intrinsic dissolution rate (IDR) was used to study the relationship between the dissolution rate and the crystalline form as it is independent of the size and the shape of the particles.51At pH 2, the intrinsic dissolution rates decreased due to the API's conversion to the HCl salt on the surfaces of dissolving disks, therefore here we focus on the intrinsic dissolution rates measured at pH 4.5. Fig. 19 shows the mean, relative intrinsic dissolution rates of the studied forms in the selected dissolution media at pH 4.5. The IDR values were calculated from the initial linear portion of the dissolution curves and were normalized to 1H2O-I.
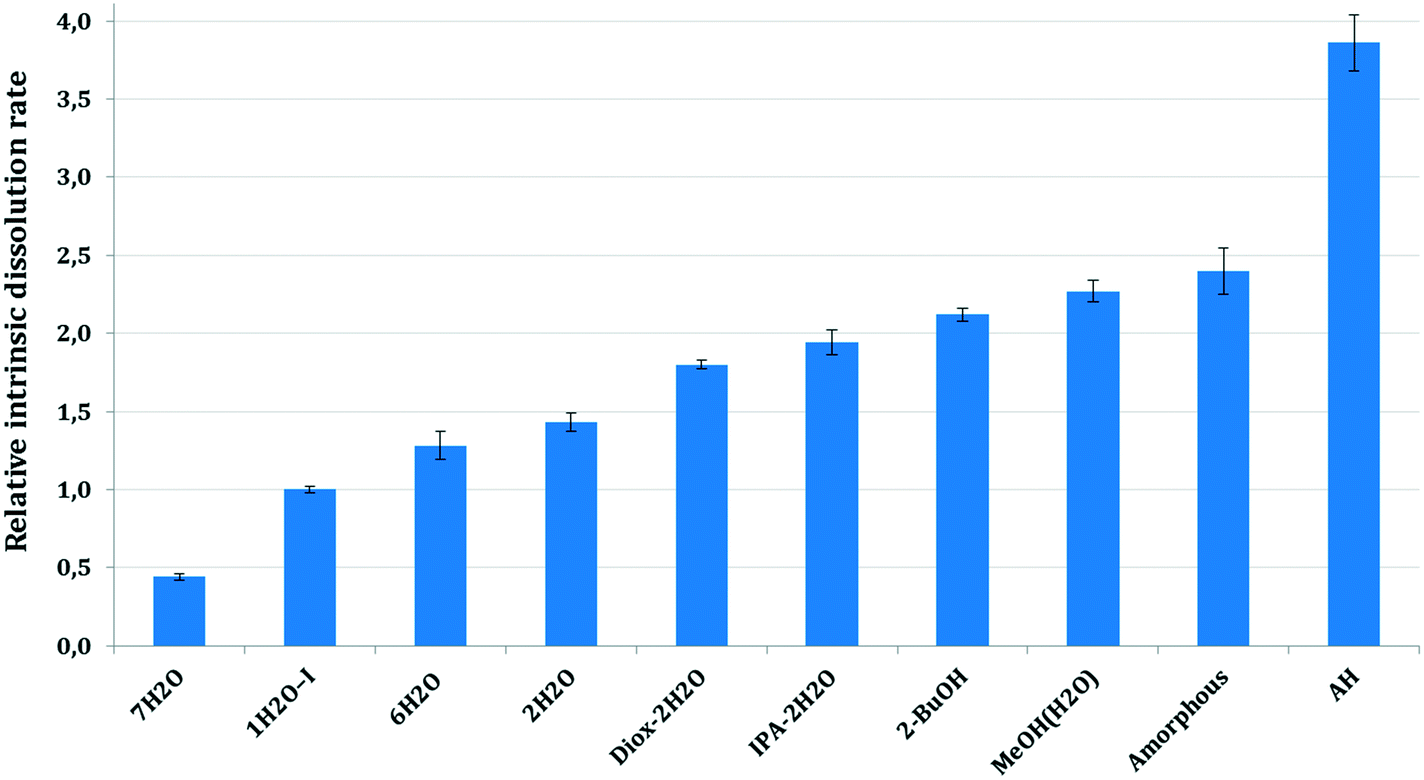 | ||
| Fig. 19 Relative intrinsic dissolution rate of bosutinib forms at pH 4.5. | ||
As it was expected, the amorphous form and AH dissolve faster than the corresponding hydrates. The hydrates possess lower thermodynamic activity in the applied media and are therefore in a more stable state than their anhydrous form.52
Solvates exhibit significantly faster dissolution than 1H2O-I. The intrinsic dissolution rate of the forms containing an organic solvent is ∼2× higher than that of 1H2O-I. The higher intrinsic dissolution rate of the solvates is caused by the negative Gibbs free energy of mixing of the organic solvent with the aqueous media. The incorporated solvent is released during the dissolution of the solvate and it contributes to the Gibbs free energy of solution, increasing the thermodynamic driving force for the dissolution process.53,54 The mixed solvates, Diox–2H2O and IPA–2H2O, showed comparable intrinsic dissolution rates in accordance with their analogous crystal structures. The pure solvate, 2-BuOH, dissolves slightly faster, but the difference between the IDR of mixed and pure solvates is not significant.
