
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA family of heterotetrameric clusters of chloride species and halomethanes held by two halogen and two hydrogen bonds†
Daniil M.
Ivanov
a,
Alexander S.
Novikov
a,
Galina L.
Starova
a,
Matti
Haukka
b and
Vadim Yu.
Kukushkin
*a
aInstitute of Chemistry, Saint Petersburg State University, 7/9 Universitetskaya Nab., 199034 Saint Petersburg, Russian Federation. E-mail: v.kukushkin@spbu.ru
bDepartment of Chemistry, University of Jyväskylä, P.O. Box 35, FI-40014 Jyväskylä, Finland
First published on 13th June 2016
Abstract
Two previously reported 1,3,5,7,9-pentaazanona-1,3,6,8-tetraenate (PANT) chloride platinum(II) complexes [PtCl{HN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(R)NCN[C(Ph)C(Ph)]CNC(R)NH}] (R = tBu 1, Ph 2) form solvates with halomethanes 1·1¼CH2Cl2, 1·1⅖CH2Br2, and 2·CHCl3. All these species feature novel complex-solvent heterotetrameric clusters, where the structural units are linked simultaneously by two C–X⋯Cl–Pt (X = Cl, Br) halogen and two C–H⋯Cl–Pt hydrogen bonds. The geometric parameters of these weak interactions were determined using single-crystal XRD, and the natures of the XBs and HBs in the clusters were studied for the isolated model systems (1)2·(CH2Cl2)2, (1)2·(CH2Br2)2, and (2)2·(CHCl3)2 using DFT calculations and Bader's AIM analysis. The evaluated energies of the weak interactions are in the range 0.9–3.0 kcal mol−1. The XBs and HBs in the reported clusters are cooperative. In the cases of (1)2·(CH2Cl2)2 and (1)2·(CH2Br2)2, the contribution of the HBs to the stabilization of the system is dominant, whereas for (2)2·(CHCl3)2 contributions of both types of the non-covalent interactions are almost the same. Crystal packing and other forces such as, e.g. dipole–dipole interactions, also affect the formation of the clusters.
C(R)NCN[C(Ph)C(Ph)]CNC(R)NH}] (R = tBu 1, Ph 2) form solvates with halomethanes 1·1¼CH2Cl2, 1·1⅖CH2Br2, and 2·CHCl3. All these species feature novel complex-solvent heterotetrameric clusters, where the structural units are linked simultaneously by two C–X⋯Cl–Pt (X = Cl, Br) halogen and two C–H⋯Cl–Pt hydrogen bonds. The geometric parameters of these weak interactions were determined using single-crystal XRD, and the natures of the XBs and HBs in the clusters were studied for the isolated model systems (1)2·(CH2Cl2)2, (1)2·(CH2Br2)2, and (2)2·(CHCl3)2 using DFT calculations and Bader's AIM analysis. The evaluated energies of the weak interactions are in the range 0.9–3.0 kcal mol−1. The XBs and HBs in the reported clusters are cooperative. In the cases of (1)2·(CH2Cl2)2 and (1)2·(CH2Br2)2, the contribution of the HBs to the stabilization of the system is dominant, whereas for (2)2·(CHCl3)2 contributions of both types of the non-covalent interactions are almost the same. Crystal packing and other forces such as, e.g. dipole–dipole interactions, also affect the formation of the clusters.
Introduction
Halogen bonds (XBs) are extremely important in crystal engineering and various type of XB have been intensively studied in recent years.1–4 The XB is applied in pharmaceutical chemistry insofar as biologically active species featuring halide substituents can be co-crystallized with many organic and inorganic fragments forming XBs.5 The recent applications of XBs include the stabilization of energetic materials (e.g. explosives) in the solid state6 and the molecular design of materials with tunable photophysical properties.7A XB is commonly treated as an electrostatic attraction between electron-rich centers and areas of electropositive potential, the so-called σ-holes, existing on the surfaces of covalently bonded halogen atoms.1–4,8–11 The electron-rich centers can be heteroatoms such as halogens or π-systems. Two types of short halogen–halogen contact are usually discussed in the literature (Fig. 1).1,3,12 Type I is believed to depend on the effects of close packing, whereas type II is due to a classic XB because a halogen atom with a 90° angle provides its lone pair for interaction and the other one provides its σ-hole. The type II interactions are more useful in crystal engineering due to the relatively strict geometrical requirements, whereas both angles around the halogen atoms in the type I interaction (which is actually not a XB, see ref. 13) can be in a wide range from 180° to 90° and less, and it hardly allows the construction of supramolecular structures.
 | ||
| Fig. 1 Two types of halogen–halogen short contact. | ||
Although the vast majority of XB studies involve metal-free organic species,1,2,14 utilization of some organometallic compounds for these purposes are also reported.15,16 The combination of unique redox, magnetic, and optical properties of metal complexes with adjustable organic moieties is very useful for the design of new materials.
