
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intramolecular H⋯S interactions in metal di-(isopropyl)dithiocarbamate complexes†
Alexander
Angeloski
 a,
James M.
Hook
b,
Mohan
Bhadbhade
c,
Anthony T.
Baker
d and
Andrew M.
McDonagh
*a
a,
James M.
Hook
b,
Mohan
Bhadbhade
c,
Anthony T.
Baker
d and
Andrew M.
McDonagh
*a
aSchool of Mathematical and Physical Sciences, University of Technology Sydney, Ultimo, 2007, Australia. E-mail: andrew.mcdonagh@uts.edu.au
bNMR Facility, Mark Wainwright Analytical Centre, The University of New South Wales, Sydney, 2052, Australia
cSchool of Chemistry, The University of New South Wales, Sydney, 2052, Australia
dCollege of Science, Health and Engineering, La Trobe University, Melbourne, 3086, Australia
First published on 16th August 2016
Abstract
Networks of C–H⋯S interactions have been discovered within the molecular structure of sodium di-(isopropyl)dithiocarbamate pentahydrate with the formula Na(C7H14NS2)·5H2O, revealed by single crystal X-ray diffraction. These interactions have also been investigated by ab initio and Hirshfeld surface analyses which show that the electron density is not symmetrical about the molecule. NMR spectroscopy in solution and solid the state showed temperature dependent restricted rotation of the isopropyl groups, which is attributed to the intramolecular C–H⋯S interactions. The ubiquitous nature of C–H⋯S intramolecular interactions in this class of compound is evident in the structures of other di-(isopropyl)dithiocarbamate complexes deposited in the CSD. In general, the restricted rotation in di-(isopropyl)dithiocarbamate complexes can be directly attributed to intramolecular C–H⋯S interactions, which subsequently influence the geometry in association with steric repulsion factors.
1. Introduction
Noncovalent interactions can exert significant influence on the geometry of molecules and their associated crystal structures.1 Hydrogen bonding is an important noncovalent interaction where the focus has traditionally been on strong acceptors such as oxygen and nitrogen.2–7 In contrast, sulfur is considered a weak acceptor, and the influence of H⋯S hydrogen bonds on molecular structure has received considerably less attention.8–11 Such interactions can influence side chain geometries and secondary structure in biological systems (such as those involving the amino acid cysteine).12–16 Thus, an understanding of the nature of H⋯S interactions is of fundamental importance. Here, we examine a low molecular weight compound to illustrate the influence of H⋯S interactions upon both solid and solution phase geometries. We demonstrate that the H⋯S interaction has a profound effect in both regimes that effectively breaks the symmetry of the molecule and creates a restricted rotation about several of the covalent bonds within the molecule.Dithiocarbamate complexes have been studied extensively for their ability to coordinate a range of transition and main block elements, and for their interesting and useful properties.17,18 In particular, there is a significant amount of data pertaining to di-(isopropyl)dithiocarbamate (dipdtc) compounds. The structure of the dipdtc ligand is such that the C2NCS2 atoms lie within a plane. It has been known for some time that there is a disruption to the symmetry of dipdtc complexes whereby the methine hydrogens are oriented in different directions relative to the C2NCS2 plane (Fig. 1). This orientation results in the inequivalence of the two isopropyl groups, which has been ascribed to a steric interaction hindering rotation about the N–Cisopropyl bonds based on NMR experiments in solution.19–23 The NMR data reveal thermodynamic parameters associated with this restricted rotation but in isolation, these studies do not elucidate the origin of the inequivalence of the two isopropyl groups.
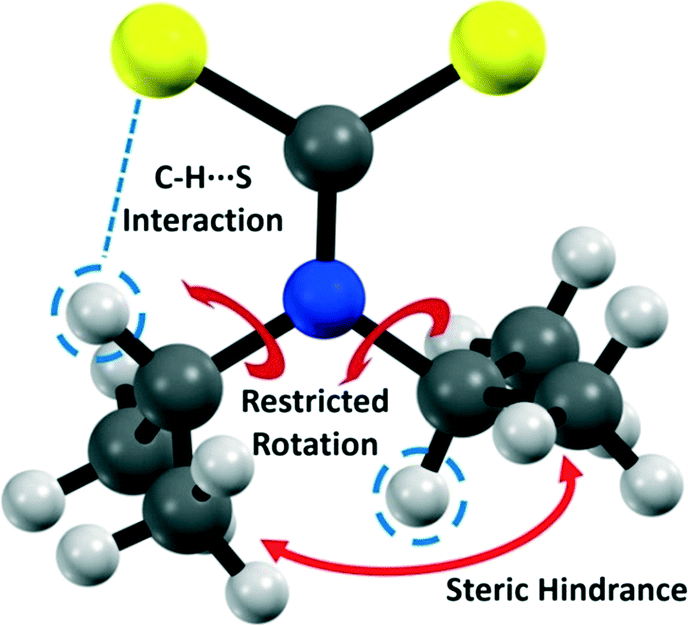 | ||
| Fig. 1 Structure of the dipdtc anion showing the important interactions. | ||
In this work, we have redetermined24 the crystal structure of sodium di-(isopropyl)dithiocarbamate pentahydrate, Na(dipdtc)·5H2O, with greater accuracy to allow a detailed examination of intramolecular C–H⋯S interactions. The compound was also analysed using variable temperature solution state NMR spectroscopy, which shows features associated with restricted rotation about the N–Cisopropyl bonds. Using a combination of theoretical calculations and experimental observations, we elucidate the origin of the inequivalence of the two isopropyl groups and restricted rotation. Furthermore, we show that these C–H⋯S interactions are ubiquitous throughout metal dipdtc complexes for which structural determinations have been deposited.