7H2O showed the slowest intrinsic dissolution. The order of the hydrates' IDR is not inversely proportional to their stoichiometries, because it does not reflect only the thermodynamic activity in the applied media. The crystal lattice forces in the solid state are equally important. Unfortunately, we were not able to record the intrinsic dissolution rate of 1H2O-II as it transformed to 7H2O too fast. The relative IDRs for 1H2O-II (0.45) and 7H2O (0.44) are identical and the conversion was confirmed by offline Raman spectroscopy.
The intrinsic dissolution rate of the amorphous form and AH is not accurate. In the case of the amorphous API, slight gel formation was observed which probably hindered the diffusion from the surface of the disc and therefore the real intrinsic dissolution rate is expected to be higher. In the case of AH, the initial part of the dissolution curve was steep, the dissolution was really fast within the first two minutes, then the rate decreased (see the ESI†). These results were obtained in a reproducible manner. Such a curvature, a change in the slope of the curve is generally an indicative of phase transformation, when the intrinsic dissolution rate decreases with time due to precipitation of a more stable form. In this case, no transformation occurred and the structure of the residue was confirmed by offline Raman spectroscopy and XRPD. Our assumption is that even though the force used was sufficient to form a non-disintegrating disk in the case of the other solid forms, the properties of the AH bulk powder did not enable adequate compaction. The disintegration of the surface layer of the disc could result in the observed high initial dissolution rate. Therefore the second, less steep part of the curve was applied in our calculations, which is not precise, as the smoothness of the surface probably damaged to some extent.
The intrinsic dissolution rates of the solid forms in descending order are:
AH > amorphous > MeOH(H2O) > 2-BuOH > IPA–2H2O > Diox–2H2O > 2H2O > 6H2O > 1H2O-I > 7H2O.
Intrinsic dissolution rate investigations reflect the lattice energies to some extent, but one must consider the negative Gibbs free energy of mixing the released organic solvent and water and therefore the solvates' dissolution rate is inevitably higher. The observed high intrinsic dissolution rate of AH is plausible as during solvation the previously unsatisfied functional groups can engage in hydrogen bonding.
The dissolution results are in correlation with the water activity results, as in the applied media 7H2O is the most stable form, hence it showed the slowest dissolution. Interestingly, the other hydrates despite having higher stoichiometry exhibit faster intrinsic dissolution than 1H2O-I. 1H2O-II, 2H2O and 6H2O disappeared in the competitive slurry experiments as they are in higher energy states (have higher solubility). Consequently, they dissolved and were replaced by the stable hydrates, 1H2O-I and 7H2O.