XBs involving metal complexes form networks,16 where anions are XB donors and their counterions are XB acceptors. These networks were found in halogen-containing pyridinium halometallates or cyanometallates (XC5H5N)2[MX′4] (where M is a divalent metal ion, e.g., CoII, PtII, or PdII, X is a halogen except F, and X′ is a halide or cyanide ligand).16–18 Similar networks19,20 have also been found in the associations of substituted ammonium bromometallates (nPr4N)[CuBr2], (nBu4N)2[MBr4] (M = Zn, Cd, Co), and (nBu4N)2[Pt2Br6] with bromoform, tetrabromomethane, and 1,1-dibromo-1,1,2,2-tetrafluoroethane serving as XB donors. 3D-Networks formed by XBs were also observed in the associations of (nBu4N)2[PtBr4Cl2] and (nBu4N)2[Pt2Br10] with dibromine.21 Another large group of hybrid systems22,23 is represented by complexes trans-[MX2(4-X′py)2] forming intermolecular XBs between the halide ligands X− and halogen substituents X′ in the pyridine ligands. In addition, palladium NCN24 and PCP25 pincer complexes featuring halide ligands were co-crystallized with diiodine, 1,4-diiodotetrafluorobenzene, and 1,4-diiodooctafluorobutane displaying networks and chains formed by XBs.
In most reports focused on crystal engineering involving XBs, strong iodine centered XB donors were explored, which contain the large σ-holes due to electron withdrawing substituents.1,3 In contrast to a substantial number of these works, a few reports20,26–28 have shown that even simple halomethanes can be effectively used in the design of structures held by XBs. In particular, we reported26 that chloroform can act as both a XB and hydrogen bond (HB) donor forming the heterotetrameric clusters (Cl−)2·(CHCl3)2 (Fig. 2) in the solid state. The term “heterotetrameric cluster” means a symmetrical supramolecular associate formed by two pairs of different species (dimer of a dimer). This term was previously suggested for designation of neutral clusters with halomethanes containing four molecules linked by two HBs and two XBs.27,28
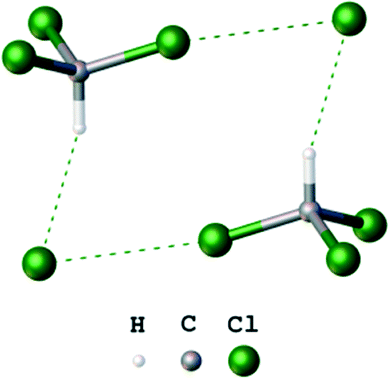 | ||
| Fig. 2 Ball-and-stick model of the heterotetrameric cluster (Cl−)2·(CHCl3)2.26 | ||
In this study, we demonstrate that relevant heterotetrameric structures could be obtained using crystallization of the annulated triazapentadiene systems, viz. 1,3,5,7,9-pentaazanona-1,3,6,8-tetraenate (PANT) platinum(II) chloride complexes (Fig. 3, A), with the halomethanes CH2Cl2, CH2Br2, and CHCl3 (Fig. 3, B). The heterotetramers display the previously unreported H2ClC–Cl⋯Cl–Pt, H2BrC–Br⋯Cl–Pt, and HCl2C–Cl⋯Cl–Pt halogen and the C–H⋯Cl–Pt hydrogen bonds. These experimental data along with the results of a detailed inspection of the large amount of available literature/CCDC data open up a new family of heterotetrameric clusters held simultaneously by two C–X⋯Cl XBs and two C–H⋯Cl HBs with halomethanes. All our results are discussed in the sections that follow.
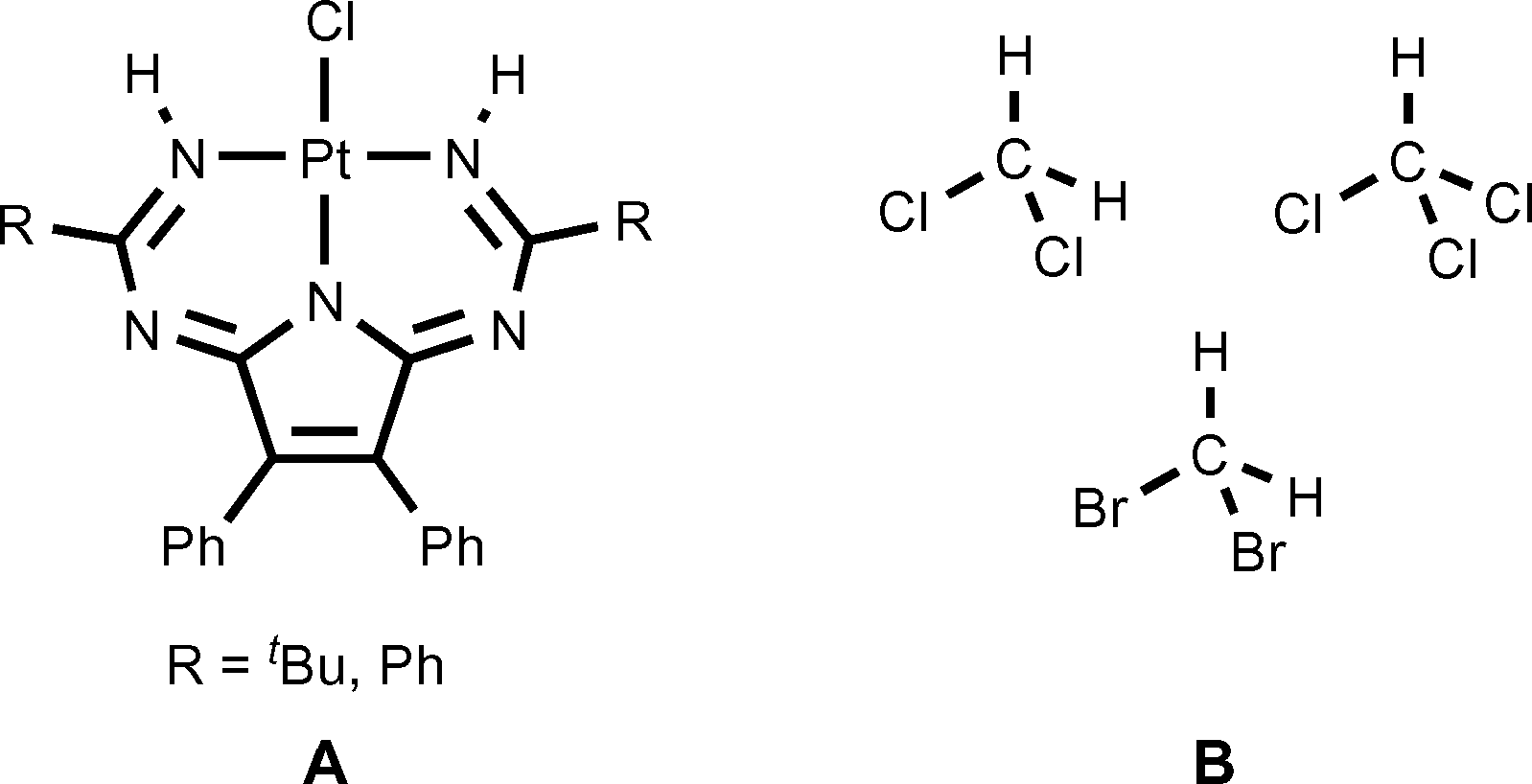 | ||