2. Experimental
Reagents and instruments
Chemicals and solvents used in synthetic procedures were analytical grade and purchased from Sigma Aldrich and used as received. Millipore water (18.4 MΩ cm−1) was used in synthetic procedures. A high resolution mass spectrum was acquired using an Agilent 6510 Q-TOF with a mobile phase of 70% acetonitrile, 30% water.Synthesis
Sodium di-(isopropyl)dithiocarbamate was prepared as described by Angeloski et al.25 An aqueous solution of sodium hydroxide (10.00 g in 40 mL) was cooled with stirring to 5 °C. Diisopropylamine (25 mL) was added followed by diethyl ether (75 mL). The solution was maintained between 5–10 °C whilst carbon disulfide (25 mL) was added dropwise. A precipitate formed upon addition and the resultant mixture was stirred for 20 minutes. The crude material was collected by vacuum filtration, and the filter cake purified by recrystallization using layer diffusion of ether into a methanolic solution of the crude material. The colourless crystals were collected by vacuum filtration, washed with warm diethyl ether and dried in vacuo to produce 15.34 g of sodium di-(isopropyl)dithiocarbamate (31%). HRMS (M + H)+ for NaNS2C7H14 calculated: 200.0538; found: 200.0549. 1H NMR (600.1 MHz, CD3CN, 258 K): δ 6.21 (sept, J = 6.8 Hz, 1H, H2), 3.79 (sept, J = 6.9 Hz, 1H, H5), 1.63 (d, J = 6.8 Hz, 6H, C6H3, C7H3), 1.06 (d, J = 6.9 Hz, 6H, C3H3, C4H3). 13C NMR (150.9 MHz, CD3CN, 258 K): δ 212.4 (C1), 56.1 (C2), 50.0 (C5), 20.9 (C6, C7), 19.6 (C3, C4).NMR spectroscopy and kinetic analysis
Variable temperature proton (1H), carbon (13C), 2D exchange spectroscopy (EXSY, with mixing time 100 ms), heteronuclear single quantum correlation (HSQC) and heteronuclear multiple bond correlation (HMBC) nuclear magnetic spectroscopy of solutions was performed using a Bruker Avance III NMR spectrometer fitted with a BBFO Plus solution state probe. The frequency was 600.1 MHz for 1H, 150.9 MHz for 13C and 60.8 MHz for 15N experiments. A Bruker BVT3000 VT unit was used in conjunction with a BCU-Xtreme cooler to accurately adjust the sample temperature between 293 and 258 K with deuterated acetonitrile as the solvent. Instrument broadening was accounted for using acetonitrile residual proton resonances. Using this data, experimental rate constants were calculated at each temperature using the signal FWHM for the methyl peak at 1.63 ppm. Arrhenius and Eyring activation parameters were obtained using generalized least squares linear regressions of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k vs. 1/T and log(k/T) vs. 1/T respectively. A solid state 1H NMR spectrum was acquired using a Bruker Avance III 700 MHz solid state NMR spectrometer. Samples were loaded into a 1.3 mm zirconia rotor and a MAS rate of 60 kHz was adopted using a Bruker MAS 2 unit.
k vs. 1/T and log(k/T) vs. 1/T respectively. A solid state 1H NMR spectrum was acquired using a Bruker Avance III 700 MHz solid state NMR spectrometer. Samples were loaded into a 1.3 mm zirconia rotor and a MAS rate of 60 kHz was adopted using a Bruker MAS 2 unit.
Crystallographic analysis
Crystals of Na(dipdtc)·5H2O suitable for analysis were prepared by layer diffusion of ether into a methanolic solution. A suitable crystal was selected under a polarising microscope (Leica M165Z), mounted on a MicroMount (MiTiGen) consisting of a thin polymer tip with a wicking aperture. The X-ray diffraction measurements were carried out on a Bruker Kappa-II CCD diffractometer at 150 K by using IμS Incoatec Microfocus Source with Mo-Kα radiation (λ = 0.710723 Å). The structure was solved using charge flipping and the full matrix least squares refinement was performed using ShelXl26 in Olex2.27 Heavy atoms were refined isotropically until R-factor convergence, and then an anisotropic model was applied. Hydrogen atoms were located using a difference Fourier plot, and restrained to neutron diffraction distances where required for water molecules.CrystalExplorer28 was used to generate Hirshfeld surfaces29–31 representing dnorm and electron deformation density. The latter surface was calculated using TONTO32 which is integrated into CrystalExplorer, and the experimental geometry was used as the input. The electron deformation density was mapped on the Hirshfeld surface using the 6-311G(d,p) basis set with the Density Functional Theory.
Crystal structure retrieval
Previous determinations of metal dipdtc crystal structures were retrieved from the Cambridge Structural Database (CSD)33 with the following specifications: R ≤ 5%, no disorder, no errors, no powder structures and complete 3D coordinates. This produced a total of 32 crystal structures, which were sorted by hand to remove duplicates and non-relevant structures. A final number of 28 crystal structures were retrieved. Lists of the crystal structure CSD reference codes are available in the ESI.†Evaluation of intramolecular contacts
