
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural analysis and optical properties of the Bi2−xYxWO6 system
S.
Pasternak
a,
D.
Levy
b,
Y.
Paz
a and
B.
Pokroy
*b
aDepartment of Chemical Engineering and the Russell Berrie Nanotechnology Institute, Technion – Israel Institute of Technology, 3200003 Haifa, Israel
bDepartment of Materials Science and Engineering, Technion – Israel Institute of Technology, 3200003 Haifa, Israel. E-mail: bpokroy@tx.technion.ac.il
First published on 22nd June 2016
Abstract
Photocatalytic conversion of solar energy into chemical energy has attracted considerable interest for several decades. One compound already reported as a visible-light-active photocatalyst for water splitting is BiYWO6, a member of the Bi2−xYxWO6 family of compounds. The structural and optical properties of other members of this family have not been reported to date. In this work, we synthesized various compositions of Bi2−xYxWO6, studied their optical properties, and report their structural parameters obtained by utilizing powder diffraction coupled with Rietveld refinement.
Introduction
Mixed oxides of bismuth have been widely studied as candidates for the visible-light-photocatalytic removal of contaminants1–7 and as photoanodes in photoelectrochemical cells.8–11 To a large extent, this interest originates from their relatively narrow bandgaps owing to the mixing of Bi 6s and O 2p orbitals, which leads to a cathodic shift in the position of the valence band.12In the context of light utilization, the Aurivillius family of layered perovskites is among the most interesting groups. The members of this family of ceramics have been studied mainly because of their dielectric and ferroelectric properties, which facilitate their use as resonators and antennae.13,14 One specific sub-group of this family is characterized by Bi2−xLnxWO6 stoichiometry, where Ln is a lanthanide-series element and x is usually between 0.3 and 1.3.15 A decade ago, the originally reported structure of the Bi2−xLnxWO6 system was re-established by means of modern powder X-ray and neutron diffraction, with Bi0.7Yb1.3WO6 taken as a representative compound of the series.16 The structure was shown to be monoclinic with an A2/m space group and to consist of both edge and corner-sharing WO6 octahedra.17
The finding that BiYWO6 can serve as an active water-splitting photocatalyst under visible light,18 making it possible to store solar energy as hydrogen fuel, sparked new interest in this material. Traditionally, this compound has been synthesized by high-temperature methods such as solid-state reaction19 and microwave-assisted solid-state reaction.20 These methods lead to low specific surface area, making the compound impractical for catalytic applications. BiYWO6 was recently reported to be synthesized also by the sol–gel and hydrothermal methods, exhibiting photocatalytic activity under visible-light irradiation.21 The facile sol–gel preparation procedure paved the way for the use of film electrodes made of Bi2−xYxWO6 as photoanodes for water oxidation. The optical and electrical properties of the resulting compounds are expected to depend on their composition, which thus affects their photocatalytic properties. Among the Bi2−xYxWO6 compounds, BiYWO6 is the only one whose crystallographic data have been reported to date.20 BiYWO6 was shown to have the same monoclinic structure as the high-temperature phase of Bi2WO6 and other bismuth tungstates containing ytterbium, neodymium and lanthanum17 such as Bi0.7Yb1.3WO6. The orthorhombic structure (Bi2WO6 at room temperature) and the monoclinic structure (Bi2WO6 at high temperature and Bi2−xMxWO6, M = Yb, Nb, La) consist of alternating layers of BiO2 and WO6 (Fig. 1) but differ with respect to the connectivity of the WO6 octahedra: in the orthorhombic structure, all the basal corners are shared by the WO6 octahedra, whereas in the monoclinic structure the octahedra tilt at 45° and share one edge and two corners forming chains of two corner-sharing octahedra parallel to the b-axis. This difference affects the Bi2O2 layer. In the orthorhombic structure, all of the Bi cations have the same chemical environment so that the two Bi crystallographic sites differ only in their symmetry. In contrast, in the monoclinic structure, the two Bi crystallographic sites have different environments, as illustrated in Fig. 1. From this figure, it is possible also to highlight that the monoclinic structure is more flexible in the Bi2O2 layer; thus, it can more easily accommodate cations with different dimensions.
