
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A stimuli-responsive Au(I) complex based on an aminomethylphosphine template: synthesis, crystalline phases and luminescence properties†
Igor D.
Strelnik
ab,
Vladislav V.
Gurzhiy
c,
Vladimir V.
Sizov
b,
Elvira I.
Musina
a,
Andrey A.
Karasik
a,
Sergey P.
Tunik
*b and
Elena V.
Grachova
 *b
*b
aA. E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center of Russian Academy of Sciences, Arbuzov str. 8, 420088 Kazan, Russia
bInstitute of Chemistry, St. Petersburg State University, Universitetskii pr. 26, 198504 St. Petersburg, Russia. E-mail: bird231102@mail.ru
cInstitute of Earth Sciences, St. Petersburg State University, Universitetskaya nab. 7/9, 199034 St. Petersburg, Russia
First published on 16th August 2016
Abstract
Herein we report the synthesis of a stimuli-responsive binuclear Au(I) complex based on the 1,5-bis(p-tolyl)-3,7-bis(pyridine-2-yl)-1,5-diaza-3,7-diphosphacyclooctane ligand, which is a novel template for the design of luminescent metal complexes. In the solid state, the complex obtained gives three different crystalline phases, which were characterized by XRD analysis. It was also found that the crystalline phases can be reversibly interconverted by recrystallization or solvent vapour treatment. The emission of these phases varies in the 500–535 nm range. Quite unexpectedly, the emission energy of these phases is mostly determined by the non-covalent interactions of the solvent molecules with the ligand environment, which have nearly no effect on the Au–Au interactions in the chromophoric centre. The complex obtained demonstrates thermo/solvatochromism to display greenish emission in a DCM matrix and blue emission in an acetone matrix at 77 K, in contrast to the blue emission of the phase containing a DCM molecule and greenish-yellow emission of the acetone solvate in a crystal cell at room temperature. The potentially important role of co-crystallized solvent molecules in the ligand-based emission of the complex obtained is supported by DFT calculations.
Introduction
Luminescence of stimuli-responsive materials can be used in a wide range of smart technologies including applications in sensing trace amounts of organic vapours.1 It is well known that luminescence of solid state samples and, in particular, crystalline materials depends strongly on the local environment of chromophores2 and crystallinity3,4 of the corresponding phases. The Au(I) complexes are among the most promising candidates for the design of this type of materials due to a large variety of supramolecular structures, which display sensitivity to temperature,5 organic vapours,3,4,6 ions7 and mechanical stress8 to give a response in luminescence intensity or in the wavelength of emission. These reactions onto external stimuli are provoked by variations in the structure of materials mainly related to noncovalent interactions in the solid phase, such as the formation/breaking of aurophilic interactions, hydrogen bonding, π–π stacking, etc.,9 which affect the chromophoric centre characteristics. Phosphine or polyphosphine ligands are excellent templates for construction of polynuclear Au(I) complexes due to the stability of the P–Au bond. Recently, we reported on the synthesis and characterization of mixed metal gold–copper alkynyl–phosphine complexes, the amorphous phase of which undergoes crystallization upon exposure to the vapours of polar organic solvents (methanol, THF, and acetone) to give a substantial blue shift and increase in the emission intensity.4 It is important to note that the effects cannot be completely explained by the direct interaction of the chromophore with the co-crystallized solvent but has been assigned to the cooperative effect of the phase crystallinity.The cyclic aminomethylphosphines in common and 1,5-diaza-3,7-diphoshpacyclooctanes in particular are convenient templates for the targeted design of Au(I) luminescent complexes due to easy functionalization of the framework at the heteroatoms of the cycle and their ability to vary framework conformation.10 Insertion of functional groups into the cyclic systems allows obtaining the ligands with desired properties (chirality,11 water-solubility,12 chromophoricity13) or enhanced reactivity. The conformation of the aminomethylphosphine cycle in the complexes depends on the metal oxidation state, solvent and temperature.14 For the copper subgroup metals, the 1,5-diaza-3,7-diphosphacyclooctanes preferably exist in “crown” conformation with nearly co-directed lone electron pairs at the phosphorus atoms. This conformation allows the disposition of the Au(I) ions in close proximity to each other, which in principle makes the formation of an aurophilic bond possible that in turn makes 1,5-diaza-3,7-diphosphacyclooctanes prospective ligands for the design of stimuli-responsive Au(I) complexes.
In an attempt to search for stimuli responsive materials of this sort, herein we present a study of an Au(I) chloride complex based on pyridyl substituted 1,5-diaza-3,7-diphosphacyclooctane.13 This is the first example of the application of 1,5-diaza-3,7-diphosphacyclootanes as a template for the design of luminescent complexes. The complex obtained shows a clear dependence of the luminescence properties on the nature of co-crystallized solvent (dichloromethane, acetone) and structure of the crystalline phases. It was also found that both crystallization from solution and solid phase recrystallization upon exposure to the solvent vapours cause identical effects that may be used in preparation of highly selective vapour sensing devices.