Atoms from the retrieved structures were relabelled to be in accordance with the labelling scheme in Fig. 2. The presence of an intramolecular C–H⋯S interaction was preliminarily assessed by calculation of the S1⋯H2 distance. Quantitative measurements were performed using the heavy atom structural parameters to avoid uncertainty due to hydrogen atom treatment. The structures were sorted by hand to retrieve the following parameters: C1–N1–C2 and C1–N1–C5 bond angles. The C1–N1, C2–S1 and C5–S2 (Fig. 2) distances were also extracted. Bond angles were kept to two decimal places, and bond lengths to three. All statistical analyses were conducted using the statistical analysis software, R 3.2.5.34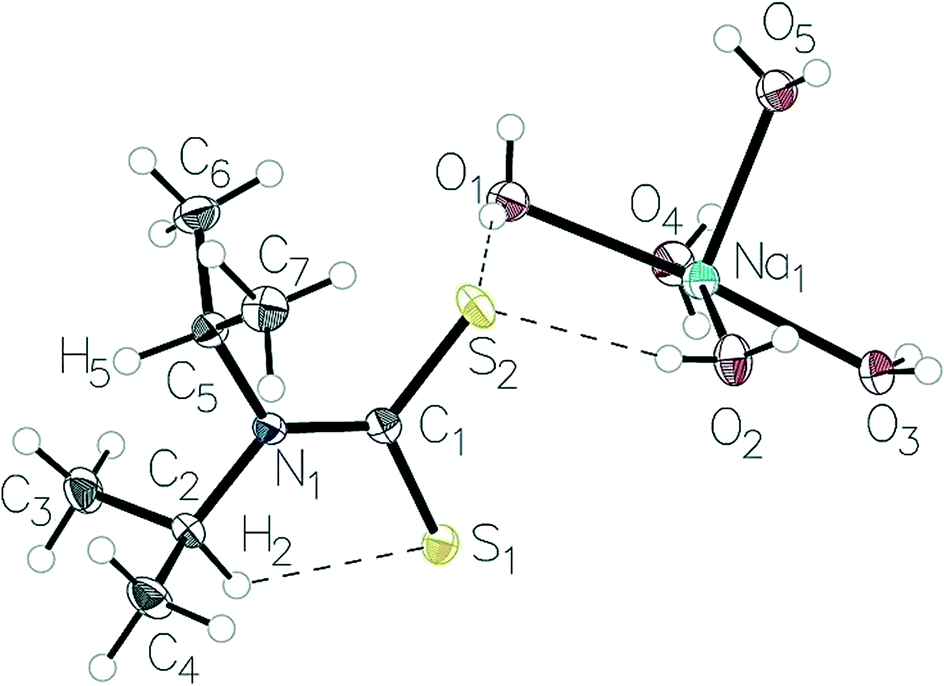 | ||
| Fig. 2 The structure the Na(dipdtc)·5H2O asymmetric unit showing the atom-labelling scheme and thermal displacement ellipsoids at 50% probability. | ||
3. Results and discussion
X-Ray structure determination
Relevant crystal data, selected bond lengths and angles are given in Tables 1 and 2 (see ESI† for complete data). These values are in good agreement with the previously reported structure (CSD-Refcode BUNPIJ), with bond lengths falling in the ranges expected on the analysis of the literature.24,36 The asymmetric unit, Fig. 2, contains one molecular anion together with one sodium cation (Na1) co-ordinated to five water molecules (O1–O5) (the sixth octahedral site is occupied by O3 on the symmetry related water molecule O3 (1 − x, 1 − y, 1 − z)). The co-ordination sphere of sodium is distorted octahedral with deviations from ideal octahedral geometry of less than 5°. The Na–O distances range from 2.3859(15) Å to 2.4492(14) Å. There is no direct S2–Na1 bond; Na1 is located more than 4 Å from S2. Within the dipdtc anion, the maximum deviations from the least-squares plane through C2–C5–S2–S1 (with a r.m.s. deviation of 0.008 Å) are 0.009 Å for C2 and C5, and 0.008 Å for S1 and S2. The S2–C1 and S1–C1 bond lengths are inequivalent with S1–C1 [1.7484(16) Å] elongated compared to S2–C1 [1.7145(17) Å]. The N1–C2 and N1–C5 bond lengths are equal within experimental error and all bond lengths are similar to those in other M(dipdtc) structures, where M = Pb, Hg, Ni, In, Co, Be, Cu and Au.37–44| Chemical formula | 2(C7H14NS2)·H20Na2O10 |
| M r | 578.76 |
| Crystal system, space group | Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Temperature (K) | 150 |
| a, b, c (Å) | 5.9472(11), 7.7189(16), 17.425(3) |
| α, β, γ (°) | 92.183(8), 95.095(8), 106.851(8) |
| V (Å3) | 760.8(3) |
| Z | 1 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.38 |
| Crystal size (mm) | 0.41 × 0.09 × 0.04 |
| Absorption correction | Multi-scan SADABS2014/5 (ref. 35) was used for absorption correction. wR2(int) was 0.1663 before and 0.0594 after correction. The ratio of minimum to maximum transmission is 0.9019. The λ/2 correction factor is 0.00150 |
| Diffractometer | Bruker APEX-II CCD |
| T min, Tmax | 0.672, 0.746 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 18709, 3304, 2666 |
| R int | 0.060 |
| (sinθ/λ)max (Å−1) |
0.639 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.032, 0.070, 1.04 |
| No. of reflections | 3304 |
| No. of parameters | 241 |
| No. of restraints | 11 |
| H-Atom treatment | All H-atom parameters refined |
| Δ〉max, Δ〉min (e Å−3) | 0.30, −0.25 |
| Symmetry code: (i) −x + 1, −y + 1, −z + 1. | |||
|---|---|---|---|
| S1–C1 | 1.7484(16) | Na1–O4 | 2.3891(13) |
| S2–C1 | 1.7145(17) | Na1–O1 | 2.4492(14) |
| N1–C2 | 1.492(2) | Na1–O2 | 2.4207(13) |
| N1–C1 | 1.345(2) | Na1–O5 | 2.3859(15) |
| C2–C3 | 1.524(2) | Na1–O3i | 2.4278(14) |
| C5–C6 | 1.522(2) | Na1–O3 | 2.3879(14) |
| C2–N1–C5 | 113.38(12) | O4–Na1–O1 | 92.33(5) |
| C1–N1–C2 | 122.01(13) | O4–Na1–O2 | 162.67(5) |
| C1–N1–C5 | 124.59(14) | O4–Na1–O3i | 84.60(5) |
| N1–C2–C3 | 111.51(13) | O2–Na1–O1 | 95.54(5) |
| N1–C2–C4 | 111.23(13) | O2–Na1–O3i | 80.40(5) |
| C4–C2–C3 | 112.74(15) | O5–Na1–O4 | 96.85(5) |
| S2–C1–S1 | 118.11(9) | O5–Na1–O1 | 88.03(5) |
| N1–C1–S1 | 120.25(12) | O5–Na1–O2 | 98.83(5) |
| N1–C1–S2 | 121.63(12) | O5–Na1–O3 | 97.38(5) |
| N1–C5–C7 | 113.60(13) | O5–Na1–O3i | 175.04(5) |
| N1–C5–C6 | 113.02(13) | O3–Na1–O4 | 85.89(5) |
| C6–C5–C7 | 113.36(15) | O3–Na1–O1 | 174.47(5) |
| O3–Na1–O2 | 84.83(5) | O3i–Na1–O1 | 87.17(5) |
| Na1–O3–Na1i | 92.55(5) | O3–Na1–O3i | 87.45(5) |
From the molecular assembly diagram, Fig. 3, a layered supramolecular motif is evident parallel to the crystallographic b direction (see ESI† for images showing packing diagrams viewed along b and c directions). The layered structure is stabilised by van der Waals interactions between the alkyl groups on the anion.