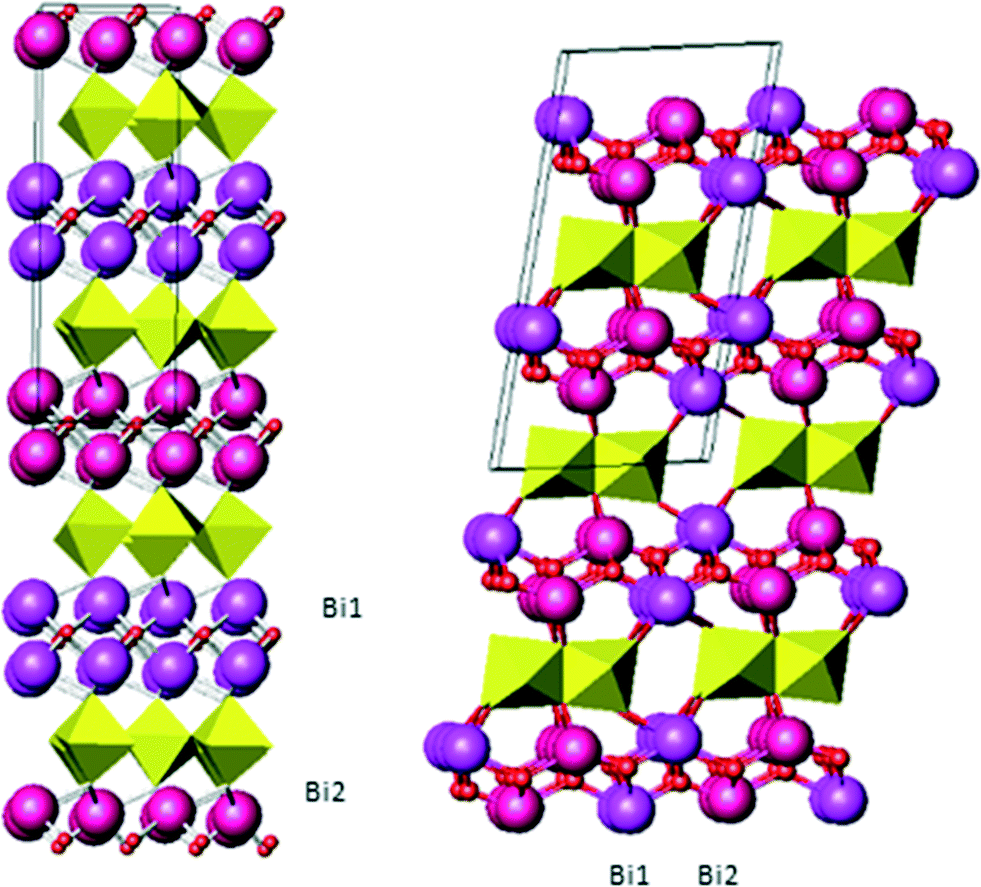 | ||
| Fig. 1 Structures of the orthorhombic Bi2WO6 (left) and monoclinic BiYWO6 (right). | ||
In this work, using the sol–gel method, we synthesized different Bi2−xYxWO6 (BYW) compounds corresponding to x values of 0.3, 0.5, 0.7, 0.85 and 1 and report their optical, morphological and structural properties for the first time.
Experimental
Synthesis
For a typical synthesis, stoichiometric amounts of Bi2O3 (99.99%, Alfa Aesar), Y2O3 (99.99%, Strem Chemicals) and 5(NH4)2O·12WO3·5H2O (99.999%, Alfa Aesar) were dispersed in 200 mL of aqueous solution containing 6.3 g of pentetic acid (DTPA, Strem Chemicals) and 6 mL of concentrated (25% by weight) NH4OH. The dispersions were heated to 80 °C and stirred vigorously for 24 h to obtain transparent solutions. After heating in wide petri dishes at 100 °C for 1 h to evaporate most of the water and ammonia, these BYW-precursor solutions formed sticky gels containing the DTPA and BYW precursors. The gels were collected and heated in air at high temperatures (800–1000 °C) for 4 h to form yellow powders with different stoichiometric ratios of bismuth and yttrium.Characterization
The X-ray powder diffraction (XRPD) patterns of all BYW samples were collected by using a Rigaku SmartLab X-ray diffractometer in Bragg–Brentano geometry with Cu Kα radiation. The diffracted beam was monochromatized by silicon single crystals and collected by using a D/tex linear detector. The XRPD data were collected over a 2θ range of 10° to 120° with a step size of 0.02°.High-resolution electron microscopy images were obtained using a Zeiss Ultra Plus High Resolution Field Emission Gun Scanning Electron Microscope (FEGSEM). Energy-dispersive X-ray spectroscopy (EDS) spectra were acquired using an Oxford X-MaxN Silicon drift detector (SDD). The diffuse reflectance spectra of the powders were recorded using a Shimadzu UV 2600 spectrophotometer equipped with an ISR-2600Plus Integrating Sphere Attachment. The Raman spectra were collected using a micro-Raman spectrometer (iHR550, Horiba) with a green laser (λ = 514 nm).
Diffuse reflectance measurements were performed by putting a small amount of sample (50 mg) on top of a BaSO4 tablet and then pressing. Pure BaSO4 was used as a baseline because its reflectance is close to 100% in the examined range (200 nm < λ < 700 nm).
Results
Fig. 2 shows the SEM images of the different compounds. All synthesized particles were submicron in size and exhibited uniform morphologies. Backscattered electron analysis revealed no evidence of inhomogeneity or the presence of additional phases in the samples. | ||
| Fig. 2 High-resolution SEM images of the samples. (A) Bi1.7Y0.3WO6, (B) Bi1.5Y0.5WO6, (C) Bi1.3Y0.7WO6, (D) Bi1.15Y0.85WO6, and (E) BiYWO6. Scale bar, 1 μm. | ||
Chemical compositions of the studied samples were determined by EDS microanalysis. Calculated compositions of the samples are presented in Table 1. All compositions comply, within a small experimental error (indicated in the table), with the nominal stoichiometry. The systematic underestimation of the yttrium content can be explained by the relatively high errors arising from the necessity for deconvolution of the yttrium Lα-line and the tungsten Mα-line.