Results and discussion
Synthesis and structural characterization of complex 1
The ligand used in this study is a cyclic bisphosphine with a pyridyl function bound directly to the coordination centres of the ligand (P-atoms). 1,5-Bis(p-tolyl)-3,7-bis(pyridine-2-yl)-1,5-diaza-3,7-diphosphacyclooctane (PNNP ligand) was synthesized according to the previously described method.13 The one-stage reaction shown in Scheme 1 gives the binuclear phosphine–chloride Au(I) complex (1) in high yield.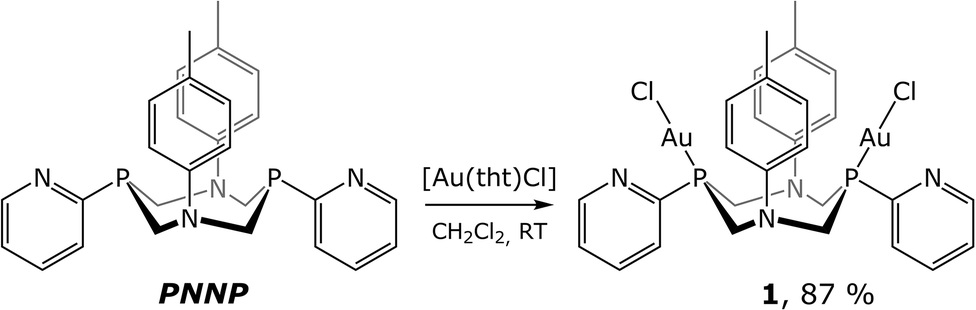 | ||
| Scheme 1 Synthesis of the complex 1. | ||
In solution, the compound obtained was characterized using 31P and 1H NMR spectroscopy and mass-spectrometry. The 31P{1H} NMR spectrum of 1 displays a singlet resonance at 9.9 ppm generated by the equivalent phosphorus atoms of the PNNP ligand that is compatible with the idealized C2 symmetry group of the structural pattern shown in Scheme 1. The phosphorus signal is considerably down-field shifted compared to that of the free ligand (−44.8 ppm) that clearly indicates ligand coordination to the Au(I) centre. The 1H NMR spectrum of 1 (see Fig. S1†) also agrees well with this structural pattern. In the low field region (6.1–8.8 ppm) of the proton spectrum, a clearly resolved set of the signals corresponding to the aromatic protons of the pyridyl and p-tolyl groups of the diphosphine is displayed. The pyridyl function of this ligand remains uncoordinated in solution because the signal of the ortho-pyridyl proton, which is highly sensitive to N-coordination, displays nearly the same chemical shift as that found for the free ligand (8.78 for 1vs. 8.73 for PNNP). The p-tolyl protons in the spectrum of 1 give AB systems in the 6.95–7.0 ppm range. The protons of the P–CH2–N fragments appear in the 4.5–5.0 ppm interval as two dd signals of an ABX system that is a clear indication of the diastereotopic positions of the hydrogen atoms.13 The 2JHH 15.6 Hz and 2JPH 2.0 Hz coupling constants of the axial methylene protons indicate the ligand “crown” conformation similar to that of the free aminomethylphosphine frameworks. The high field area of these spectra displays the singlet resonance of the methyl protons of the p-tolyl group at about 2.3 ppm.
Thus, the number of signals in the phosphorus and proton spectra, their multiplicity and relative intensity are completely compatible with the structural pattern shown in Scheme 1. The ESI+ mass spectrum of 1 contains the signal corresponding to a monocation [M − Cl−]+. The m/z values and isotopic pattern of the signal agree exactly with the calculated spectrum of this ion (Fig. S2†).