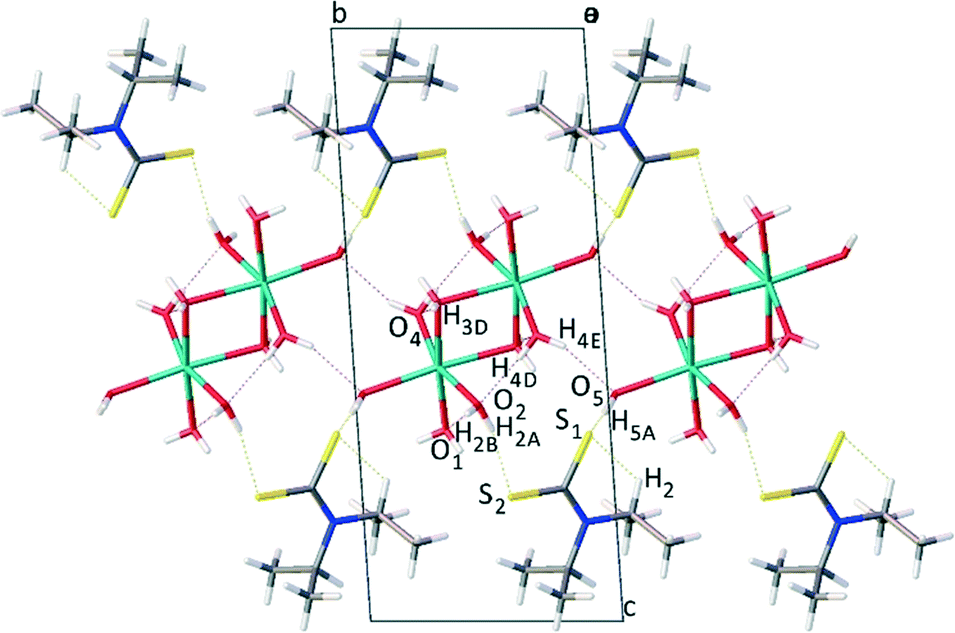 | ||
| Fig. 3 Molecular packing diagram as viewed along the a axis, showing layered morphology of Na(dipdtc)·5H2O. | ||
Sodium ions are positioned between layers of symmetrically equivalent ligand anions, forming a two dimensional Na–Na array oriented parallel to the crystallographic b direction. The sodium ions are separated by a distance of 3.4804(12) Å, significantly longer than the sum of ionic radii of 2.32 Å. Alternating sodium ions are linked by bridging water molecules at O3, forming a four membered Na1–O3–Na1–O3 ring with vertices of 92.55(5)° for Na1–O3–Na1 and 87.45(5)° for O3–Na1–O3. That is, dinuclear entities [Na2(OH2)10] exist in which the distorted NaO6 octahedra share an edge. The array of Na cations is stabilised by a network of hydrogen bonding45 between alternate and neighbouring water molecules where distances and angles are in agreement with literature values for O–H⋯O hydrogen bonds.46–48 The ligand anions are linked by O–H⋯S hydrogen bonds2,49 to the water molecules associated with the array of sodium cations. These hydrogen bonding contacts are summarised in Table 3.
| D–H⋯A | D–H (Å) | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) |
|---|---|---|---|---|
| Symmetry codes: (i) x, y + 1, z; (ii) x + 1, y, z; (iii) −x + 1, −y + 1, −z + 1; (iv) −x + 1, −y + 2, −z + 1. | ||||
| C2–H2⋯S1 | 0.998(16) | 2.392(16) | 3.0264(17) | 120.8(12) |
| C7–H7A⋯S2 | 0.921(19) | 2.714(17) | 3.270(2) | 119.7(13) |
| C6–H6A⋯S2 | 0.938(19) | 2.741(17) | 3.280(2) | 117.4(13) |
| O2–H2A⋯S2 | 0.85(2) | 2.41(2) | 3.2336(15) | 165(2) |
| O5–H5A⋯S1i | 0.83(2) | 2.53(2) | 3.2831(14) | 151(2) |
| O3–H3D⋯O4ii | 0.83(2) | 2.02(2) | 2.8407(18) | 171(2) |
| O2–H2B⋯O1ii | 0.84(2) | 1.97(2) | 2.8123(18) | 175(2) |
| O4–H4D⋯O2iii | 0.83(2) | 1.95(2) | 2.7796(19) | 176(2) |
| O4–H4E⋯O5iv | 0.83(2) | 2.08(2) | 2.8751(19) | 160(2) |
Contacts involving intermolecular H⋯S, H⋯O and H⋯H interactions were examined using Hirshfeld surface analysis.29–31,50,51 The majority of interactions within the structure are dominated by van der Waals H⋯H interactions (∼65%) followed by O–H⋯S (∼17%) and O–H⋯O (∼13%) interactions. The intermolecular contacts are highlighted in the map of dnorm on the Hirshfeld surface, Fig. 4. The dark red regions are due to hydrogen bonding whilst the blue and white regions reflect H⋯H interactions. C–H⋯S interactions are also evident.