| Nominal stoichiometry | Bismuth (wt%) | Yttrium (wt%) | Tungsten (wt%) | EDS stoichiometry |
|---|---|---|---|---|
| Bi1.7Y0.3WO6 | 60 ± 3.9 | 4.5 ± 1.2 | 34 ± 2.4 | Bi1.8Y0.1WO6 |
| Bi1.5Y0.5WO6 | 51 ± 2.5 | 15 ± 1.1 | 34 ± 1.4 | Bi1.5Y0.4WO6 |
| Bi1.3Y0.7WO6 | 45 ± 1.0 | 21 ± 0.3 | 34 ± 0.6 | Bi1.3Y0.6WO6 |
| Bi1.15Y0.85WO6 | 39 ± 2.4 | 26 ± 1.4 | 35 ± 2.1 | Bi1.1Y0.8WO6 |
| BiYWO6 | 35 ± 0.5 | 31 ± 0.9 | 34 ± 0.8 | BiY0.9WO6 |
Fig. 3(A) presents the reflection spectrum of one of the prepared samples (Bi1.7Y0.3WO6). All of the other samples were measured in the same way. The measured reflectance values in the vicinity of the absorption onset were roughly 40% to 60%. To extract the bandgap values of the various samples, the wavelength-dependent absorption coefficient was calculated using the modified Kubelka–Munk transformation , where R is the measured fractional reflection.22 Bandgap energies were calculated according to the method proposed by Tauc.23 In the absence of data regarding the nature of the optical transition, the value of (F(R))n was plotted against photon energy both under the assumption of a direct transition (Fig. 3(B), n = 2) and under the assumption of an indirect transition (Fig. 3(C), n = 0.5). The best fit was obtained for the indirect transition relationship, giving bandgap values consistent with the onset of absorption (2.4–2.5 eV). The calculated bandgap values are listed in Table 2.
, where R is the measured fractional reflection.22 Bandgap energies were calculated according to the method proposed by Tauc.23 In the absence of data regarding the nature of the optical transition, the value of (F(R))n was plotted against photon energy both under the assumption of a direct transition (Fig. 3(B), n = 2) and under the assumption of an indirect transition (Fig. 3(C), n = 0.5). The best fit was obtained for the indirect transition relationship, giving bandgap values consistent with the onset of absorption (2.4–2.5 eV). The calculated bandgap values are listed in Table 2.
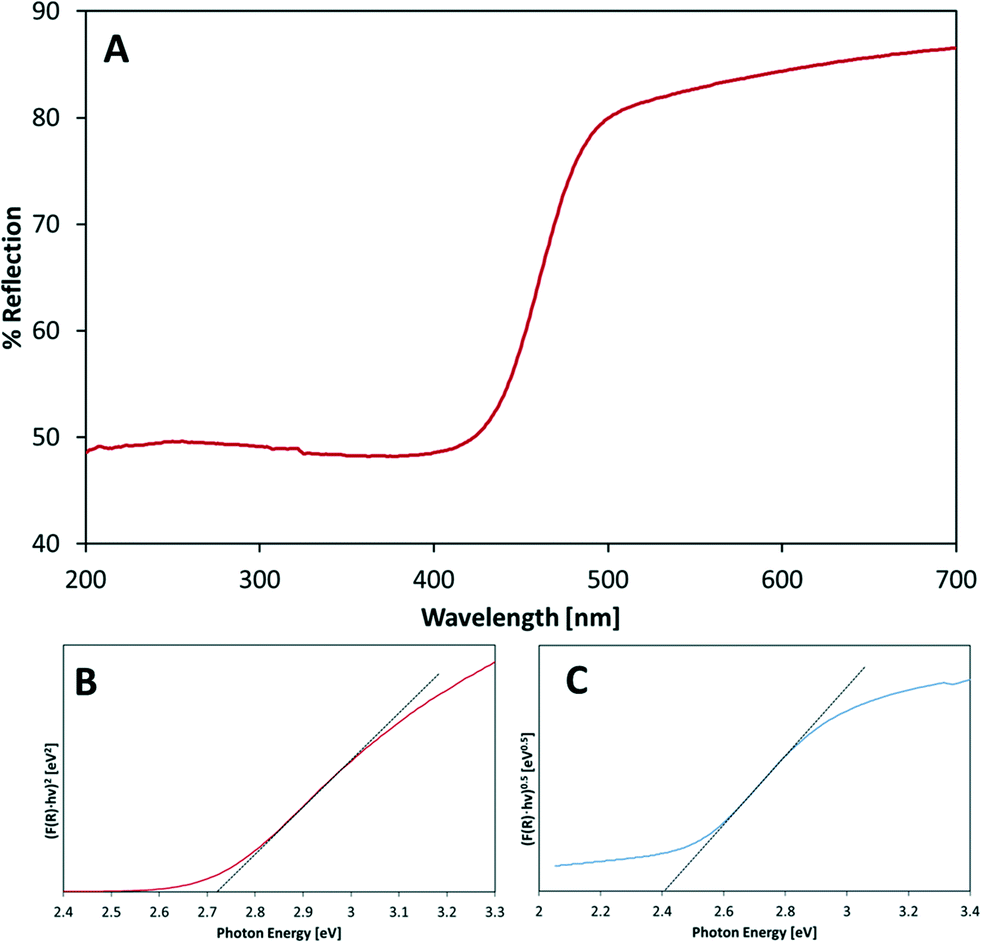 | ||
| Fig. 3 UV-vis measurement data for Bi1.7Y0.3WO6. (A) Diffuse reflectance spectrum. (B) Tauc plot assuming a direct optical transition. (C) Tauc plot assuming an indirect optical transition. | ||
| Bi1.7Y0.3WO6 | Bi1.5Y0.5WO6 | Bi1.3Y0.7WO6 | Bi1.15Y0.85WO6 | BiYWO6 | |
|---|---|---|---|---|---|
| Indirect bandgap/eV | 2.40 | 2.45 | 2.48 | 2.50 | 2.43 |
| Direct bandgap/eV | 2.72 | 2.69 | 2.78 | 2.80 | 2.71 |
X-ray diffractograms (XRDs) of the various compounds are shown in Fig. 4, together with the tick marks of the monoclinic cell as reported for BiNdWO6.17 Neither superstructures nor impurity peaks can be seen in the XRDs. The shift to smaller angles (bigger d-spacing) observed with the increase in the bismuth content can be attributed to the insertion in the structure of the Bi3+ (cation radius,24 1.17 Å) cation, whose radius is larger than that of Y3+ (cation radius,24 1.02 Å).