Solid state structure of complex 1
The single crystal X-ray diffraction study of the solid phases‡ (for details see the Notes and References section, the ESI† and Table S1) obtained upon crystallization of 1 from various solvents revealed that crystallization from dichloromethane (1a), acetone (1b) and a mixed solvent system (dichloromethane/acetone = 20/1) (1c) gives three different phases, which display very similar molecular structural patterns but vary considerably in packing of the crystal cell (see Fig. 1, S3†). It is also worth noting that interconversion between the a, b, c phases of 1 may occur not only through crystallization from the corresponding solvents but also by treatment of the solid phase samples with the solvent vapours as shown in Scheme 2. | ||
| Fig. 1 Top: Molecular view of 1a–c (colour legend: gold, yellow; nitrogen, blue; phosphorous, red; oxygen, magenta; chlorine, green; carbon, grey; hydrogen, light grey). Bottom: Crystal packing of 1a–c showing “host–guest” aggregates with acetone molecules (the crystalline phase 1a contains no acetone). | ||
 | ||
| Scheme 2 Interconversion of the complex 1 crystalline phases upon crystallization or solvent vapour exposition: i crystallization from acetone; ii crystallization from the DCM/acetone mixture; iii crystallization from DCM; iv acetone vapour treatment. | ||
The 1a–c crystals contain molecules of the solvent (dichloromethane – 1a, acetone – 1b and 1c), which occupy essentially different positions in the crystal cell and fill available voids in the crystal lattice. It has been also found that in the solid state a molecule of 1 demonstrates conformational isomerism related either to the formation of aurophilic bonding in 1a or to its ability to form “host–guest” aggregates with the solvent molecules in 1b and 1c (see Fig. 1.) The PNNP ligand in the complex molecule in 1a–c crystals has a “crown” conformation with the C2 symmetry of the structural pattern. This conformation and axial orientation of p-tolyl substituents at nearly planar nitrogen atoms give a “basket” shape cavity, which may be contracted because of aurophilic bonding (1a) or exist in open form (1b and 1c) that makes hosting of a solvent molecule possible. In the crystal cell of 1a, the solvent (DCM) does not display short intermolecular contacts with the complex and occupies the positions outside the “basket” cavity that allows intramolecular aurophilic bonding (Au–Au distance of 3.313 Å; Table 1) due to the flexible backbone of the PNNP ligand.
| 1a | 1b | 1c | |
|---|---|---|---|
| Au–Au (intramolecular) | 3.313 | 5.388 | 5.956 |
| P–P (intramolecular) | 3.760 | 4.337 | 4.454 |
| N–N (intramolecular) | 4.053 | 3.561 | 3.413 |
| Cl–Cl (intramolecular) | 3.679 | 6.664 | 7.460 |
| Distance between p-tolyl centroids | 8.289 | 6.868 | 6.932 |
| Cl–Hacetone shortest distance | — | 2.826 | 3.280 |
| p-Tolylcentroid–Hacetone shortest distance | — | 3.169 | 3.365 |
| Oacetone–HCH2 of PNNP | — | 2.476 | — |
| Torsion angle of pyridine rings | 80.21 | 36.26 | 0 |
| Au–P–P angles | 83.59 | 103.62 | 109.83 |
In the 1b and 1c crystalline phases, the intramolecular Au⋯Au distances are substantially longer (5.388 and 5.956 Å, respectively), which indicate the absence of aurophilic interactions in the open “basket” conformation. The open mode of the “basket” cavity in 1b and 1c is “ready-to-host” an acetone molecule and to form a “host–guest” system with rather strong hydrogen (H–O, H–Cl) bonding between acetone and a molecule of 1. However the interaction of the hosted solvent with the complex molecule is essentially different in 1b and 1c phases. In 1b, one can observe a hydrogen bond network formed by the relatively short contacts of the methylene protons of the PNNP ligand and an oxygen atom of acetone (2.426 Å) together with bonding of a coordinated chloride ion and methyl protons of acetone (2.876 Å) (see Fig. 1, top and Table 1). It was found that the methyl protons of acetone are located close to the p-tolyl fragment and the shortest distance between the centroid of a phenyl ring and these protons is 3.169 Å. These interactions give infinite chains of {1-acetone} aggregates (see Fig. 1, bottom). In the crystal phase of (1c, one of the solvent molecules is disposed outside the “basket” whereas the other sits in the cavity but the distances between acetone and coordinated chlorides and between acetone and a phenyl ring are substantially longer (3.280 and 3.365 Å, respectively) than those in 1b and no contacts between acetone oxygen and the PNNP protons were found. These crystallographic peculiarities proved to be a key reason for substantial variations in the photophysical properties of the crystalline phases of 1 (see Fig. 2). It is worth noting that the 1a and 1c phases are somewhat similar in the absence of a strong and highly organized hydrogen bond network found in 1b that in turn results in similarity of their photophysical characteristics and made them substantially different from those found for 1b. The formation of different crystalline phases of 1 provides a unique opportunity to study the effect of molecular conformation, packing arrangement and “host–guest” interactions on the solid state luminescence properties.