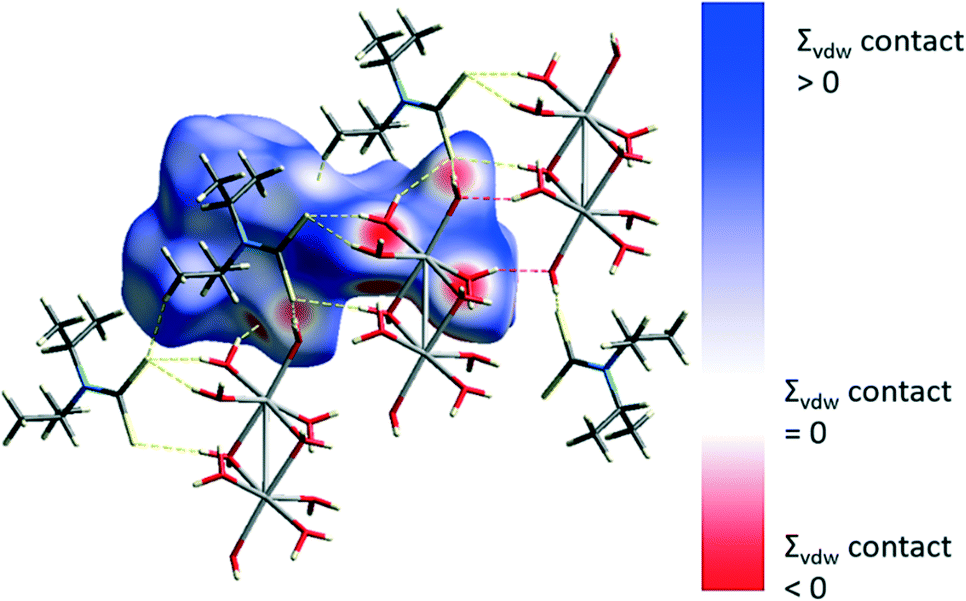 | ||
| Fig. 4 Hirshfeld surface and O–H⋯S hydrogen bonds (yellow) and O–H⋯O hydrogen bonds (red). | ||
Of significance to the current work are the intramolecular non-bonding interactions involving sulfur. The S1⋯H2 intramolecular interaction has a distance of ∼2.4 Å (Table 3), which as we show here, exerts influence throughout the entire molecular structure. The C1–N1–C2 angle is 2.58 (13)° smaller than the C1–N1–C5 angle and the C2–S1 distance is 0.034 Å shorter than the C5–S2 distance (Table 4). The S1⋯H2 interaction also results in a pair of interactions between S2 and the methyl hydrogens attached to C6 and C7 of ∼2.7 Å (Table 3). That is, the S1⋯H2 interaction creates an inequivalence of the two isopropyl groups within the dipdtc anion. The intramolecular C–H⋯S dihedral angles and lengths are similar to those involving cysteine (117.4° and 2.51 Å) and methionine (119.0° and 2.74 Å) residues interacting within proteins.12
| Parameter | Mean ± standard error | Current study |
|---|---|---|
| C2–S1 distance | 3.024(6) Å | 3.0264(17) Å |
| C5–S2 distance | 3.147(8) Å | 3.1014(18) Å |
| S2–C1–S1 angle | 111.81(71)° | 118.11(9)° |
| C1–N1 distance | 1.321(2) Å | 1.345(2) Å |
| C1–N1–C5 angle | 123.78(11)° | 124.59(13)° |
| C1–N1–C2 angle | 120.11(22)° | 122.01(13)° |
Electrostatic deformation density and topological features of the intramolecular C–S⋯H interaction.
A 3D electrostatic deformation density map (Fig. 5) shows the electron density about the dipdtc anion. The lone pair electron density (LPED) about S1 and S2 is of particular interest as this may influence hydrogen bond directionality and the capacity of S1 and S2 to form multiple H bonds.2 Our calculations show that the S atom LPED adopts a toroidal geometry, in agreement with previous analyses of sulfur-containing compounds.2 Upon closer examination (Fig. 6), the LPED about S1 and S2 is somewhat distorted as a result of S⋯H interactions. In addition to intermolecular interactions with H2O, electron density is directed from S1 towards H2 while S2 directs electron density towards H6 and H7. A region of charge depletion (shown in red) about H2 is oriented toward the LPED about S1. This attractive behaviour between S1 and H2 is the origin of the unsymmetrical geometry of the isopropyl groups about N1.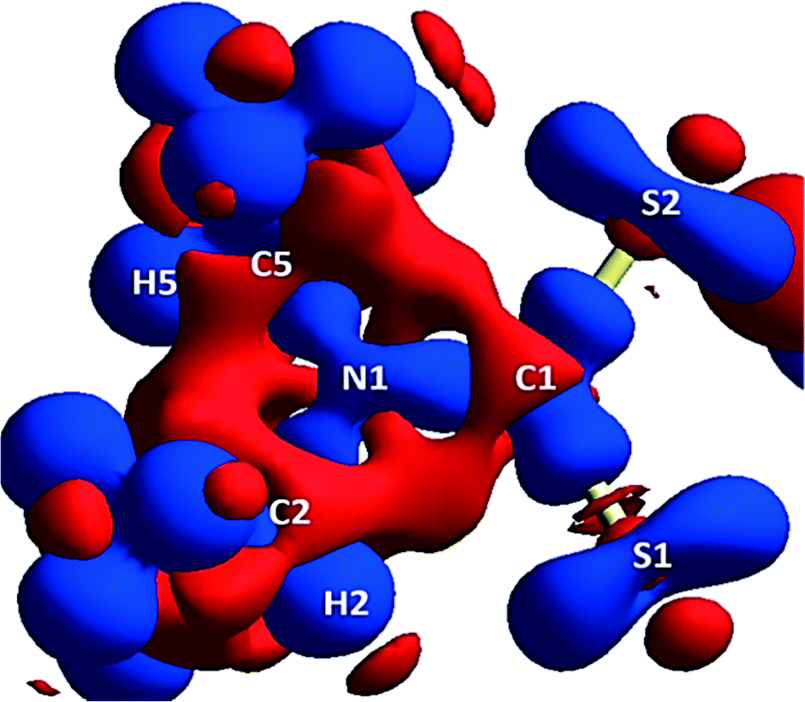 | ||
| Fig. 5 Theoretical 3D electrostatic densities of the Na(dipdtc)·5H2O anion at the B3LYP 6-311G(d,p) level. Areas of lower density are shown in red, and areas of higher density are shown in blue. | ||
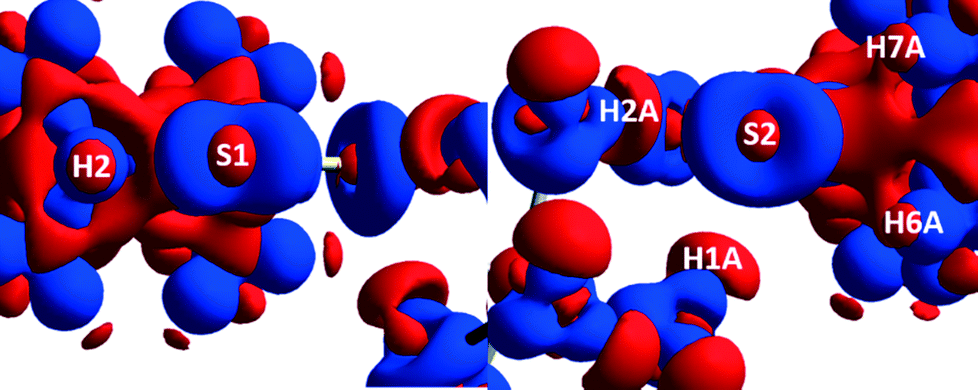 | ||
| Fig. 6 3D Static deformation densities of the sulfur tori (viewed down the C–S bond). | ||
NMR Spectroscopy
The crystallographic intramolecular C–H⋯S interactions yield two inequivalent isopropyl groups which are also manifested in solution, evidenced by two sets of signals in solution state NMR spectra. 1H and 13C NMR spectra of Na(dipdtc)·5H2O recorded using deuterated acetonitrile at room temperature revealed broad proton and carbon resonances associated with the methyl and methine groups. Two broad signals for the methyl protons and two broad signals for the methine protons were observed, consistent with two inequivalent environments (ESI†). That is, H2 is maintained in synchronous gear-like arrangement relative to H5 such that the steric repulsion between the associated methyl groups is minimised, Fig. 1. A DOSY experiment confirmed that the signals arose from only a single molecular species (ESI†).The methine protons, H2 and H5, exhibit chemical shifts of 6.2 and 3.8 ppm, respectively. We attribute the large difference in chemical shifts to the influence of the C–H⋯S interaction upon the local electron density about H2, whereby H2 is significantly more deshielded than H5. Similarly, the signal assigned to the methyl protons associated with C6 and C7 appears at 1.6 ppm while the signal assigned to protons on C3 and C4 appears at 1.1 ppm. Thus, while the influence of the C–H⋯S interaction is still apparent, the difference in chemical shifts between the methyl protons on the two inequivalent isopropyl groups is less than that between H2 and H5 due to the greater distance between the methyl protons and sulfur. In the 13C NMR spectrum recorded at 293 K, two sets of signals are also observed, which are significantly broadened. 