 | ||
| Fig. 4 X-ray diffraction patterns of the studied samples. | ||
To determine the effect of yttrium insertion on the bismuth tungstate structure, the diffractograms were refined by means of the Rietveld method.25 The software we used was GSAS-II.26 Since no structure has been reported for these compositions, we chose the structure of the BiNdWO6 reported by Berdonosov et al.17 as a model on which to base the refinements performed on our diffraction data, but substituted the Y cation for Nd. This structure is monoclinic (A2/m, s.g. 12) with the tungsten cation in single-octahedral coordination, and the Bi and the Nd/Y cations are split on two different crystallographic sites in pseudo-octahedral coordination.
The structure reported by Berdonosov et al.16 was determined by neutron diffraction and was characterized by disordered oxygen atoms. As the neutron diffraction technique is more sensitive than XRD to small deviations in the position of the oxygen atoms, the structures refined by the latter technique are less affected by oxygen disorder. To confirm this statement, both the ordered and the disordered structures were refined using the collected diffractogram of BiYWO6. The results confirmed that the disordered structure does not improve the refinement results relative to the results obtained with the ordered one. Therefore, to simplify the description of the structure, we chose the ordered structure for the refinement process in all diffractograms of the studied compositions.
For Rietveld refinement, we refined the background by using 15-term Chebyshev polynomial functions and described the peak profile by refining the three Caglioti formula terms (U, V, W) and the microstrain parameter for the Gaussian and the Lorentzian contribution, respectively. To correct the small error in displacement of the sample in the diffractometer, we refined the sample displacement parameter. We also refined the cell edges, the isotropic thermal parameters and the positions of each atom. To determine the distribution of bismuth and yttrium cations in the two bismuth sites, we refined the fractions occupied by these atoms on the two crystallographic sites. The occupation parameters were constrained so as to obtain full occupancy over the two crystallographic sites, and the Bi/Y ratio was taken as constant according to the nominal ratio of each sample. The main characteristics of these structures are shown in Table 3, and the plot of the refined diffraction pattern of the BiYWO6 phase is described in Fig. 5. As expected, the increasing quantity of bismuth in the structure caused the cell parameters to increase (Fig. 6). The expansion of the cell edges as a function of the quantity of bismuth demonstrated linear behavior up to 1.5 bismuth atoms per unit formula (x = 0.5). Above this value, the expansion of the cell edges was saturated, especially along the c-axis.