| Fig. 3 Structures of the [PtCl(PANT)] complexes (A) and the halomethanes (B) employed in this work. | ||
Results and discussion
Synthesis and crystallization
In accordance with our recently reported procedure,29 the PANT platinum(II) chloride complexes were synthesized by coupling of the nitrile ligands in trans-[PtCl2(RCN)2] with 2,3-diphenylmaleimidine (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5, CH2Cl2, RT) followed by purification of the formed [PtCl(PANT)] complexes using column chromatography.
:2.5, CH2Cl2, RT) followed by purification of the formed [PtCl(PANT)] complexes using column chromatography.
Two PANT species (R = tBu 1, Ph 2) were found to co-crystallize with halomethanes (CHCl3, CH2Cl2, and CH2Br2) forming heterotetrameric complex–halomethane clusters (Table 1). Isostructural Cl/Br exchange was detected for clusters 1 with CH2Cl2 and CH2Br2 and corresponding crystalline phases demonstrate close cell parameters and analogous packing features (for detailed information see ESI†).
| R | Nos | Solvent system | Solvates |
|---|---|---|---|
| t Bu | 1 | CH2Cl2 | 1·1¼CH2Cl2 |
| CH2Br2 | 1·1⅖CH2Br2 | ||
| Ph | 2 | CHCl3 | 2·CHCl3 |
The C–X⋯Cl–Pt halogen bonds
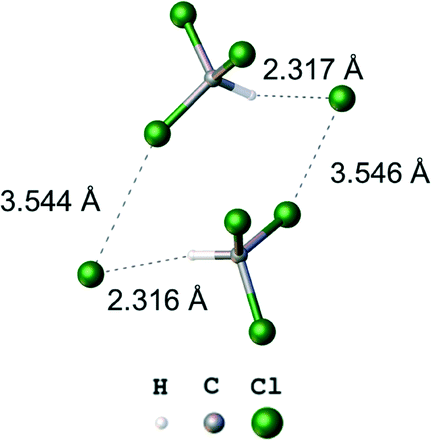
In general, a C–X⋯Cl–Pt (X = Cl, Br) XB occurs when the distance between the appropriate X atoms is less than the sum of their van der Waals (vdW) radii. However, one should take into consideration that several data sets for vdW radii based on different approaches have been suggested for the same atoms/ions and the Cl and Br vdW radii vary from 1.75 to 1.80 Å and from 1.85 to 1.90 Å, respectively, depending on the system used. Currently the most widely used vdW radii system was proposed by Bondi30 with the smallest values so far suggested. It is commonly accepted that X atoms are involved in non-covalent interactions when the X⋯X distance is shorter than the sum of the Bondi's vdW radii. However, it has also been argued that Bondi's vdW radii are too small by a systematic deviation and too close to the corresponding covalent radii.31 A possible alternative for the Bondi's vdW radii is the data set proposed by Rowland,32 which was obtained by inspection of a large amount of statistical data, and it is believed that Rowland's values fit better for the investigation of HBs33 and XBs34 in the solid phase and in calculated model systems35 and we used these radii for the analysis of the obtained experimental data.The C–X⋯Cl–Pt (X = Cl, Br) short contacts (3.447(2) Å and 3.5012(9) for Cl⋯Cl, and 3.330(2) for Cl⋯Br) that are less than the sum of Rowland's32 vdW radii (2Rw(Cl) = 3.52 Å, Rw(Cl) + Rw(Br) = 3.63 Å) were found in each cluster (Table 2). The corresponding angles around the chloride ligands are close to 90° and around solvent halogen atoms are close to 180°. These data indicate that the short contacts are due to the XB and complex molecules behave as XB acceptors of type II (Fig. 1).