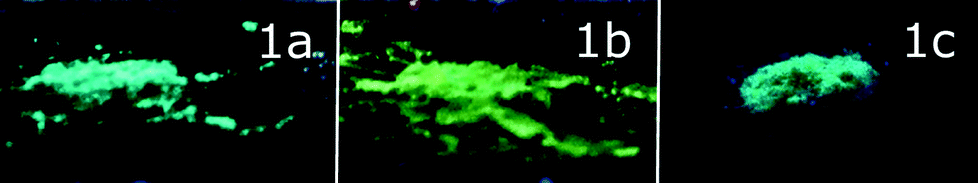 | ||
| Fig. 2 1a–c phases under UV lamp irradiation (λexc = 365 nm). | ||
Photophysical properties of complex 1 in the solid phase
It was found that complex 1 displays very weak emission in solution at room temperature but shows appreciable luminescence in solution at 77 K and in the solid state, in the latter case, the emission characteristics being determined by the variations in the structure of the crystalline phase. The luminescence spectra of free PNNP in the solid state and those of solid state samples of 1 are shown in Fig. 3, and the emission characteristics are given in Table 2.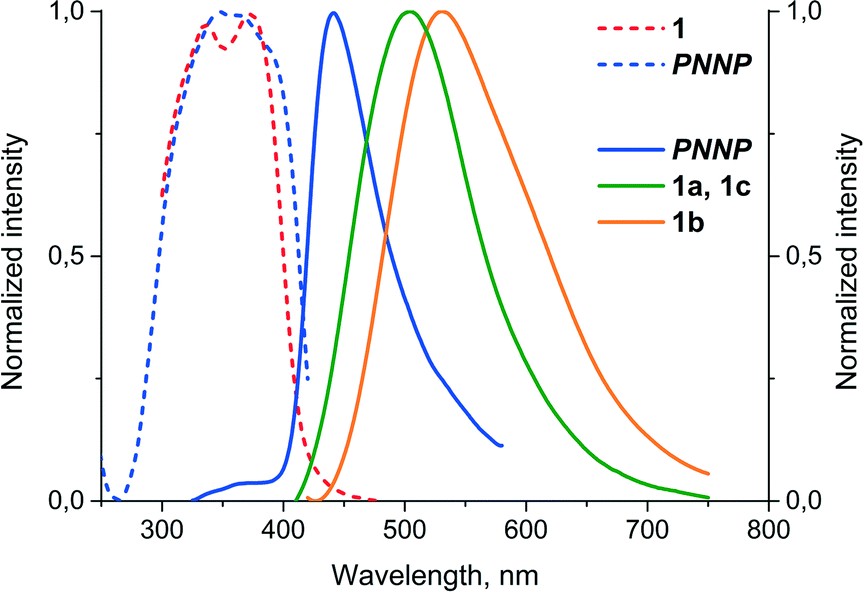 | ||
| Fig. 3 Normalized excitation (dash lines) and emission (solid lines) spectra of free PNNP and the crystalline phases of 1 (λexc = 340 nm, room temperature). | ||
| λ exc,a nm | λ em,b nm | τ, ns | Φ, % | |
|---|---|---|---|---|
| a Excitation spectra were monitored at the maximum of the corresponding emission bands (440 nm for free PNNP; 500 nm for 1, 1a, 1c; 535 nm for 1b). b λ exc = 340 nm (room temperature) and λexc = 365 nm (77 K). c DCM matrix at 77 K. d Acetone matrix at 77 K. | ||||
| PNNP | 340 | 440 | 0.3(0.49); 81(0.42) | <1 |
| 1a | 335, 375 | 500 | 47(0.51); 441(0.49) | 1.2 |
| 1b | 335, 375 | 535 | 198(0.42); 925(0.58) | 1.5 |
| 1c | 335, 375 | 500 | 33(0.34); 191(0.66) | 1.2 |
| 1 | 520 | |||
| 1 | 490 | |||
The room temperature solid state emission of free PNNP is a typical fluorescence with a Stokes shift of about 80 nm and lifetime in the nanosecond domain. In contrast to the ligand luminescence, the emission of 1a–c displays Stokes shifts of ca. 180 nm and excited state lifetimes in the microsecond domain, which are indicative of the triplet origin of the emission observed, i.e. phosphorescence.
The solid-state samples of 1a–c display an identical clear-cut excitation band with two maxima at 335 and 375 nm, which point to an essentially similar mechanism of excitation, which starts from the electron transfer between the orbitals of the same nature. The luminescence bands of 1a and 1c also coincide to give green emission at 500 nm; the emission maximum of 1b is red shifted by 35 nm, changing the colour of emission into a greenish-yellow one. The identical emission bands of 1a and 1c, in spite of aurophilic interactions in 1a and its absence in 1c, clearly point to the mostly intraligand (IL) nature of the emissive excited state. The effect of a solvent on luminescence in the cases of 1a and c is negligible because there is nearly no interaction between solvent molecules and coordinated PNNP, which is evidently the chromophoric centre of the {complex–solvent} aggregate (see the DFT calculations section vide infra). Thus, even a relatively weak interaction of an acetone molecule (short Oacetone–H (CH2 of PNNP) contacts) with coordinated PNNP in 1b results in substantial variation of the emission characteristics. This observation indicates a crucial influence of the local solvent interaction with the chromophoric centre on the photophysical parameters of complex 1 in the crystalline phases. The cooperative electronic effect of the host–guest interaction is evidently increased by additional hydrogen bonding between acetone protons and coordinated chloride in the conjugated infinite {complex–solvent} chain (see Fig. 1). It has to be also noted at this point that the PXRD patterns of 1a–c (Fig. S3 and Tables S2–S4†) are essentially similar to those calculated from the single crystal data, which makes discussion of the photophysical characteristics in relation to structural parameters revealed by XRD studies possible. The presence of some signals of another phase may be assigned to the loss of the solvent upon powder sample preparation.