| Sample | X = 0.3 | X = 0.5 | X = 0.7 | X = 0.85 | X = 1 | |
|---|---|---|---|---|---|---|
| a [Å] | 8.204(1) | 8.1772(7) | 8.163(2) | 8.139(1) | 8.130(2) | |
| b [Å] | 3.79452(2) | 3.7812(1) | 3.7704(3) | 3.7583(2) | 3.7523(3) | |
| c [Å] | 16.225(1) | 16.1897(8) | 16.150(2) | 16.093(4) | 16.047(2) | |
| β [deg] | 102.141(4) | 102.205(4) | 102.263(6) | 102.263(5) | 102.341(7) | |
| V [Å3] | 493.80(7) | 489.26(4) | 485.7(1) | 481.05(9) | 478.3(1) | |
| Bi/Y1 | x | 0.9295(3) | 0.9275(3) | 0.9253(3) | 0.9239(3) | 0.9229(4) |
| z | 0.3329(2) | 0.3317(2) | 0.3309(2) | 0.3307(2) | 0.3302(2) | |
| Occ. Bi | 0.840(9) | 0.765(8) | 0.685(9) | 0.616(9) | 0.56(1) | |
| Bi/Y2 | x | 0.3965(4) | 0.3978(3) | 0.3974(5) | 0.3963(5) | 0.3969(6) |
| z | 0.3182(2) | 0.3181(2) | 0.3172(2) | 0.3167(2) | 0.3166(2) | |
| Occ. Bi | 0.859(9) | 0.735(2) | 0.615(9) | 0.534(9) | 0.44(1) | |
| Bi/Y | Uiso × 100 (Å2) | 0.62(5) | 0.94(5) | 0.5(6) | 1.12(6) | 0.4(7) |
| W | x | 0.2986(3) | 0.2990(3) | 0.2986(3) | 0.2972(3) | 0.2972(4) |
| z | 0.4980(2) | 0.4993(2) | 0.4992(2) | 0.4985(2) | 0.4983(2) | |
| Uiso × 100 (Å2) | 0.92(9) | 1.62(9) | 1.1(1) | 1.17(1) | 0.8(1) | |
| O1 | x | 0.106(3) | 0.114(4) | 0.130(5) | 0.118(4) | 0.123(5) |
| z | 0.236(2) | 0.255(2) | 0.279(2) | 0.258(2) | 0.264(3) | |
| O2 | x | 0.356(4) | 0.357(4) | 0.359(4) | 0.355(4) | 0.357(5) |
| z | 0.233(2) | 0.238(2) | 0.242(3) | 0.235(2) | 0.240(3) | |
| O3 | x | 0.684(4) | 0.672(3) | 0.673(4) | 0.671(4) | 0.665(5) |
| z | 0.485(2) | 0.472(2) | 0.459(2) | 0.473(2) | 0.471(2) | |
| O4 | x | 0.513(4) | 0.502(4) | 0.500(5) | 0.505(4) | 0.505(5) |
| z | 0.574(2) | 0.555(2) | 0.552(2) | 0.592(2) | 0.561(2) | |
| O5 | x | 0.152(4) | 0.164(3) | 0.182(4) | 0.174(4) | 0.173(4) |
| z | 0.569(2) | 0.578(2) | 0.553(3) | 0.577(3) | 0.579(3) | |
| O6 | x | 0.173(4) | 0.166(4) | 0.161(4) | 0.179(4) | 0.181(5) |
| z | 0.400(2) | 0.397(2) | 0.389(2) | 0.392(2) | 0.392(2) | |
| O (All) | Uiso × 100 (Å2) | 2.2(4) | 3.2(4) | 3.8(5) | 4.1(5) | 2.7(5) |
| R wp | 6.85% | 6.20% | 6.88% | 5.96% | 8.33% | |
| GoF | 3.46 | 2.86 | 2.96 | 4.36 | 4.21 | |
| RF2 | 4.68% | 4.35% | 5.17% | 4.40% | 6.13% |
 | ||
| Fig. 5 Graph of the Rietveld refinement on BiYWO6. The observed diffractogram is represented by red dots, the calculated diffractogram by a black line, the difference between them (Io − Ic) by a green line, and the background by a blue line. | ||
 | ||
| Fig. 6 Cell edges and volume versus yttrium fraction. | ||
It is interesting to observe that below x = 0.5 the cell volume is larger than the volume of Bi2WO6 (487.63 Å3),27 owing to the fact that the monoclinic structure of the mixed phase is less dense than the orthorhombic structure of the pure phase.
From a structural point of view, the change in the Bi/Y ratio does not induce significant structure distortion. Nevertheless, we noticed small variations in the two Bi–O and W–O polyhedra. The average Bi–O and W–O distances are plotted in Fig. 7 together with those of the pure Bi2WO6 phase reported by Knight.27
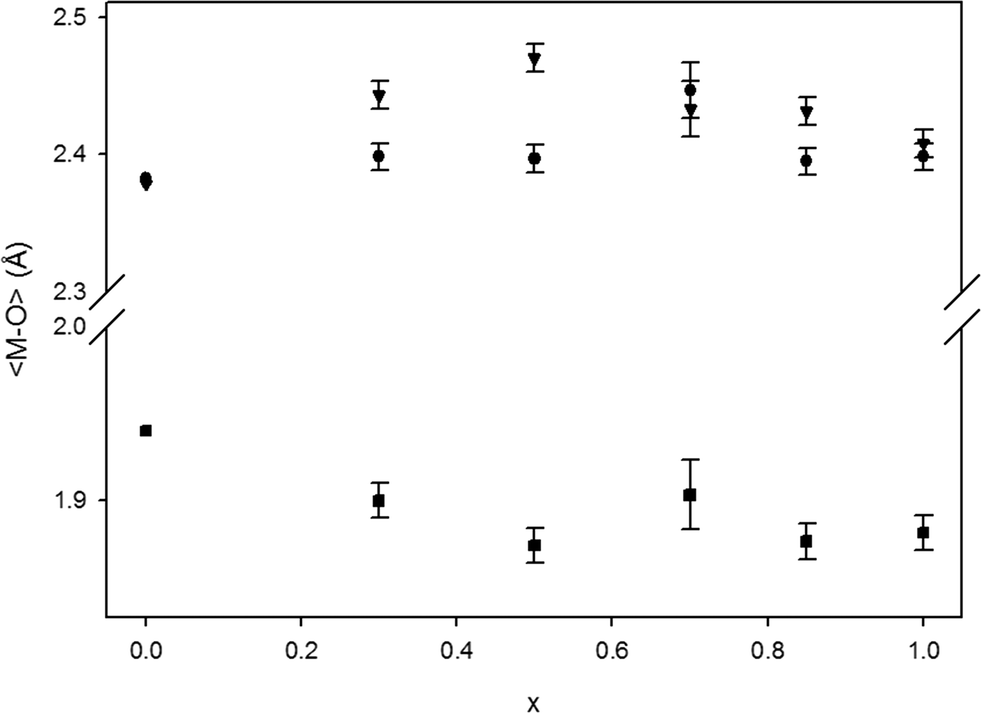 | ||
| Fig. 7 Average M–O distances. The data for x = 0 were taken from published data.27 Black dots, triangles and squares refer to Bi1–O, Bi2–O and W–O bond lengths, respectively. | ||
Although XRDs are not highly sensitive to the position of the oxygen atoms, leading to some errors in calculation of the cation–oxygen distances, we can still discuss trends in the data shown in Fig. 7. Despite the fact that the preferred site for bismuth cations in all samples is the Bi/Y1 crystallographic site, the Bi/Y1–O distance is relatively less affected by the bismuth–yttrium ratio, and the Bi/Y2–O distance increases as “x” changes from 1 to 0.5 and then decreases. In addition, the W–O distance in the same range of composition is not affected by the insertion of bismuth cations.