2D-EXSY spectra recorded at 293 K contain H2/H5 cross peaks with intensity equal to the source peaks indicating a chemical exchange process at room temperature (ESI†).
Similarly, the high speed MAS solid state 1H NMR spectrum (ESI†) contains methyl resonances in the range of 0–2 ppm and a signal assigned to the methine C–H⋯S proton H2 at 6.1 ppm. A signal at ∼3.7 ppm is assigned to the methine proton H5, which is partially obscured by a broad peak associated with water molecules.
Variable temperature 1H NMR (Fig. 7) data also provide kinetic insights into the rotation about the C2–N1 and C5–N1 bonds. A 2D-EXSY spectrum recorded at 258 K exhibits H2/H5 cross peaks of lower intensity than those recorded at 293 K, characteristic of reduced exchange between H2 and H5, and consistent with a greater restriction of rotation of the isopropyl groups at this temperature. Upon cooling from 293 K to 258 K, the broad signals observed in the 1D 1H NMR spectrum resolve into their respective splitting patterns (septets for H2/H5 and doublets for the methyl protons), which is also consistent with the slowing rate of concerted rotation of the isopropyl groups. From the VT NMR data recorded using acetonitrile as solvent, a value of 63 kJ mol−1 for ΔG‡ was calculated at 283 K and at 263 K, a value of 56 kJ mol−1 was obtained. While the latter value for ΔG‡ agrees with data reported by others20 using dichloromethane as solvent, our values for ΔH‡ (27 kJ mol−1), ΔS (and −117 J mol−1 K−1) and activation energy (30 kJ mol−1) are all somewhat smaller than those reported.20 This indicates that the barrier to rotation may be lower in acetonitrile although quantitative comparison to the literature data20 is problematic as the exact composition of the previously reported compound was uncertain.
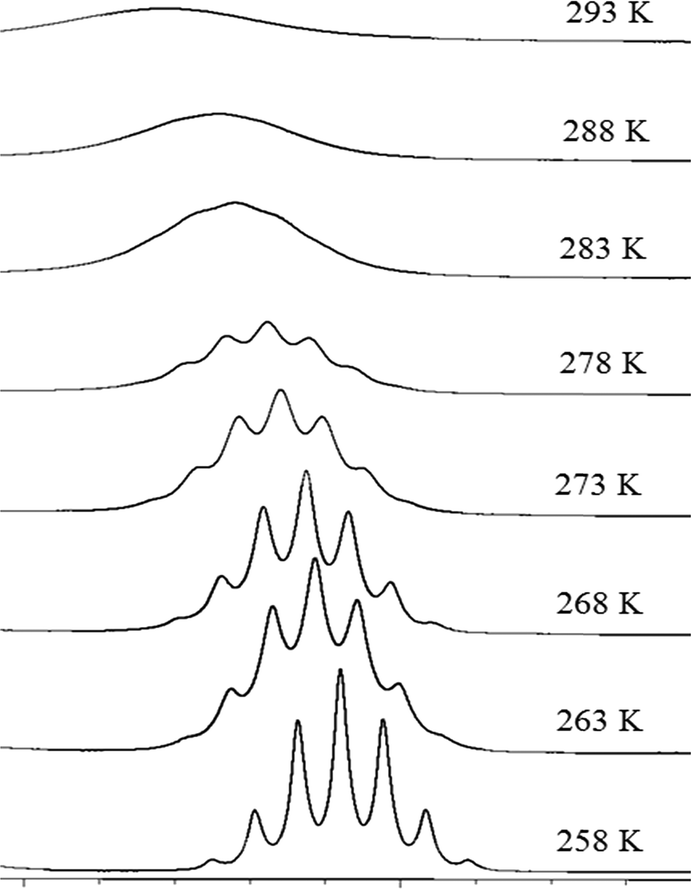 | ||
| Fig. 7 Selected portion of the 600 MHz 1H spectra for Na(dipdtc)·5H2O in CD3CN, showing the change in H2 resonance as a function of temperature. | ||
Database survey of intramolecular C–H⋯S interactions in metal di(isopropyl)dithiocarbamate complexes
Having examined the CH⋯S interactions in Na(dipdtc)·5H2O, we sought to find crystallographic evidence for this interaction in other dipdtc complexes. A survey of the Cambridge Structural Database was undertaken. A total of 28 suitable structure determinations were selected. All structures were planar about the S2CNC2 moiety and importantly, showed evidence of intramolecular S1⋯H2 interactions that induce differences between the two isopropyl groups. Table 4 summarises some relevant structural parameters.The C2 to S1 distances, x (Fig. 8), were chosen for comparison in preference to H2 to S1 distances as the former are more accurate data. Values for x are tightly clustered about the mean of 3.024 Å. The shortest distance is 2.941 Å (Nd(dipdtc)3phen) and the longest is 3.117 Å (Co(dipdtc)3). With the exception of Co(dipdtc)3, all values of x fall within the 2.941–3.054 Å (a range of 0.113 Å). In comparison, the C5–S2 distances (y) varied more, and ranged from 3.072 to 3.219 Å (a range of 0.147 Å) as shown in Fig. 9. The mean x distance for the 28 structures is significantly shorter (by 0.123 ± 0.01 Å) than that of y (t = −12.233, df = 49.412, p < 2.2 × 10−16).