| Cluster | Pt–Cl⋯X–C | d(X⋯Cl), Å | ∠(C–X⋯Cl),° | ∠(X⋯Cl–Pt),° | E b | E b |
|---|---|---|---|---|---|---|
| a E b = −V(r)/2.38 b E b = 0.429G(r).39 c Comparison between the sum of Rowland's32 vdW radii and conventional halogen bond angle. | ||||||
| (1)2·(CH2Cl2)2 | C1S–Cl1S⋯Cl1A–Pt1A | 3.447(2) | 171.8(3) | 108.41(7) | ||
| 3.50 | 168.4 | 109.9 | 0.9 | 1.4 | ||
| 3.36 | 170.8 | 108.9 | 1.6 | 1.6 | ||
| (1)2·(CH2Br2)2 | C1S–Cl1S⋯Cl1A–Pt1A | 3.330(2) | 172.0(4) | 107.98(8) | ||
| 3.17 | 171.0 | 106.2 | 2.5 | 2.7 | ||
| (2)2·(CHCl3)2 | C1S–Cl3S⋯Cl1–Pt1 | 3.5012(9) | 172.16(7) | 87.902(17) | ||
| 3.61 | 169.7 | 83.8 | 0.9 | 1.1 | ||
| *Comparisonc | 3.52 (Cl⋯Cl) | 180 | 90 | |||
| 3.63 (Cl⋯Br) | ||||||
Solvates 1·1¼CH2Cl2, 1·1⅖CH2Br2 (Fig. 4), and 2·CHCl3 (Fig. 5) form independent heterotetrameric clusters comprised of two solvent molecules and two molecules of the complexes linked by two XBs and two hydrogen bonds (HBs). In 1, the geometric parameters of these heterotetramers are very similar. Structures 1·CH2Br2 and 2·CHCl3 demonstrate the first examples of the H2BrC–Br⋯Cl–Pt and the HCl2C–Cl⋯Cl–Pt XBs, and only the H2ClC–Cl⋯Cl–Pt contact was previously observed.36
 | ||
| Fig. 4 The halogen and hydrogen bonds in the isostructural heterotetramers from 1·1¼CH2Cl2 (a) and 1·1⅖CH2Br2 (b). Thermal ellipsoids are shown with 50% probability. | ||
 | ||
| Fig. 5 The halogen and hydrogen bonds in the heterotetramer from 2·CHCl3. Thermal ellipsoids are shown with 50% probability. | ||
Theoretical consideration of the XBs and HBs in the heterotetrameric clusters
Analysis of the crystallographic data shows that the clusters exhibit short contacts, which may be caused by non-covalent interactions such as HBs and XBs and/or by the crystal packing effects. In addition, a significant limitation of the X-ray crystallography is the impossibility of determining the exact positions of light atoms (first of all hydrogen atoms) and this greatly complicates the interpretation of experimental information. In order to clarify the situation, we carried out theoretical DFT calculations so that the nature of the weak interactions can be analyzed in detail by using computational methods.We focused on the study of isolated heterotetrameric clusters from 1·1¼CH2Cl2, 1·1⅖CH2Br2, and 2·CHCl3 (Fig. 6, structures (1)2·(CH2Cl2)2, (1)2·(CH2Br2)2, and (2)2·(CHCl3)2, respectively) taking into account that if the crystal packing effects are significant, the structures should change substantially on going from the solid state to the gas phase during the geometry optimization procedure. Otherwise the geometries expectedly are preserved in the isolated form.37
 | ||
| Fig. 6 Views of the optimized heterotetrameric clusters (1)2·(CH2Cl2)2 (a), (1)2·(CH2Br2)2 (b), and (2)2·(CHCl3)2 (c). | ||
This commonly accepted approach helps to exclude the crystal packing effects from consideration and to investigate the short contacts only within the clusters.
The results of our theoretical calculations are summarized in Tables 2, 3 and S5.† In the case of (1)2·(CH2Cl2)2, we found an asymmetric distortion of the structure and the emergence of two new short contacts N–H⋯Cl–CH2Cl. Both HCl2C–H⋯Cl–Pt contacts are shortened (by 0.19 and 0.11 Å) one of the H2ClC–Cl⋯Cl–Pt contacts is very slightly elongated (by 0.05 Å), and another one is slightly shortened (by 0.09 Å). In the case of (1)2·(CH2Br2)2, the symmetrical structure of the cluster is preserved and both the HBr2C–H⋯Cl–Pt contacts are significantly elongated (by 0.51 Å), two new short contacts N–H⋯Br–CH2Br are formed, and the H2BrC–Br⋯Cl–Pt contacts are shortened (by 0.16 Å).
| Cluster | C–H⋯Cl | d(Cl⋯H), Å | d(Cl⋯C), Å | ∠(C–H⋯Cl), ° | E b | E b |
|---|---|---|---|---|---|---|
| a E b = −V(r)/2.38 b E b = 0.429G(r).39 c Comparison between the sum of Rowland's32 vdW radii and the minimal hydrogen bond angle. | ||||||
| (1)2·(CH2Cl2)2 | C1S–H1SA⋯Cl1A′ | 2.809 | 3.708(8) | 154.6 | ||
| 2.63 | 3.595 | 147.316 | 2.2 | 2.4 | ||
| 2.70 | 3.666 | 147.033 | 1.9 | 1.9 | ||
| (1)2·(CH2Br2)2 | C1S–H1SB⋯Cl1A′ | 2.786 | 3.696(12) | 156.5 | ||
| 3.304 | 4.211 | 141.279 | — | — | ||
| (2)2·(CHCl3)2 | C1S–H1S⋯Cl1′ | 2.795 | 3.4989(17) | 127.8 | ||
| 2.476 | 3.410 | 142.624 | 2.8 | 3.0 | ||
| *Comparisonc | 2.86 | 3.53 | 120 | |||
For (2)2·(CHCl3)2, both Cl3C–H⋯Cl–Pt contacts are shortened (by 0.31 Å) and the HCl2C–Cl⋯Cl–Pt contacts are slightly elongated (by 0.11 Å) on going from the solid state to the gas phase, whereas the length of the HCl2C–Cl⋯Ph contacts remains virtually unchanged. In all three cases, the ∠(C–X⋯Cl) and ∠(X⋯Cl–Pt) angles (X = Cl, Br) are changed insignificantly and the largest deviation from the experimental values (4°) was observed in the case of the ∠(Cl⋯Cl–Pt) angle in (2)2·(CHCl3)2.