It can be also highlighted that there is a small, but not negligible, preference of the bismuth cation to occupy the Bi/Y1 site with respect to the other site (Table 3). A possible explanation can be that the Bi/Y1 and Bi/Y2 sites are different: the latter site is in the middle of the double octahedron chain of WO6, while the former site bonds two of these chains (see Fig. 1). Thus, the Bi/Y1 site can better arrange a bigger cation without changing the bond length than the Bi/Y2 site because it is more flexible. Moreover, the presence of a cavity between the two polyhedron chains perpendicular to the a-axis can explain why this axis expands less (<0.2%) than the other two as the bismuth content increases: along the a-axis, the structure can better arrange a bigger cation around the cavity.
The collected Raman spectra are described in Fig. 8, and the calculated peak positions are shown in Table 4. The spectra were fitted with Gaussian-type functions and their intensities were denoted as strong (St), shoulder (Sh), weak (W) and very weak (Vw). The peak assignment and our discussion of the results are based on the work of Maczka et al.,28 which describes the vibrational spectra on the basis of lattice dynamics calculation. The spectra can be divided into five principal regions. The peaks below 180 cm−1 represent the translation of Bi3+/Y3+ and W6+ ions, while the peaks located between 180 cm−1 and 370 cm−1 represent the Bi/Y–O bending modes of oxygen in (Bi/Y)O6 polyhedra, and those between 370 cm−1 and 590 cm−1 represent the bending of WO6 polyhedra and the out-of-plane bending–stretching of Bi/Y–O bonds. The last two regions (640–720 cm−1 and 760–904 cm−1) are associated with the symmetric and antisymmetric stretching of the W–O bond.
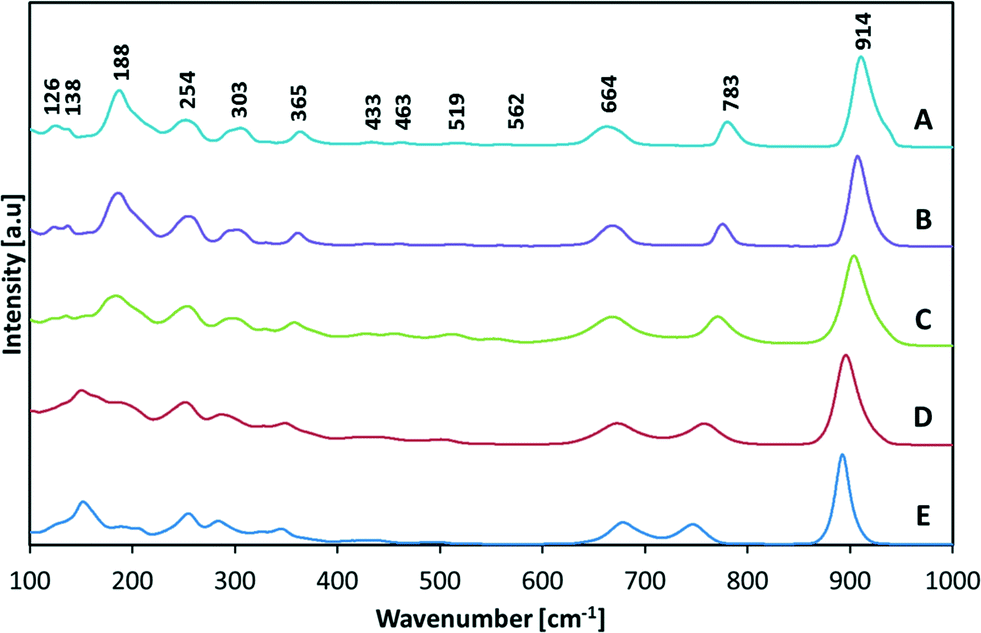 | ||
| Fig. 8 Intensity-normalized Raman spectra of BYW powders of various compositions: BiYWO6 (A), Bi1.15Y0.85WO6 (B), Bi1.3Y0.7WO6 (C), Bi1.5Y0.5WO6 (D) and Bi1.7Y0.3WO6 (E). | ||
| X = 0.3 | X = 0.5 | X = 0.7 | X = 0.85 | X = 1 | Assignment | |
|---|---|---|---|---|---|---|
| 893 (21) | 898 (30) | 906 (34) | 909 (23) | 914 (27) | St | Asymmetric stretching modes of WO6 octahedrons (apical O) |
| 747 (28) | 757 (39) | 772 (34) | 776 (16) | 783 (21) | St | Symmetric stretching modes of WO6 octahedrons (apical O) |
| 717 (30) | 721 (29) | 728 (30) | — | — | Sh | |
| 679 (36) | 675 (54) | 667 (57) | 668 (29) | 664 (41) | St | Asymmetric stretching modes of WO6 octahedrons (equatorial O) |
| — | — | 555 (14) | 559 (25) | 562 (16) | Vw | Bending modes of WO6 octahedrons and stretching + bending modes of (Bi,Y)O6 polyhedra |
| 496 (23) | 499 (32) | 512 (20) | 517 (23) | 519 (23) | Vw | |
| 434 (27) | 444 (26) | 456 (20) | 460 (13) | 463 (14) | Vw | |
| 411 (17) | 419 (23) | 428 (25) | 431 (18) | 433 (16) | Vw | |
| 345 (15) | 351 (28) | 361 (23) | 362 (17) | 365 (22) | W | Bending modes of oxygen in (Bi,Y)O6 polyhedra |
| 328 (13) | 327 (5) | 330 (10) | 331 (10) | 332 (7) | Vw | |
| 285 (20) | 292 (21) | 299 (22) | 301 (22) | 303 (25) | W | |
| 253 (26) | 250 (25) | 252 (22) | 254 (22) | 254 (22) | W | |
| 200 (23) | 194 (26) | 186 (33) | 189 (30) | 188 (27) | St | |
| 153 (21) | 149 (18) | 140 (14) | 137 (7) | 138 (7) | W | Translation of Bi3+/Y3+ and W6+ ions |
| 129 (18) | 131 (13) | 122 (7) | 125 (12) | 126 (13) | W | |