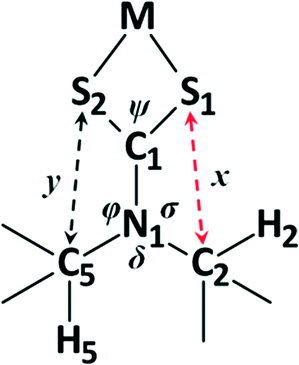 | ||
| Fig. 8 Depiction of the important distances and angles discussed in the text. | ||
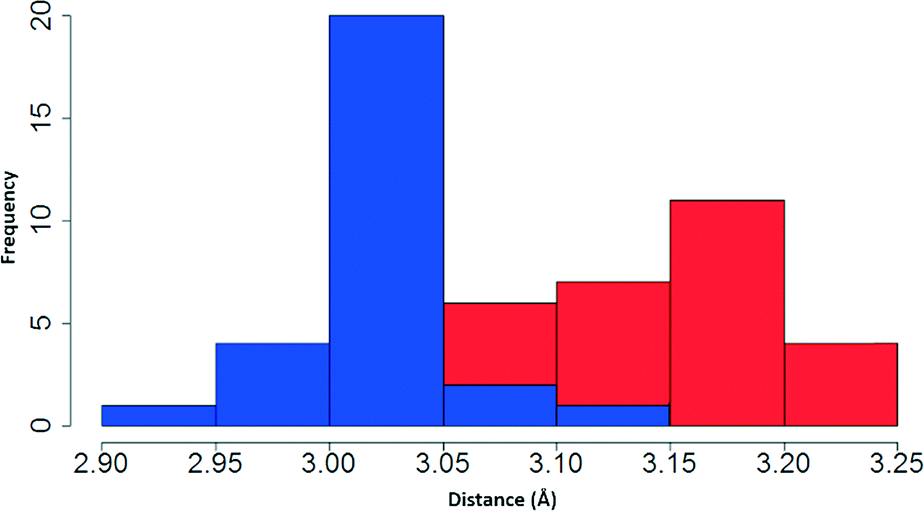 | ||
| Fig. 9 Stacked histogram showing distribution of distances x (blue) and y (red). | ||
Unsurprisingly, the S1–C1–S2 angles (ψ) are dependent on the coordinated metal24 and span the range 106.47 to 117.31°. The angle C1–N1–C2 (σ) ranges from 117.0 to 122.3° while the angle C1–N1–C5 (φ) has a range of 122.1 to 124.9° and for any structure, σ < φ (t = −5.058, df = 40.716, p < 2.2 × 10−16). As planarity is maintained across the S2CNC2 moiety for all structures, φ + δ + σ = 360° and δ > 114° in all cases.
Our interpretation of these data is as follows. The angle ψ is dependent upon the coordinated metal while the distance x is a consequence of the C–H⋯S interaction. There were no statistically significant pairwise correlations between x and ψ, nor between x and any other structural parameter. Thus, the relationships σ ≠ φ and x ≠ y are a consequence of the C–H⋯S interaction. The C–H⋯S interaction restricts the rotation of the corresponding isopropyl group about the C2–N1 bond. This, in turn, influences the rotation of the isopropyl group about the C5–N1 bond (through steric effects). Previously published VT NMR-based analysis of the unusual geometry of metal dipdtc complexes attributed the cause of restricted rotation to purely steric effects19–23 but here we have shown that the underlying cause of the restricted rotation is the intramolecular C–H⋯S interaction, an electronic effect.
Conclusions
We have shown that C–H⋯S intramolecular interactions are present within the molecular structure of Na(dipdtc)·5H2O. Theoretical calculations constrained by the experimentally derived structural data indicate that these interactions arise from the interaction between the lone pair electron density on sulfur and regions of electron depletion about a methine hydrogen.Inequivalent chemical environments about the methine protons produced by the C–H⋯S intramolecular interaction were observed using NMR spectroscopy in solution and solid samples. Variable temperature solution state NMR spectroscopy was used to probe the restricted rotation of the isopropyl groups about the N–C single bonds revealing an energy barrier for this rotation of 30 kJ mol−1. The Gibbs free energy of the transition state (63 kJ mol−1) is in agreement with previous studies of restricted rotation in dipdtc structures.
An analysis of 28 similar structures using the CSD revealed the presence of intramolecular C–H⋯S interactions. In all of the analysed structures, the heavy atom geometries supported the presence of these interactions. In all cases, the relevant intramolecular C–S distances were shorter and less variable where intramolecular C–H⋯S interactions were present. There are no significant correlations between the steric factors of the structure and the C–H⋯S intramolecular interaction.
Thus, the restricted rotation in metal dipdtc structures is directly attributable to the intramolecular C–H⋯S interaction, which subsequently influences the geometry in association with steric repulsion factors between methyl groups. We propose that these interactions are worthy of further examination in a wider range of compounds such as those found in biological systems (proteins, peptides) where bonds are subject to restricted rotation in proximity to sulfur atoms.
Acknowledgements
We acknowledge Mr Leinad Diaz and Dr Ronald Shimmon for laboratory assistance and the Mark Wainwright Analytical Centre, UNSW, for access to single crystal XRD, solid and solution state NMR spectrometers. We also acknowledge Dr Parthapratim Munshi for assistance with CrystalExplorer software.References
- M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama and H. Suezawa, CrystEngComm, 2009, 11, 1757–1788 RSC.
- B. M. Francuski, S. B. Novaković and G. A. Bogdanović, CrystEngComm, 2011, 13, 3580 RSC.