Photophysical properties of complex 1 in frozen solvents
Upon freezing in liquid nitrogen, the solution of 1 starts to intensely exhibit luminescence and the emission colour being different in various solvents (see Fig. 4 and Table 2). The frozen dichloromethane solution of 1 displays a yellow-greenish emission, which is 20 nm red-shifted relative to the emission of 1a and c while in frozen acetone the band maximum is 45 nm blue-shifted relative to the room temperature emission of 1b. The complex in neat acetone solution evidently tends to form an aggregate with the solvent molecule, in particular at low temperature. Thus, the observed blue shift in the emission of the frozen solution compared to the room temperature band of 1b is a typical phenomenon due to stabilization of the ground state at 77 K. The red shift in the emission band of 1 in frozen dichloromethane compared to that of 1a at room temperature may be assigned to more effective relaxation of the triplet excited state for an unconstrained molecule in the solvent matrix compared to an analogous process in the solid phase where strong intermolecular interactions in the crystal cell do not allow deep intramolecular rearrangement. The emission switch-on/switch-off upon temperature variations is reversible and warming the frozen solution results in luminescence quenching, while refreezing recovers the emission.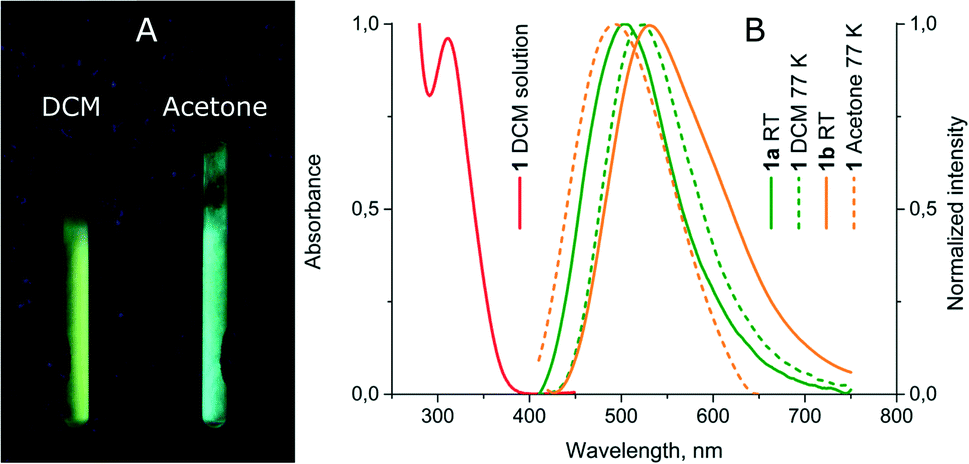 | ||
| Fig. 4 (A) Photos of luminescence of 1 in frozen DCM and acetone solutions under UV lamp irradiation; (B) normalized absorption and emission spectra of 1. | ||
It is worth noting that the concentration of 1 in solution does not affect the emission characteristics both at room and low temperatures, which is indicative of the absence of complex aggregation and/or formation of an exciplex in solution.
DFT calculations
The absorption spectrum of 1 in solution resembles that of a free PNNP ligand, for which TDDFT calculations show two bands with maxima around 355 nm and 250 nm. For PNNP, three transitions (364 nm, 359 nm, and 352 nm) corresponding to electron transfer from p-tolyl fragments to pyridyl rings dominate in the lower-energy band. For 1, the positions of the low-energy and high-energy bands remain unchanged, suggesting that the photophysical behaviour of the complex is largely determined by the properties of the ligand. The energy of the lowest triplet state of 1 obtained from TDDFT calculations was 391 nm compared to 382 nm for free PNNP. The nature of this state is predominantly defined by the two highest occupied orbitals and two lowest virtual orbitals of 1. For the isolated complex 1, which can be considered as a simplified model of 1a, the two highest occupied orbitals (HOMO and HOMO−1) are mostly localized at the p-tolyl fragments of the ligand. However, noticeable contributions from gold and phosphorous atoms can also be found. Both LUMO and LUMO+1 have large contributions from the pyridyl fragments, though LUMO also involves orbitals of both gold atoms, which form an intramolecular aurophilic bond (see Fig. S5†).Addition of one or two acetone molecules located at the same position, as found in 1b and 1c crystalline phases, appears to have no immediate effect on the computed spectral properties of 1. However, the insertion of an acetone molecule between p-tolyl fragments leads to the loss of aurophilic contact. As a result, the contribution of gold atoms into the highest occupied orbitals is noticeably lower than for an isolated complex, while the lowest vacant orbitals no longer indicate the presence of Au–Au bonding (see Fig. S6 and S7†).