Additional information on the nature of the short contacts in (1)2·(CH2Cl2)2, (1)2·(CH2Br2)2, and (2)2·(CHCl3)2, can be obtained using the topological analysis of the electron density distribution (AIM method).40 This approach has already been successfully used by us upon studies of non-covalent interactions and properties of coordination bonds in various transition metal complexes and organic compounds.37,41–46 The low magnitude of the electron density and the positive values of the Laplacian and energy density in appropriate bond critical points (3, −1) (Table S5†) are typical for XBs47–50 and also for some HBs51 indicating that the interaction is weak.52,53 We have defined the energies of these contacts according to the procedures proposed by Espinosa et al.38 and Vener et al.39 (1–3 kcal mol−1), and one can state that these bonds may be classified as very weak mainly due to the electrostatic and dispersion interactions. The strength of the XBs decreases in the series: (1)2·(CH2Br2)2 > (1)2·(CH2Cl2)2 > (2)2·(CHCl3)2. Bond critical points (3, −1) for very long HBr2C–H⋯Cl–Pt contacts in (1)2·(CH2Br2)2 were not found.
We evaluated the vertical total energies of the heterotetrameric clusters dissociation (Ev) through the “halogen” and “hydrogen” contacts for (1)2·(CH2Cl2)2, (1)2·(CH2Br2)2, and (2)2·(CHCl3)2; corresponding values of Ev are given in Table 4. The cooperativity of the XBs and HBs was recognized for the studied heterotetramers. Thus, to quantify the relative contributions of these non-covalent interactions in the stabilization of the heterotetrameric clusters, we calculated the vertical total energies of their dissociation (Ev) through the “halogen” and “hydrogen” contacts for (1)2·(CH2Cl2)2, (1)2·(CH2Br2)2, and (2)2·(CHCl3)2, the corresponding values of Ev are given in Table 4. For (1)2·(CH2Cl2)2 the contribution of the HBs to the stabilization of the system prevail nearly twice over the contribution of XBs, for (1)2·(CH2Br2)2 – approximately by one third, and for (2)2·(CHCl3)2 contributions of both types of non-covalent interaction are almost the same. The existence of (1)2·(CH2Cl2)2 and (1)2·(CH2Br2)2 is determined mainly by HBs, whereas for (2)2·(CHCl3)2 both types of non-covalent interaction are equally essential. In the case of (2)2·(CHCl3)2, the non-covalent interactions make the major contribution to the stabilization of the cluster, whereas for (1)2·(CH2Cl2)2 and especially for (1)2·(CH2Br2)2 the role of the XBs and HBs in stabilization of these supramolecular systems is, although smaller, still quite significant.
| Cluster | Dissociation | E v |
|---|---|---|
| (1)2·(CH2Cl2)2 | Through the XB | 12.09 |
| Through the HB | 21.58 | |
| (1)2·(CH2Br2)2 | Through the XB | 30.77 |
| Through the HB | 38.88 | |
| (2)2·(CHCl3)2 | Through the XB | 8.94 |
| Through the HB | 8.26 |
Thus, the results of our theoretical calculations reveal that the crystal packing noticeably affects the geometrical features of the heterotetrameric clusters and also that the XBs and HBs in these supramolecular associates are relatively weak.
The formation of such ordered structures as the heterotetramers can be accompanied by a significant entropy decrease and stabilization of the system from the energy viewpoint. In order to prove that, we tried to carry out a geometry optimization procedure for the two model metal-free systems, viz. the “heterotetramer” and the “heterotrimer plus one outlying chloroform molecule”, (Cl−)2·(CHCl3)2 and (Cl−)2·(CHCl3) + CHCl3, respectively (M06-2X/6-311++G(d,p) level of theory). We located the minima for the heterotetramer (Cl−)2·(CHCl3)2 (Fig. 7) on the potential energy surface. However, several attempts to find an appropriate stationary point for the (Cl−)2·(CHCl3) + CHCl3 system with the outlying chloroform molecule led to the separation of this model system into two fragments Cl−·(CHCl3), which move away from each other upon the geometry optimization (Fig. 8); we finished the optimization when the distance between these fragments exceeded 13 Å. A possible rationale for this phenomenon is an electrostatic repulsion between the two chloride anions. In conjunction with the found minima for the heterotetrameric system (Cl−)2·(CHCl3)2, our results suggest that the presence of two Cl− close to each other in the crystal lattice can be provided through the formation of bridges with CHCl3. These neutral species sufficiently shield the negatively charged anions thus providing their coexistence in the close proximity.