Packing efficiency
One measure of how tightly a molecule packs in a crystal lattice is the packing efficiency, measured by the packing coefficient.20,55This quantity reflects the percentage of void space in molecular crystals. Packing coefficients were calculated by the following equation:
| Ck = ZVmolVcell−1, |
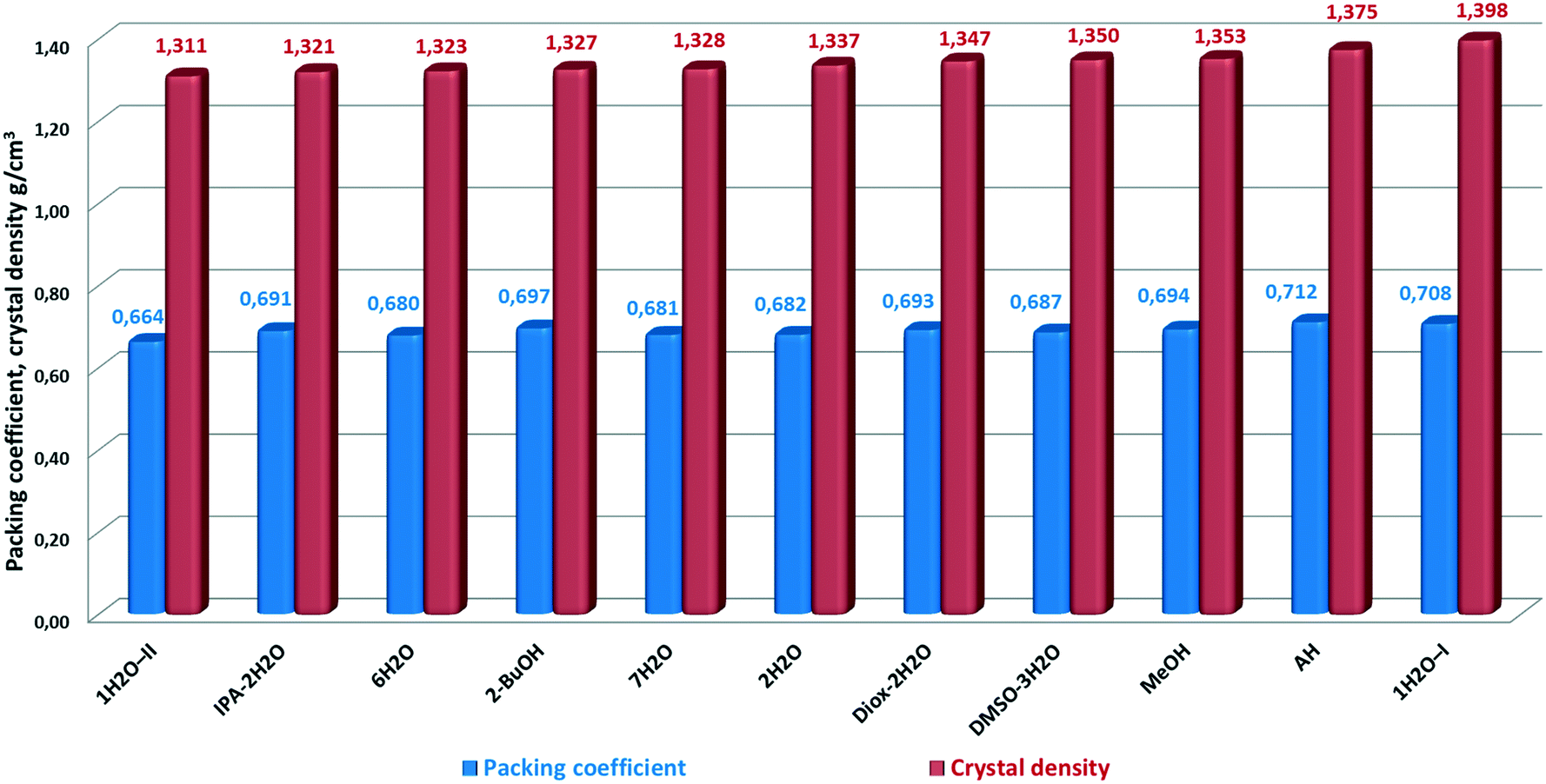 | ||
| Fig. 20 Packing coefficient and crystal density of bosutinib forms. | ||
AH has the highest packing efficiency – however it seems contradictory for a disordered structure – and the second highest density. This is surprising as the observed inability to form unsolvated crystals from common solvents could imply that the molecules cannot pack efficiently with themselves. The results indicate that energetically efficient packing is achieved also for AH, consisting of bosutinib molecules only and thus, the lattice energy does not rely solely on the presence of strong H-bonds. Bosutinib cannot readily give rise to unsolvated forms. Only one anhydrous form was discovered, which can only be obtained by non-traditional methods; offering a loophole to crystallize in the absence of solvents providing H-bonds.