For the acetone molecule located between the p-tolyl fragments in 1b and 1c, the smallest distance between methyl hydrogens and a chloride ion is at least 3 Å. Therefore, this solvent molecule is unlikely to be capable of having strong interactions with the complex. This conclusion is supported by the amount of charge transfer from the acetone molecule to the complex, which does not exceed 0.05e (as estimated from Mulliken atomic charges). In contrast to that, the acetone molecule located on the other side of the PNNP ring between the pyridyl fragments has a more favourable orientation, forming O–H contacts with interatomic distances of 2.2–2.5 Å. As a result, the charge transfer to the complex for this molecule is close to 0.1e, which is comparable to the effect of some coordinating solvents. For the aggregate with two acetone molecules, which was used to model the 1b phase, the total charge transfer from solvent molecules is 0.14e., i.e., three times larger than for the system modelling the 1c phase. Stronger interactions with acetone molecules together with the effect induced by cooperative interactions in the infinite chain {complex–solvent}n may be responsible for the observed difference in emission of the 1b and 1c phases.
Thus, the results of model DFT calculations, which take into account local interactions of an isolated complex molecule with the solvents found in the solid state, support the ligand-based mechanism of emission and point to the potentially important role of solvent molecules co-crystallized with the complex in the 1b and 1c phases. However, the study of a molecule of complex 1 in solution (with or without additional solvent molecules) is unable to provide an accurate description of the solid-state photophysical behaviour for two primary reasons. First of all, constrained geometry optimizations carried out for a single molecule of 1 cannot provide a correct description of excited state relaxation in the solid state. Secondly, the modelling of media effects under the framework of the PCM approach is unlikely to give an adequate account of the influence of the crystalline environment on the electronic structure of 1. Therefore, dedicated solid-state quantum chemical calculations are necessary to obtain a fully quantitative description of the emissive properties of the complex.
Conclusions
In conclusion, we demonstrated that a binuclear Au(I) complex based on a flexible aminomethylphosphine template could exhibit stimuli-responsive luminescence properties. Different crystalline phases of the Au(I) complex were obtained using slight modification of the crystallization conditions. Careful XRD analysis showed that molecular conformations, packing arrangements and non-covalent interactions between the binuclear Au(I) complex and a solvent (acetone) molecule are interdependent factors and their combination determines the solid-state luminescence properties. The observed reversible interconversion between the different crystalline phases of the Au(I) complex occurs as a vapour-stimulated {single crystal-to-single crystal} transformation between crystalline phases and we believe that these results may provide ideas for the design of novel stimuli-responsive luminescent materials.Acknowledgements
The authors (I. D. S., E. I. M., A. A. K.) greatly appreciate the financial support of the Russian Science Foundation, grant 15-13-30031. This work was carried out using equipment of core facilities of St. Petersburg State University Research Park: Centre for Magnetic Resonance, Centre for Optical and Laser Materials Research, Centre for Chemical Analysis and Materials Research, the X-ray Diffraction Centre, and the Thermogravimetric and Calorimetric Research Centre.Notes and references
- G.-J. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2011, 40, 2541 RSC; J. Carlos Lima and L. Rodriguez, Chem. Soc. Rev., 2011, 40, 5442 RSC; X. Zhang, B. Li, Z.-H. Chen and Z.-N. Chen, J. Mater. Chem., 2012, 22, 11427 RSC; O. S. Wenger, Chem. Rev., 2013, 113, 3686 CrossRef CAS PubMed; Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
- R. C. Ropp, Luminescence and the Solid State, Elsevier Science, 2004 RSC; H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC; T. Lasanta, J. M. Lopez-de-Luzuriaga, M. Monge, M. E. Olmos and D. Pascual, Chem. – Eur. J., 2013, 19, 4754 CrossRef CAS PubMed.
- I. O. Koshevoy, Y.-C. Chang, A. J. Karttunen, M. Haukka, T. Pakkanen and P.-T. Chou, J. Am. Chem. Soc., 2012, 134, 6564 CrossRef CAS PubMed.
- J. R. Shakirova, E. V. Grachova, A. S. Melnikov, V. V. Gurzhiy, S. P. Tunik, M. Haukka, T. A. Pakkanen and I. O. Koshevoy, Organometallics, 2013, 32, 4061 CrossRef CAS.