 | ||
| Fig. 7 Calculated cluster (Cl−)2·(CHCl3)2. | ||
 | ||
| Fig. 8 Attempted calculation of (Cl−)2·(CHCl3) + CHCl3. | ||
Retrieved CCDC data for the heterotetrameric clusters
The heterotetrameric clusters stabilized simultaneously by two C–X⋯Cl XBs and two C–X⋯Cl HBs (Fig. 9) were not described in the literature except for only one example (Cl−)2·(CHCl3)2 reported by us.25 Therefore we inspected available CCDC data for these heterotetrameric clusters to clarify consistent patterns of their formation. | ||
| Fig. 9 Schematic representation of the heterotetrameric clusters with R–Cl (cluster A) or Cl− (cluster B) as both the HB and XB acceptors and R′2CHCl as the XB and HB donors. | ||
The first type of cluster is stabilized by two C–X⋯Cl–R XBs and two C–X⋯Cl–R HBs (Fig. 9, cluster A) incorporating a neutral RCl species as the Lewis base. We also acquired data on analogous clusters where free chloride anions behave as both XB and HB acceptors (Fig. 9, cluster B). Noticeably, in each cluster the XBs and HBs alternate with each other. Other variants of bridging molecules in the clusters with two HBs and two XBs were not found.
In the case of free chloride anions as both the HB and XB acceptors, 8 type B clusters (Fig. 9) with chloroform and 3 clusters with dichloromethane were found and all of them exhibited a center of symmetry. Only one cluster in structure UPEROX was described in our previous work.26
 | ||
| Fig. 10 ChemDraw (top) and ball-and-stick (bottom) models of the tetrameric cluster from KAVDUG. | ||
Experimental
Crystallization and XRD experiments
Compounds 1 and 2 were synthesized as previously described.29 Solvents were obtained from commercial sources and used as received. Crystals 1·1¼CH2Cl2, 1·1⅖CH2Br2, and 2·CHCl3 were grown using the slow evaporation of 1 and 2 solutions in corresponding solvents.A suitable crystal of 1·1¼CH2Cl2 was studied on an Xcalibur, Eos diffractometer. The crystal was kept at 100(2) K during data collection. Using Olex2 1.2,58 the structure was solved with the ShelXT59 structure solution program using Direct Methods and refined with the ShelXL-2014 (ref. 60) refinement package using Least Squares minimisation.
A suitable crystal of 1·1⅖CH2Br2 was studied on a SuperNova, Dual, Cu at zero, Atlas diffractometer. The crystal was kept at 100(2) K during data collection. Using Olex2,58 the structure was solved with the ShelXS60 structure solution program using Direct Methods and refined with the ShelXL60 refinement package using Least Squares minimisation. The unit cell of 1·1⅖CH2Br2 also contains disordered molecules of CH2Br2 (total potential solvent accessible void volume is 305 Å3; electron count per cell is 206 electrons) that have been treated as a diffuse contribution to the overall scattering without specific atom positions using SQUEEZE/PLATON.61
A suitable crystal of 2·CHCl3 was immersed in cryo-oil, mounted in a Nylon loop, and measured at a temperature of 100 K. The X-ray diffraction data was collected on a Bruker Smart Apex II diffractometer using Mo Kα radiation (λ = 0.71073 Å). The SAINT62 programme package was used for cell refinement and data reduction. The structure was solved using direct methods using a SHELXS-97 (ref. 60) programme. A numerical absorption correction (SADABS)63 was applied to the data. Structural refinements were carried out using SHELXL-97.60
Computational details
The full geometry optimization of (1)2·(CH2Cl2)2, (1)2·(CH2Br2)2, and (2)2·(CHCl3)2 has been carried out at the DFT theory level using the M06 functional64 with the help of the Gaussian-0965 program package. The experimental X-ray geometries were used as starting points for the theoretical geometry optimization procedure. The calculations were carried out using a quasi-relativistic Stuttgart pseudopotential that described 60 core electrons and the appropriate contracted basis set66 for the platinum atoms and the 6-31G(d) basis set for other atoms. The M06 functional is much less time-consuming than the MP2 method used by us previously,26,67 at the same time, the M06 functional describes reasonably weak dispersion forces and non-covalent interactions.68,69 No symmetry operations have been applied. The Hessian matrix was calculated analytically to prove the location of the correct minima (no imaginary frequencies). The topological analysis of the electron density distribution with the help of the atoms in molecules (AIM) method developed by Bader40 has been performed by using the Multiwfn program (version 3.3.4).70Conclusions
The results of this work could be considered from the following perspectives. Firstly, we report here on a new family of heterotetrameric clusters held simultaneously by two C–X⋯Cl XBs and two C–H⋯Cl HBs with halomethanes. This family has now been extended from a single associate of CHCl3 with uncomplexed Cl− (previously reported by us26) to a large set of heterotetramers incorporating (i) not only CHCl3, but also rather weak XB/HB donors such as CH2Cl2 and CH2Br2, (ii) neutral chloride platinum(II) complexes, where XB/HB-accepting chloride is bound to a platinum center, and (iii) organic molecules, RCl, acting as a simultaneous XB/HB donor and XB/HB acceptor. Secondly, our theoretical calculations indicate that the XBs and HBs in the reported clusters are cooperative. In the cases of (1)2·(CH2Cl2)2 and (1)2·(CH2Br2)2, the contribution of the HBs to the stabilization of the system is dominant, whereas for (2)2·(CHCl3)2 the contributions of both types of non-covalent interaction are almost the same. Another theoretical study conducted in this work for the simple model systems, (Cl−)2·(CHCl3)2 and (Cl−)2·(CHCl3) + CHCl3, gives an idea that the halomethane in the heterotetramers shields the chloride anions thus preventing their Coulomb repulsion and the destruction of these clusters. Thirdly, two clusters, (1)2(CH2Cl2)2 and (1)2(CH2Br2)2, were found to be isostructural, which indicates a possibility of the CH2Cl2/CH2Br2 exchange with preservation of the geometrical parameters. Such isostructural exchange can be useful for the prediction of crystal structures and in the design of a series of clusters held using weak interactions and exhibiting close geometrical features. We expect that all these findings can find an application in crystal engineering and materials science, and we hope that our results open up an avenue to the generation of other similar heterotetramers and works in this direction are under way in our group.Acknowledgements
D. M. I. and A. S. N. are grateful to the Russian Foundation for Basic Research for the support of their studies (grant 16-33-00212). V. Y. K. thanks Saint Petersburg State University for research grant (12.38.225.2014). XRD studies were performed at the Center for X-ray Diffraction Studies (Saint Petersburg State University), whereas the structure determination of 2·CHCl3 was performed at the University of Jyväskylä.Notes and references
-
A. Karpfen, A. C. Legon, W. T. Pennington, T. W. Hanks, H. D. Arman, P. Metrangolo, G. Resnati, T. Pilati, S. Biella, S. V. Rosokha, J. K. Kochi, D. W. Bruce and M. Fourmigué, in Halogen Bonding: Fundamentals and Applications, ed. P. Metrangolo and G. Resnati, Springer-Verlag Berlin, Berlin, 2008, vol. 126, pp. 1–221 Search PubMed
.
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772–3783 RSC
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS PubMed
- K. B. Landenberger, O. Bolton and A. J. Matzger, J. Am. Chem. Soc., 2015, 137, 5074–5079 CrossRef CAS PubMed
- V. V. Sivchik, A. I. Solomatina, Y.-T. Chen, A. J. Karttunen, S. P. Tunik, P.-T. Chou and I. O. Koshevoy, Angew. Chem., Int. Ed., 2015, 54, 14057–14060 CrossRef CAS PubMed
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Cryst. Growth Des., 2014, 14, 2697–2702 CAS
- A. Bundhun, P. Ramasami, J. S. Murray and P. Politzer, J. Mol. Model., 2013, 19, 2739–2746 CrossRef CAS PubMed
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed
- X. J. Mu, Q. T. Wang, L. P. Wang, S. D. Fried, J. P. Piquemal, K. N. Dalby and P. Y. Ren, J. Phys. Chem. B, 2014, 118, 6456–6465 CrossRef CAS PubMed
- F. F. Awwadi, R. D. Willett, K. A. Peterson and B. Twamley, Chem. – Eur. J., 2006, 12, 8952–8960 CrossRef CAS PubMed
- P. Metrangolo and G. Resnati, IUCrJ, 2014, 1, 5–7 CrossRef CAS PubMed
- F. H. Allen, P. A. Wood and P. T. A. Galek, Acta Crystallogr., Sect. B: Struct. Sci., 2013, 69, 379–388 CrossRef CAS PubMed
- L. Brammer, G. M. Espallargas and S. Libri, CrystEngComm, 2008, 10, 1712–1727 RSC
- R. Bertani, P. Sgarbossa, A. Venzo, F. Lelj, M. Amati, G. Resnati, T. Pilati, P. Metrangolo and G. Terraneo, Coord. Chem. Rev., 2010, 254, 677–695 CrossRef CAS
- G. M. Espallargas, L. Brammer and P. Sherwood, Angew. Chem., Int. Ed., 2006, 45, 435–440 CrossRef CAS PubMed
- L. Brammer, G. M. Espallargas and H. Adams, CrystEngComm, 2003, 5, 343–345 RSC
- S. V. Rosokha, J. Lu, T. Y. Rosokha and J. K. Kochi, Chem. Commun., 2007, 3383–3385 RSC
- S. V. Rosokha and M. K. Vinakos, Cryst. Growth Des., 2012, 12, 4149–4156 CAS
- M. Berkei, J. F. Bickley, B. T. Heaton and A. Steiner, Chem. Commun., 2002, 2180–2181 RSC
- F. Zordan, L. Brammer and P. Sherwood, J. Am. Chem. Soc., 2005, 127, 5979–5989 CrossRef CAS PubMed
- G. M. Espallargas, F. Zordan, L. A. Marin, H. Adams, K. Shankland, J. van de Streek and L. Brammer, Chem. – Eur. J., 2009, 15, 7554–7568 CrossRef PubMed
- A. M. Mills, J. A. M. van Beek, G. van Koten and A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, m304–m306 Search PubMed