In general, solvents are incorporated to stabilize the structures through decreasing the void space and/or providing hydrogen bonding to surrounding API molecules. Most compounds have contributions from both of these driving forces.20
In the case of bosutinib, the stabilization effect of the solvates is not achieved by decreasing the void space as the solvent free structure is closely packed. The solvents are not utilized to fill the void space in the structures, but to satisfy previously unused hydrogen bonding capabilities in the host molecule.
1H2O-I has the highest density in correspondence with its superior stability. The density of 1H2O-I, 1.398 g cm−3, is significantly higher than that of 1H2O-II, 1.311 g cm−3. Thus, this density difference suggests stronger intermolecular interactions taking place in 1H2O-I, which supports the proposed explanation of the higher stability of this form. 1H2O-II showed the lowest packing efficiency and density and immediately converted to 7H2O in the dissolution media and in the solid state at high RH. Interestingly, the biggest difference in the density and packing efficiency is between the two most closely related forms, the true polymorphs 1H2O-I and 1H2O-II. Both packing efficiency and density of 1H2O-II are 93.8% of the corresponding value of 1H2O-I.
It seems a probable explanation that the incorporated water molecules destabilize the structure of MeOH(H2O). MeOH is the most densely packed pure solvate, but as a significant amount of water is present in the structure of MeOH(H2O), it facilitates the transformation to 1H2O-I. Under ambient conditions, the methanol activity is zero while water is omnipresent; this conversion is just a matter of time.
There is no significant variance in the packing coefficients, the difference between the most and less efficiently packed structure is just 0.048. The solvated forms have lower density and packing efficiency values, but unlike AH, the solvates have a variety of intermolecular hydrogen bonding motifs.
It is worthy of note that there is no linear correlation between the packing efficiency order and the densities. This is a consequence of the conformational nature of the solid form diversity, arising from flexible torsions and bond angles.
Conclusion
This study elucidates the solid form diversity of bosutinib. We have comprehensively evaluated the formation and stability of various solid forms. Key characteristics such as crystal structure, physical stability, dissolution rate, and packing efficiency were assessed. The survey has resulted in a basic understanding of the underlying factors giving rise to discriminative solvate formation.The number and disposition of the hydrogen bond donors and acceptors in the API do not enable strong hydrogen bonding in the solvent free structure. The anhydrous form achieves lattice energy minimization by other, weak intermolecular interactions. Nor the solvated forms possess hydrogen bonds between bosutinib molecules, they are only connected through the guest molecules. This supports our hypothesis that the main contributing factor for the solvent inclusion is the effect of the extensive H-bond network established by the solvent molecules. The stabilization effect of the solvates cannot be ascribed to the decrease of the void space in the structures, as the anhydrate is closely packed. The solvents are utilised to satisfy the previously unused hydrogen bonding capabilities in the host molecule.
Bosutinib incorporates only specific solvent molecules as essential components of the crystal structure, with certain host–solvent interactions. The solvate selectivity is based on the solvents' functionality to provide strong intermolecular interactions. Multipoint hydrogen bonding is clearly a dominant factor that governs the inclusion of solvent molecules.
According to our investigations, certain forms could serve as good candidates for pharmaceutical development but would require a detailed understanding of the impact of numerous variables on the phase transformations including the formulation method, choice of excipients or packaging type.
Acknowledgements
We would like to express our sincere acknowledgement to the Development Department, Zentiva k.s., Prague for giving the possibility to complete this research. We would like to express our gratitude to Tomáš Gurgut, Josef Beranek, Marek Schöngut, Ondřej Dammer, Lukáš Krejčík and Marcela Tkadlecová for their analytical support. This work was supported by the Grant Agency of the Czech Republic, grant no. 14-03636S. X-ray diffraction was performed using instruments of the ASTRA lab established within the Operation program Prague Competitiveness -project CZ.2.16/3.1.00/24510References
-
H. G. Brittain, Polymorphism in Pharmaceutical Solids, Taylor & Francis, 2nd ed., 2009 Search PubMed
.
-
H. G. Brittain, Polymorphism in Pharmaceutical Solids, Marcel Dekker, New York, 1999 Search PubMed
- B. J. Murphy, J. Huang, J. Casteelm, A. Cobani and J. F. Krzyzaniak, Varenicline L-tartrate Crystal Forms: Characterization Through Crystallography, Spectroscopy, and Thermodynamics, J. Pharm. Sci., 2010, 99(6), 2766–2776 CrossRef CAS PubMed
- A. Y. Lee, D. Erdemir and A. S. Myerson, Crystal Polymorphism in Chemical Process Development, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 259–280 CrossRef CAS PubMed
- E. Shefter and T. Higuchi, Dissolution Behaviour of Crystalline Solvated and Nonsolvated Forms of Some Pharmaceuticals, J. Pharm. Sci., 1963, 52(8), 781–791 CrossRef CAS PubMed
- T. Threlfall, Analysis of organic polymorphs, a review, Analyst, 1995, 120, 2435–2460 RSC
-
S. K. Niazi, Handbook of Bioequivalence Testing, 2nd ed., CRC Press, 2007 Search PubMed
- Mekinist-Public assessment report, http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002643/WC500169708.pdf (accessed 22, Jan 2016)
- Forxiga-Public assessment report, http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002322/WC500136024.pdf (accessed 22, Jan 2016)
- Jevtana-Public assessment report, http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002018/WC500104766.pdf (accessed 22, Jan 2016)
- Prezista- Summary of product characteristics, http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000707/WC500041756.pdf (accessed 22, Jan 2016)
- Coumadin FDA document, http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/009218s108lbl.pdf (accessed 22, Jan 2016)
- Crixivan Scientific discussion, http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000128/WC500035727.pdf (accessed 22, Jan 2016)
- Atorvastatin Calcium Public assessment report, http://mri.medagencies.org/download/DK_H_1744_001_PAR.pdf (accessed 22, Jan 2016)
- R. K. Khankari and D. J. W. Grant, Pharmaceutical Hydrates, Thermochim. Acta, 1995, 248, 61–79 CrossRef CAS
- R. Vaidyanathan, G. J. Withbroe, C. Seadeek, K. P. Girard, S. M. Guinness and B. C. Vanderplas, A Robust, Streamlined Approach to Bosutinib Monohydrate, Org. Process Res. Dev., 2013, 17(3), 500–504 CrossRef
-
M. S. Tesconi, G. Feigelson, H. Strong and H. Wen, PCT Int. Appl., WO/2007/005462 A1 20070111, 2007 Search PubMed
- P. Bowles, F. R. Busch, K. R. Leeman, A. S. Palm and K. Sutherland, Confirmation of Bosutinib Structure; Demonstration of Controls To Ensure Product Quality, Org. Process Res. Dev., 2015, 19(12), 1997–2005 CrossRef CAS
-
V. Kiss, O. Dammer and L. Ridvan, Novel solid phases of 4-[(2,4-dichloro-5-methoxyphenyl)amino]-6-methoxy-7-[3-(4-methyl-1 -piperazinyl)propoxy]-3-quinolinecarbonitrile, International Publication Number, WO2015149727 A1, 2014 Search PubMed
- A. J. Matzger, C. P. Price and G. D. Glick, Dissecting the Behaviour of a Promiscuous Solvate Former, Angew. Chem., Int. Ed., 2006, 45(13), 2062–2066 CrossRef PubMed
-
(a) A. L. Gillon, N. Feeder, R. J. Davey and R. Storey, Hydration in Molecular Crystals - A Cambridge Structural Database Analysis, Cryst. Growth Des., 2003, 3(5), 663–673 CrossRef CAS
- S. L. Price, D. E. Braun and S. M. Reutzel-Edens, Can computed crystal energy landscapes help understand pharmaceutical solids?, Chem. Commun., 2016, 52(44), 7065–7077 RSC
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, SIR92 - a program for automatic solution of crystal structures by direct methods, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. J. Watkin, Crystals, Version 14.40b, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, Mercury CSD 2.0 – new features for the visualization and investigation of crystal structures, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS
- R. Oishi-Tomiyasu, Distribution rules of crystallographic systematic absences on the Conway topograph and their application to powder auto-indexing, Acta Crystallogr., Sect. A: Found. Crystallogr., 2013, 69, 603–610 CAS
- V. Petříček, M. Dušek and L. Palatinus, Crystallographic Computing System JANA2006: General features, Z. Kristallogr., 2014, 229(5), 345–352 Search PubMed
- V. Favre-Nicolin and R. Černý, FOX, `free objects for crystallography': a modular approach to ab initio structure determination from powder diffraction, J. Appl. Crystallogr., 2002, 35, 734–743 CrossRef CAS
- B. T. Ibragimov, A Simple Correlation between the Structures of Different Crystal Modifications of a Given Host–Guest Complex and their Crystallization Temperatures, J. Inclusion Phenom. Macrocyclic Chem., 1999, 34(3), 345–353 CrossRef CAS
- B. T. Ibragimov, Further development of the general rule correlating guest space topology and guest content in polymorphs of the given inclusion compound with crystallization temperatures, CrystEngComm, 2007, 9, 111–118 RSC
- M. Campeta, B. P. Chekal, Y. A. Abramov, P. A. Meenan, M. J. Henson, B. Shi, R. A. Singer and K. R. Horspool, Development of a Targeted Polymorph Screening Approach for a Complex Polymorphic and Highly Solvating API, J. Pharm. Sci., 2010, 99(9), 3874–3886 CrossRef PubMed
- M. O. Miclaus, I. E. Kacso, F. A. Martin, L. David, M. M. Pop, C. Filip and X. Filip, Crystal Structure and Desolvation Behaviour of the Tadalafil Monosolvates with Acetone and Methyl Ethyl Ketone, J. Pharm. Sci., 2015, 104(11), 3782–3788 CrossRef CAS PubMed
- H. H. Monfared, A.-C. Chamayou, S. Khajehaand and C. Janiak, Can a small amount of crystal solvent be overlooked or have no structural effect? Isomorphous non-stoichiometric hydrates (pseudo-polymorphs): the case of salicylaldehyde thiosemicarbazone, CrystEngComm, 2010, 12, 3526–3530 RSC
- M. J. Pikal, J. E. Lang and S. Shah, Desolvation kinetics of cefamandole sodium methanolate: effect of water vapor, Int. J. Pharm., 1983, 17(2–3), 237–262 CrossRef CAS
- E. Van Gyseghem, S. Stokbroekx, H. N. de Armas, J. Dickens, M. Vanstockem, L. Baert, J. Rosier, L. Schueller and G. Van den Mooter, Solid state characterization of the anti-HIV drug TMC114: interconversion of amorphous TMC114, TMC114 ethanolate and hydrate, Eur. J. Pharm. Sci., 2009, 38(5), 489–497 CrossRef CAS PubMed
- T. Gelbrich and M. B. Hursthouse, A versatile procedure for the identification, description and quantification of structural similarity in molecular crystals, CrystEngComm, 2005, 7, 324–336 RSC
-
J. Rohlicek, 2016, https://sourceforge.net/projects/crystalcmp (accessed 03 August, 2016)
-
Marc Descamps, Disordered Pharmaceutical Materials, Wiley-VCH, 2016 Search PubMed
- A. T. Ng, C. Lai, M. Dabros and Q. Gao, Insight to the Thermodynamic Stability of Molecular Crystals through Crystallographic Studies of a Multipolymorph System, J. Pharm. Sci., 2014, 103(11), 3423–3431 CrossRef CAS PubMed
- A. B
![[e with combining overline]](https://www.rsc.org/images/entities/char_0065_0305.gif) rziņš, T. Rekis and A. Actiņš, Comparison and Rationalization of Droperidol Isostructural Solvate Stability: An Experimental and Computational Study, Cryst. Growth Des., 2014, 14, 3639–3648 Search PubMed