- A. J. Lees, Chem. Rev., 1987, 87, 711 CrossRef CAS; E. J. Fernandez, M. C. Gimeno, A. Laguna, J. M. Lopez-de-Luzuriaga, M. Monge, P. Pyykko and D. Sundholm, J. Am. Chem. Soc., 2000, 122, 7287 CrossRef; R. L. White-Morris, M. M. Olmstead, F. Jiang, D. S. Tinti and A. L. Balch, J. Am. Chem. Soc., 2002, 124, 2327 CrossRef PubMed; V. W.-W. Yam, C.-L. Chan, C.-K. Li and K. M.-C. Wong, Coord. Chem. Rev., 2001, 216–217, 173 CrossRef; Y.-A. Lee, J. E. McGarrah, R. J. Lachicotte and R. Eisenberg, J. Am. Chem. Soc., 2002, 124, 10662 CrossRef PubMed; M. A. Omary, M. A. Rawashdeh-Omary, M. W. A. Gonser, O. Elbjeirami, T. Grimes, T. R. Cundari, H. V. K. Diyabalanage, C. S. P. Gamage and H. V. R. Dias, Inorg. Chem., 2005, 44, 8200 CrossRef PubMed; E. M. Gussenhoven, J. C. Fettinger, D. M. Pham, M. M. Malwitz and A. L. Balch, J. Am. Chem. Soc., 2005, 127, 10838 CrossRef PubMed; E. J. Fernandez, A. Laguna, J. M. Lopez-de-Luzuriaga, M. Monge, M. Montiel, M. E. Olmos and M. Rodriguez-Castillo, Organometallics, 2006, 25, 3639 CrossRef; J. M. Lopez-de-Luzuriaga, M. Monge, M. E. Olmos, D. Pascual and M. Rodriguez-Castillo, Inorg. Chem., 2011, 50, 6910 CrossRef PubMed; Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508 RSC; I. O. Koshevoy, C.-L. Lin, A. J. Karttunen, M. Haukka, C.-W. Shih, P.-T. Chou, S. P. Tunik and T. A. Pakkanen, Chem. Commun., 2011, 47, 5533 RSC; J. Liang, Z. Chen, J. Yin, G.-A. Yu and S. H. Liu, Chem. Commun., 2013, 49, 3567 RSC.
- M. A. Mansour, W. B. Connick, R. J. Lachicotte, H. J. Gysling and R. Eisenberg, J. Am. Chem. Soc., 1998, 120, 1329 CrossRef CAS; J. Lefebvre, R. J. Batchelor and D. B. Leznoff, J. Am. Chem. Soc., 2004, 126, 16117 CrossRef PubMed; D. B. Leznoff and J. Lefebvre, Gold Bull., 2005, 38, 47 CrossRef; E. J. Fernandez, J. M. Lopez-de-Luzuriaga, M. Monge, M. E. Olmos, R. C. Puelles, A. Laguna, A. A. Mohamed and J. P. Fackler Jr., Inorg. Chem., 2008, 47, 8069 CrossRef PubMed; C. E. Strasser and V. J. Catalano, J. Am. Chem. Soc., 2010, 132, 10009 CrossRef PubMed; C. Jobbagy, T. Tunyogi, G. Palinkas and A. Deak, Inorg. Chem., 2011, 50, 7301 CrossRef PubMed.
- X. He and V. W.-W. Yam, Coord. Chem. Rev., 2011, 255, 2111 CrossRef CAS; Y.-P. Zhou, E.-B. Liu, J. Wang and H.-Y. Chao, Inorg. Chem., 2013, 52, 8629 CrossRef PubMed.
- Z. Assefa, M. A. Omary, B. G. McBurnett, A. A. Mohamed, H. H. Patterson, R. J. Staples and J. P. Fackler, Inorg. Chem., 2002, 41, 6274 CrossRef CAS PubMed; E. J. Fernandez, J. M. Lopez-de-Luzuriaga, M. Monge, M. E. Olmos, J. Perez, A. Laguna, A. A. Mohamed and J. P. Fackler Jr., J. Am. Chem. Soc., 2003, 125, 2022 CrossRef PubMed; V. J. Catalano and S. J. Horner, Inorg. Chem., 2003, 42, 8430 CrossRef PubMed; E. J. Fernandez, A. Laguna and J. M. Lopez-de-Luzuriaga, Coord. Chem. Rev., 2005, 249, 1423 CrossRef; J. Schneider, Y.-A. Lee, J. Perez, W. W. Brennessel, C. Flaschenriem and R. Eisenberg, Inorg. Chem., 2008, 47, 957 CrossRef PubMed; A. Deak, T. Tunyogi and G. Palinkas, J. Am. Chem. Soc., 2009, 131, 2815 CrossRef PubMed; A. M. Kuchison, M. O. Wolf and B. O. Patrick, Chem. Commun., 2009, 7387 RSC; Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878 RSC; X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376 RSC; K. Kawaguchi, T. Seki, T. Karatsu, A. Kitamura, H. Ito and S. Yagai, Chem. Commun., 2013, 49, 11391 RSC; J. Liang, Z. Chen, L. Xu, J. Wang, J. Yin, G.-A. Yu, Z.-N. Chen and S. H. Liu, J. Mater. Chem. C, 2014, 2, 2243 RSC; A. A. Penney, V. V. Sizov, E. V. Grachova, D. V. Krupenya, V. V. Gurzhiy, G. L. Starova and S. P. Tunik, Inorg. Chem., 2016, 55, 4720 CrossRef PubMed.