- P. Schuster, G. Zundel and C. Sandorfy, The Hydrogen Bond-Recent Developments in Theory and Experiment, North-Holland Pub. Co, 1976 Search PubMed.
- G. C. Pimental and A. L. McClellan, The Hydrogen Bond, W. H. Freeman and Co., San Francisco, 1960 Search PubMed.
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer-Verlag, Berlin, 1991 Search PubMed.
- P. A. Hunt, C. R. Ashworth and R. P. Matthews, Chem. Soc. Rev., 2015, 44, 1257–1288 RSC.
- A. Gavezzotti and L. L. Presti, Cryst. Growth Des., 2016, 16, 2952–2962 CAS.
- T. Steiner, Crystallogr. Rev., 2003, 9, 177–228 CrossRef CAS.
- G. A. Jeffrey, Crystallogr. Rev., 2003, 9, 135–176 CrossRef CAS.
- J. Perlstein, J. Am. Chem. Soc., 2001, 123, 191–192 CrossRef CAS.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond: In Structural Chemistry and Biology, Oxford University Press, 2001 Search PubMed.
- P. Zhou, F. Tian, F. Lv and Z. Shang, Proteins: Struct., Funct., Bioinf., 2009, 76, 151–163 CrossRef CAS PubMed.
- D. Pal and P. Chakrabarti, J. Biomol. Struct. Dyn., 2001, 19, 115–128 CAS.
- L. M. Gregoret, S. D. Rader, R. J. Fletterick and F. E. Cohen, Proteins: Struct., Funct., Bioinf., 1991, 9, 99–107 CrossRef CAS PubMed.
- M. Iwaoka and N. Isozumi, Molecules, 2012, 17, 7266 CrossRef CAS PubMed.
- S. Horowitz and R. C. Trievel, J. Biol. Chem., 2012, 287, 41576–41582 CrossRef CAS PubMed.
- A. A. Aly, A. B. Brown, T. M. I. Bedair and E. A. Ishak, J. Sulfur Chem., 2012, 33, 605–617 CrossRef CAS.
- J. J. Steggerda, J. A. Cras and J. Willemse, Recl. Trav. Chim. Pays-Bas, 1981, 100, 41–48 CrossRef CAS.
- R. M. Golding, P. C. Healy, P. W. G. Newman, A. H. White and E. Sinn, Inorg. Chem., 1972, 11, 2435–2440 CrossRef CAS.
- A. F. Lindmark and R. C. Fay, Inorg. Chem., 1983, 22, 2000–2006 CrossRef CAS.
- Y. I. Takeda, N. Watanabe and T. Tanaka, Spectrochim. Acta, Part A, 1976, 32, 1553–1556 CrossRef.
- S. Bhattacharya, B. K. Kanungo and S. Sahoo, J. Coord. Chem., 2006, 59, 371–378 CrossRef CAS.
- A. N. Bhat, R. C. Fay, D. F. Lewis, A. F. Lindmark and S. H. Strauss, Inorg. Chem., 1974, 13, 886–892 CrossRef CAS.
- I. Ymen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 874–877 CrossRef.
- A. Angeloski, A. T. Baker, M. Bhadbhade and A. M. McDonagh, J. Mol. Struct., 2016, 1113, 127–132 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer (Version 3.1), University of Western Australia, 2012 Search PubMed.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- D. Jayatilaka, D. J. Grimwood, A. Lee, A. Lemay, A. J. Russel, C. Taylor, S. K. Wolff, P. Cassam-Chenai and A. Whitton, TONTO, Available at: http://Hirshfieldsurface.net/, 2005 Search PubMed.
- F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- R. D. C. Team, R: A Language and Environment for Statistical Computing, Vienna, Austria, 2008 Search PubMed.
- Bruker, SADABS, Bruker AXS Inc, Wisconsin USA, Madison, 2001 Search PubMed.
- F. H. Allen, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, in International Tables for Crystallography, John Wiley & Sons, Ltd, 2006, DOI:10.1107/97809553602060000621.
- T. A. Rodina, A. V. Ivanov, A. V. Gerasimenko, O. V. Loseva, O. N. Antzutkin and V. I. Sergienko, Polyhedron, 2012, 40, 53–64 CrossRef CAS.
- M. Ito and H. Iwasaki, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 2720–2721 CrossRef CAS.
- M. Ito and H. Iwasaki, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 443–444 CrossRef.
- P. C. Healy, J. W. Connor, B. W. Skelton and A. H. White, Aust. J. Chem., 1990, 43, 1083–1095 CrossRef CAS.
- S. Bhattacharya, N. Seth Miss, V. D. Gupta, H. Nöth and M. Thomann, Z. Naturforsch., B: J. Chem. Sci., 1994, 49, 193 CAS.
- F. Jian, F. Bei, P. Zhao, X. Wang, H. Fun and K. Chinnakali, J. Coord. Chem., 2002, 55, 429–437 CrossRef CAS.
- H. Nöth and D. Schlosser, Chem. Ber., 1988, 121, 1711–1713 CrossRef.
- F. Jian, Z. Wang, Z. Bai, X. You, H.-K. Fun, K. Chinnakali and I. A. Razak, Polyhedron, 1999, 18, 3401–3406 CrossRef CAS.
- I. A. Baburin and V. A. Blatov, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 791–802 CrossRef CAS PubMed.
- X.-Z. Li, B. Walker and A. Michaelides, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6369–6373 CrossRef CAS.
- R. Kumar, J. R. Schmidt and J. L. Skinner, J. Chem. Phys., 2007, 126, 204107 CrossRef CAS PubMed.
- I. D. Brown, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 24–31 CrossRef.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CAS.
- E. N. M. Yusof, M. M. Jotani, E. R. T. Tiekink and T. B. S. A. Ravoof, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2016, 72, 516–521 CAS.
- C. Jelsch, K. Ejsmont and L. Huder, IUCrJ, 2014, 1, 119–128 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Kinetic data, data obtained from CSD database survey of metal dipdtc complexes, fingerprint plots for the Hirshfeld surface of Na(dipdtc)·5H2O, and relevant NMR spectra. CCDC 1488005. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce01475e |
| This journal is © The Royal Society of Chemistry 2016 |