rziņš, T. Rekis and A. Actiņš, Comparison and Rationalization of Droperidol Isostructural Solvate Stability: An Experimental and Computational Study, Cryst. Growth Des., 2014, 14, 3639–3648 Search PubMed - A. Bērziņš, A. Actiņš, Effect of Experimental and Sample Factors on Dehydration Kinetics of Mildronate Dihydrate: Mechanism of Dehydration and Determination of Kinetic Parameters, J. Pharm. Sci., 2014, 103(6), 1747–1755 CrossRef PubMed
- L. S. Taylor and P. York, Effect of particle size and temperature on the dehydration kinetics of trehalose dehydrate, J. Pharm. Sci., 1998, 87(3), 347–355 CrossRef CAS PubMed
- F. Kang, F. G. Vogt, J. Brum, R. Forcino, R. C. B. Copley, G. Williams and R. Carlton, Effect of Particle Size and Morphology on the Dehydration Mechanism of a Non-Stoichiometric Hydrate, Int. J. Pharm., 1998, 167(1–2), 215–221 Search PubMed
- F. Kang, F. G. Vogt, J. Brum, R. Forcino, R. C. B. Copley, G. Williams and R. Carlton, Model-Free Treatment of the Dehydration Kinetics of Nedocromil Sodium Trihydrate, J. Pharm. Sci., 2003, 92(7), 1367–1376 CrossRef PubMed
- M. R. Caira, G. Bettinetti, M. Sorrenti and L. Catenacci, Relationships between Structural and Thermal Properties of Anhydrous and Solvated Crystalline Forms of Brodimoprim, J. Pharm. Sci., 2007, 96(5), 996–1007 CrossRef CAS PubMed
- R. Chadha, P. Arora, R. Kaur, A. Saini, M. L. Singla and D. S. Jain, Characterization of solvatomorphs of methotrexate using thermoanalytical and other techniques, Acta Pharm., 2009, 59(3), 245–257 CrossRef CAS PubMed
- R. Chadha, P. Arora, A. Saini and D. Singh Jain, Solvated Crystalline Forms of Nevirapine: Thermoanalytical and Spectroscopic Studies, AAPS PharmSciTech, 2010, 11(3), 1328–1339 CrossRef CAS PubMed
- R. Chadha, A. Kuhad, P. Arora and S. Kishor, Characterisation and evaluation of pharmaceutical solvates of Atorvastatin calcium by thermoanalytical and spectroscopic studies, Chem. Cent. J., 2012, 6, 114 CrossRef CAS PubMed
- M. Karan, R. Chadha, K. Chadha and P. Arora, Identification, Characterization and Evaluation of Crystal Forms of Quinine Sulphate, Pharmacol. Pharm., 2012, 3, 129–138 CrossRef CAS
- D. J. W. Grant, H. Zhu and C. Yuen, Influence of water activity in organic solvent + water mixtures on the nature of the crystallizing drug phase. 1. Theophylline, Int. J. Pharm., 1996, 135, 151–160 CrossRef
-
J. I. Wells, Pharmaceutical Preformulation: The Physicochemical Properties of Drug Substances, Ellis Horwood, Ltd., Chichester, 1988 Search PubMed
- S. H. Yalkowsky, G. L. Amidon, G. Zografi and G. L. Flynn, Solubility of nonelectrolytes in polar solvents III: Alkyl p-aminobenzoates in polar and mixed solvents, J. Pharm. Sci., 1975, 64(1), 48–52 CrossRef CAS PubMed
- E. Shefter and T. Higuchi, Dissolution behaviour of crystalline solvated and nonsolvated forms of some pharmaceuticals, J. Pharm. Sci., 1963, 52, 781 CrossRef CAS PubMed
-
R. Liu, Water-Insoluble Drug Formulation, 2nd ed., CRC press, 2008 Search PubMed
-
A. I. Kitaigorodskii, Organic Chemical Crystallography, Consultants Bureau, New York, 1961 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1495576–1495586. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce01834c |
| ‡ Formerly Zentiva k.s., currently Janssen Pharmaceuticals, Inc. |
| § The DVS and TG curves of 7H2O indicate the existence of a distinct hydrate, probably a tetrahydrate, but we were not able to isolate this form. See the curves in the ESI.† |
| ¶ Since temperature affects the solubility of saturated MgCl2, temperature difference caused slight changes in the equilibrium relative humidity values. |
| This journal is © The Royal Society of Chemistry 2016 |