- C. Jobbagy and A. Deak, Eur. J. Inorg. Chem., 2014, 4434 CrossRef CAS; E. C.-C. Cheng, W.-Y. Lo, T. K.-M. Lee, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2014, 53, 3854 CrossRef PubMed; K. Fujisawa, Y. Okuda, Y. Izumi, A. Nagamatsu, Y. Rokusha, Y. Sadaike and O. Tsutsumi, J. Mater. Chem. C, 2014, 2, 3549 RSC; N. Nasser and R. J. Puddephatt, Inorg. Chim. Acta, 2014, 409, 238 CrossRef; E. R. T. Tiekink, Coord. Chem. Rev., 2014, 275, 130 CrossRef.
- A. A. Karasik, A. S. Balueva, E. I. Musina and O. G. Sinyashin, Mendeleev Commun., 2013, 23, 237 CrossRef CAS; E. I. Musina, A. A. Karasik, O. G. Sinyashin and G. N. Nikonov, Adv. Heterocycl. Chem., 2015, 117, 83 CrossRef.
- A. A. Karasik, R. N. Naumov, O. G. Sinyashin, G. P. Belov, H. V. Novikova, P. Lonnecke and E. Hey-Hawkins, Dalton Trans., 2003, 2209 RSC.
- A. A. Karasik, R. N. Naumov, R. Sommer, O. G. Sinyashin and E. Hey-Hawkins, Polyhedron, 2002, 21, 2251 CrossRef CAS.
- E. I. Musina, V. V. Khrizanforova, I. D. Strelnik, M. I. Valitov, Y. S. Spiridonova, D. B. Krivolapov, I. A. Litvinov, M. K. Kadirov, P. Lonnecke, E. Hey-Hawkins, Y. H. Budnikova, A. A. Karasik and O. G. Sinyashin, Chem. – Eur. J., 2014, 20, 3169 CrossRef CAS PubMed.
- S. K. Latypov, A. G. Strelnik, S. N. Ignatieva, E. Hey-Hawkins, A. S. Balueva, A. A. Karasik and O. G. Sinyashin, J. Phys. Chem. A, 2012, 116, 3182 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed; O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef; CrysAlisPro, Agilent Technologies, Version 1.171.36.20 (release 27-06-2012) Search PubMed; A. L. Spek, PLATON, Utrecht University, Utrecht, The Netherlands, 2005 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedure for 1 preparation; details of the transformation of the crystalline phases; details of the photophysical experiment; computational details; crystal data and structure refinement for 1a–c; bond lengths, angles and torsion angles for 1a–c. CCDC 1030853, 1027244 and 1027245. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce01272h |
| ‡ The crystal structures of 1a–c were determined by means of single crystal X-ray diffraction analysis using Rigaku Oxford Diffraction Supernova Atlas (1a) and Excalibur Eos (1b, 1c) diffractometers at a temperature of 100 K. 1a: (C28H30Au2Cl2N4P2)·(CH2Cl2)3, P2/c, a = 15.3277(5), b = 8.8720(2), c = 14.8008(6) Å, β = 112.309(4)°, V = 1862.06(12) Å3, Z = 2, R1 = 0.044, CCDC 1030853. 1b: (C28H30Au2Cl2N4P2)·(C3H6O), P2/c, a = 8.8698(2), b = 10.6900(3), c = 17.5273(5) Å, β = 96.988(3)°, V = 1649.56(8) Å3, Z = 2, R1 = 0.026, CCDC 1027244. 1c: (C28H30Au2Cl2N4P2)·(C3H6O)2, C2/m, a = 24.029(4), b = 16.9105(16), c = 12.0433(18) Å, β = 124.55(2)°, V = 4030.5(13) Å3, Z = 4, R1 = 0.035, CCDC 1027245. The structures were solved by direct methods and refined using the SHELXL-97 program incorporated in the OLEX2 program package. Absorption correction was applied in the CrysAlisPro program complex. The unit cell of 1a·3CH2Cl2 contains disordered dichloromethane molecules that have been treated as a diffuse contribution to the overall scattering without specific atom positions by SQUEEZE/PLATON.15 |
| This journal is © The Royal Society of Chemistry 2016 |