| 85 (12) | 86 (14) | 86 (20) | 87 (16) | 86 (17) | Sh | |
| 78 (8) | 77 (8) | 77 (8) | 78 (8) | 77 (8) | W |
In the strong peaks around 900 cm−1, we can observe a shoulder on the right side for samples with high yttrium content, suggesting local disorder in the W–O bond. The well-separated peaks located at 664 cm−1 and 783 cm−1 in BiYWO6 tend to shift in opposite directions as the yttrium content decreases, leading to partial overlap for x = 3. This trend towards overlapping is in line with the single peak observed when x = −0, i.e. with the higher symmetry of monoclinic Bi2WO6.29
Discussion and conclusion
We characterized the Bi2−xYxWO6 solid solution, both structurally and spectroscopically, in the x range between 0.3 and 1. As in the case of other Bi2−xLnxWO6 solid solutions, the structure was found to be monoclinic, unlike the orthorhombic phase found for Bi2WO6 at room temperature.Using XRPD alone, it is not possible to determine local disorder in the Bi sites, but it could be claimed that there is some preference by Y to remain in the Bi2 site for x > 0.3. For x = 0.3, the composition of the two Bi crystallographic sites is the same (within the experimental error). The Raman spectra confirm the presence of this disorder: all Raman peaks resulting from vibration of the W–O bond are broad and asymmetric, except for the spectrum of Bi1.7Y0.3WO6. In this last case, the peaks are sharper and more symmetric, indicating that the compositional disorder in the bismuth layer as detected by structural analysis is very small but is still large enough to prevent the formation of an orthorhombic structure. Unfortunately, we were unable to synthesize a sample with x < 0.3, which would be required in order to define the transition composition between the orthorhombic phase and the monoclinic phase.
The diffuse reflectance spectra of the powder samples, converted to absorbance spectra by the Kubelka–Munk transformation, exhibited absorption in the visible part of the spectrum at wavelengths lower than 500 nm. Bandgap values were determined by extrapolation of the linear regions in Tauc plots assuming both direct and indirect optical transitions. We estimate that the error in determining the intercept value was roughly ±0.02 eV. The differences in bandgap values between the various samples were found to be relatively insignificant in terms of the overlap with the solar spectrum. It has already been reported that the valence band of bismuth-containing oxides consists mostly of O 2p and Bi 6s orbitals, while the conduction band likely consists of Y 4d and W 5d orbitals. The presence of these orbitals in all members of the studied solid solutions may explain the relative insensitivity of the bandgap to the composition. More evidence for this phenomenon was reported for Bi2−xLuxWO6 (0.4 < x < 1), where the visible-light photoluminescence properties of the solid solution did not change significantly with the composition.30 The monoclinic russellite phase of Bi2WO6 was reported to have an indirect bandgap that approaches 2.50 eV as the calcination temperature is increased.31 The best fit for all of our samples was indeed obtained for the indirect transition, and we therefore speculate that the members of the Bi2−xYxWO6 family are also indirect bandgap semiconductors.