- M. T. Johnson, Z. Dzolic, M. Cetina, O. F. Wendt, L. Ohrstrom and K. Rissanen, Cryst. Growth Des., 2012, 12, 362–368 CAS
- P. V. Gushchin, G. L. Starova, M. Haukka, M. L. Kuznetsov, I. L. Eremenko and V. Y. Kukushkin, Cryst. Growth Des., 2010, 10, 4839–4846 CAS
- D. S. Yufit, O. V. Shishkin, R. I. Zubatyuk and J. A. K. Howard, Cryst. Growth Des., 2014, 14, 4303–4309 CAS
- D. S. Yufit, O. V. Shishkin, R. I. Zubatyuk and J. A. K. Howard, Kristallografiya, 2014, 229, 639–647 CAS
- D. M. Ivanov, P. V. Gushchin, A. S. Novikov, M. S. Avdontceva, A. A. Zolotarev, G. L. Starova, Y.-T. Chen, S.-H. Liu, P.-T. Chou and V. Y. Kukushkin, Eur. J. Inorg. Chem., 2016, 2016, 1480–1487 CrossRef CAS
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS
- A. N. Chernyshev, M. V. Chernysheva, P. Hirva, V. Y. Kukushkin and M. Haukka, Dalton Trans., 2015, 44, 14523–14531 RSC
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed
- J. Seo, I. Yoon, J. E. Lee, M. R. Song, S. Y. Lee, S. H. Park, T. H. Kim, K. M. Park, B. G. Kim and S. S. Lee, Inorg. Chem. Commun., 2005, 8, 916–919 CrossRef CAS
- A. A. Melekhova, A. S. Novikov, K. V. Luzyanin, N. A. Bokach, G. L. Starova, V. V. Gurzhiy and V. Y. Kukushkin, Inorg. Chim. Acta, 2015, 434, 31–36 CrossRef CAS
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS
- M. V. Vener, A. N. Egorova, A. V. Churakov and V. G. J. Tsirelson, Comput. Chem., 2012, 33, 2303–2309 CrossRef CAS PubMed
-
R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed
- D. M. Ivanov, Y. V. Kirina, A. S. Novikov, G. L. Starova and V. Y. Kukushkin, J. Mol. Struct., 2015, 1104, 19–23 CrossRef
- X. Ding, M. J. Tuikka, P. Hirva, V. Y. Kukushkin, A. S. Novikov and M. Haukka, CrystEngComm, 2016, 18, 1987–1995 RSC
- K. I. Kulish, A. S. Novikov, P. M. Tolstoy, D. S. Bolotin, N. A. Bokach, A. A. Zolotarev and V. Y. Kukushkin, J. Mol. Struct., 2016, 1111, 142–150 CrossRef CAS
- A. S. Novikov, M. L. Kuznetsov and A. J. L. Pombeiro, Chem. – Eur. J., 2013, 19, 2874–2888 CrossRef CAS PubMed
- A. S. Novikov and M. L. Kuznetsov, Inorg. Chim. Acta, 2012, 380, 78–89 CrossRef CAS
- D. M. Ivanov, A. S. Novikov, I. V. Ananyev, Y. V. Kirina and V. Y. Kukushkin, Chem. Commun., 2016, 52, 5565–5568 RSC
- S. J. Grabowski and E. Bilewicz, Chem. Phys. Lett., 2006, 427, 51–55 CrossRef CAS
- Y. H. Wang, Y. X. Lu, J. W. Zou and Q. S. Yu, Int. J. Quantum Chem., 2008, 108, 90–99 CrossRef CAS
- Y. X. Lu, J. W. Zou, Y. H. Wang, Y. J. Jiang and Q. S. Yu, J. Phys. Chem. A, 2007, 111, 10781–10788 CrossRef CAS PubMed
- A. Forni, J. Phys. Chem. A, 2009, 113, 3403–3412 CrossRef CAS PubMed
- I. Rozas, I. Alkorta and J. Elguero, J. Am. Chem. Soc., 2000, 122, 11154–11161 CrossRef CAS
- J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS
- P. Politzer, P. Lane, M. C. Concha, Y. G. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed
- S. Gowrisankar, H. Neumann, A. Spannenberg and M. Beller, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, m255 CAS
- D. M. L. Goodgame, D. A. Grachvogel, M. A. Hitchman, N. J. Long, H. Stratemeier, A. J. P. White, J. L. M. Wicks and D. Williams, J. Inorg. Chem., 1998, 37, 6354–6360 CrossRef CAS
- A. V. Shtemenko, A. A. Golichenko and K. V. Domasevitch, Z. Naturforsch., A: Phys. Sci., 2001, 56(b), 381–385 CAS
- M. Podsiadło, K. Dziubek and A. Katrusiak, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 595–600 Search PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
-
A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2005 Search PubMed
-
Bruker, AXS SAINT, Bruker AXS, Inc., Madison, WI, USA, 2009 Search PubMed
-
G. M. Sheldrick, SADABS - Bruker Nonius scaling and absorption correction, Bruker AXS, Inc., Madison, Wisconsin, USA, 2008 Search PubMed
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, in Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS
- P. V. Gushchin, M. L. Kuznetsov, Q. Wang, A. A. Karasik, M. Haukka, G. L. Starova and V. Y. Kukushkin, Dalton Trans., 2012, 41, 6922–6931 RSC
- L. A. Burns, A. Vazquez-Mayagoitia, B. G. Sumpter and C. D. Sherrill, J. Chem. Phys., 2011, 134, 25 CrossRef PubMed
- M. Sumimoto, Y. Kawashima, D. Yokogawa, K. Hori and H. Fujumoto, Int. J. Quantum Chem., 2013, 113, 272–276 CrossRef CAS
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Packing features of investigated solvates, description of other weak interactions, additional information for the theoretical consideration, full information for CCDC inspection, and cartesian atomic coordinates of the calculated equilibrium structures. CCDC 1456075, 1456509 and 1470271. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce01179a |
| This journal is © The Royal Society of Chemistry 2016 |