Acknowledgements
The research leading to these results received funding from the European Research Council under the European Union's Seventh Framework Program (FP/2007-2013)/ERC Grant Agreement [number 336077]. The authors also thank the Russel Berrie Nanotechnology Institute, the ISF-ICORE program and the Israel Science Foundation for their financial support. The Raman spectra were collected with the help of Prof. Moshe Paz-Pasternak of the High Pressure Laboratory, Raymond & Beverly Sackler Faculty of Exact Sciences, Tel Aviv University.References
- W. Wei, Y. Dai and B. Huang, J. Phys. Chem. C, 2009, 113, 5658–5663 CAS.
- S. Gupta and V. R. Subramanian, ACS Appl. Mater. Interfaces, 2014, 7, 18597–18608 Search PubMed.
- L. Zhang, W. Wang, L. Zhou and H. Xu, Small, 2007, 3, 1618–1625 CrossRef CAS PubMed.
- J. Yu and A. Kudo, Adv. Funct. Mater., 2006, 16, 2163–2169 CrossRef CAS.
- M. Nussbaum, N. Shaham-Waldmann and Y. Paz, J. Photochem. Photobiol., A, 2014, 290, 11–21 CrossRef CAS.
- J. Luan, B. Pan, Y. Paz, Y. Li, X. Wu and Z. Zou, Phys. Chem. Chem. Phys., 2009, 11, 6289–6298 RSC.
- Z. Zou, J. Ye, R. Abe and H. Arakawa, Catal. Lett., 2000, 68, 235–239 CrossRef CAS.
- L. Zhang and D. Bahnemann, ChemSusChem, 2013, 6, 283–290 CrossRef CAS PubMed.
- X. Zhao, Y. Wu, W. Yao and Y. Zhu, Thin Solid Films, 2007, 515, 4753–4757 CrossRef CAS.
- K. Sayama, A. Nomura and T. Arai, J. Phys. Chem. B, 2006, 110, 11352–11360 CrossRef CAS PubMed.
- X. Jing, B. Huang, X. Yang, J. Wei, Z. Wang, P. Wang, L. Zheng, Z. Xu, H. Liu and X. Wang, Appl. Surf. Sci., 2008, 255, 2651–2654 CrossRef CAS.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- G. N. Rocha, L. F. L. Melo, P. B. A. Fechine, P. V. S. Silva, M. A. S. da Silva and A. S. B. Sombra, in Microwave & Optoelectronics Conference (IMOC), 2011 SBMO/IEEE MTT-S International, IEEE, 2011, pp. 642–646 Search PubMed.
- H. Ait Ahsaine, A. Taoufyq, L. Patout, M. Ezahri, A. Benlhachemi, B. Bakiz, S. Villain, F. Guinneton and J.-R. Gavarri, J. Solid State Chem., 2014, 218, 124–130 CrossRef CAS.
- A. Watanabe, Mater. Res. Bull., 1980, 15, 1473–1477 CrossRef CAS.
- P. S. Berdonosov, D. O. Charkin, V. A. Dolgikh, S. Y. Stefanovich, R. I. Smith and P. Lightfoot, J. Solid State Chem., 2004, 177, 2632–2634 CrossRef CAS.
- P. S. Berdonosov, D. O. Charkin, K. S. Knight, K. E. Johnston, R. J. Goff, V. A. Dolgikh and P. Lightfoot, J. Solid State Chem., 2006, 179, 3437–3444 CrossRef CAS.
- H. Liu, J. Yuan, W. Shangguan and Y. Teraoka, J. Phys. Chem. C, 2008, 112, 8521–8523 CAS.
- N. Santha, P. Koshy, M. T. Sebastian and R. Ratheesh, J. Mater. Sci.: Mater. Electron., 2002, 13, 229–233 CrossRef CAS.
- G. N. Rocha, L. F. L. Melo, M. C. Castro Jr, A. P. Ayala, A. S. de Menezes and P. B. A. Fechine, Mater. Chem. Phys., 2013, 139, 494–499 CrossRef CAS.
- S. Pasternak and Y. Paz, J. Photochem. Photobiol., A, 2016, 318, 14–24 CrossRef CAS.
- A. A. Christy, O. M. Kvalheim and R. A. Velapoldi, Vib. Spectrosc., 1995, 9, 19–27 CrossRef CAS.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi, 1966, 15, 627–637 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- H. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- B. Toby and R. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- K. S. Knight, Mineral. Mag., 1992, 56, 399–409 CAS.
- M. Maczka, J. Hanuza, W. Paraguassu, A. Gomes SouzaFilho, P. Tarso CavalcanteFreire and J. MendesFilho, Appl. Phys. Lett., 2008, 92, 112911 CrossRef.
- A. Kania, A. Niewiadomski and G. Kugel, Phase Transitions, 2013, 86, 290–300 CrossRef CAS.
- H. Ait ahsaine, M. Ezahri, A. Benlhachemi, B. Bakiz, S. Villain, J.-C. Valmalette, F. Guinneton, M. Arab and J.-R. Gavarri, RSC Adv., 2015, 5, 96242–96252 RSC.
- J. Yu, J. Xiong, B. Cheng, Y. Yu and J. Wang, J. Solid State Chem., 2005, 178, 1968–1972 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |