
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynchronised photoreversion of spirooxazine ring opening in thin crystals to uncover ultrafast dynamics†
Khalid M.
Siddiqui‡
ab,
Gastón
Corthey‡
 a,
Stuart A.
Hayes
a,
Andreas
Rossos
a,
Daniel S.
Badali
a,
Rui
Xian
a,
R. Scott
Murphy
c,
Benjamin J.
Whitaker
b and
R. J. Dwayne
Miller
*ade
a,
Stuart A.
Hayes
a,
Andreas
Rossos
a,
Daniel S.
Badali
a,
Rui
Xian
a,
R. Scott
Murphy
c,
Benjamin J.
Whitaker
b and
R. J. Dwayne
Miller
*ade
aMax Planck Institute for the Structure and Dynamics of Matter, Center for Free Electron Laser Science, Luruper Chaussee 149, 22761, Hamburg, Germany. E-mail: dwayne.miller@mpsd.mpg.de
bSchool of Chemistry, University of Leeds, Leeds LS2 9JT, UK
cDepartment of Chemistry and Biochemistry, Research and Innovation Centre, University of Regina, 3737 Wascana Parkway, Regina, SK S4S 0A2, Canada
dHamburg Center for Ultrafast Imaging, University of Hamburg, Luruper Chaussee 149, 22761 Hamburg, Germany
eDepartments of Chemistry and Physics, University of Toronto, 80 St. George Street, Toronto, ON M5S3H6, Canada
First published on 22nd June 2016
Abstract
Reversibility is an important issue that prevents ultrafast studies of chemical reactions in solid state due to product accumulation. Here we present an approach that makes use of spectrally-selected, post-excitation, ultrashort laser pulses to minimise photoproduct build-up, i.e. recover before destroy. We demonstrate that this method enabled us to probe the ultrafast dynamics of the ring opening reaction of spironaphthooxazine thin crystals by means of transient absorption spectroscopy. By extension, this approach should be amenable to other photochromic systems and use with structural probes.
While chemistry happens predominantly in solution, chemical reactions occurring in the solid state can be exploited to fabricate dynamic devices relevant for today's needs.1 Solid-state reactions can be more selective and more efficient than in solution2 and offer more controllable parameters making solids the preferred medium for some industrial applications. On a more fundamental level, solid-state media presents a unique platform for mechanistic studies of chemical reactions using spectroscopic and diffraction techniques which constitutes an important first step towards their control. Ultrafast spectroscopies3,4 employing pump-probe methods have been at the forefront of the techniques that have been applied for this purpose. In a time-resolved pump-probe experiment, a system under study is excited (pumped) by a laser pulse and probed at a user-defined delay later by another pulse (not necessarily a laser pulse) and the cycle is repeated until the dynamics of interest are covered. Such techniques have been widely applied to solutions5 and in the gas phase6,7 but, while many examples exist for studies of phase transitions in molecular crystals,8–11 time-resolved studies of photochemical reactions in solid state are lacking in comparison. This can be ascribed to the requirement that must be met for any ultrafast pump-probe experiment, namely that of reversibility: the system under investigation must return to its initial state between pump laser shots within the period of the repetition rate used. If this condition is not fulfilled, the measured time-resolved signal would contain contributions from species other than the actual ground state of the system. This is easily ensured for measurements in solution or gas phase by flowing the sample, thus replacing it completely between laser shots. On the other hand, for systems in the solid state this is not possible and, therefore, samples are either translated or replaced after a few laser shots. In either case, a large, homogeneous sample area, or many high quality samples must be available for probing, which may not always be trivial or practical. One approach to tackle the reversibility issue is to employ a scheme that uses a dual echelon grating design12 to split the probe pulse into many delayed replicas thereby enabling the dynamics to be covered in a single pump laser shot.13 A limitation of this scheme is that the probe cannot be wavelength-resolved and therefore acquiring data over a broad spectral range requires several experiments to be performed. Despite this limitation, such single-shot experiments should be the choice for completely irreversible samples, i.e. those that are irreparably damaged after a single pump shot. For systems with limited reversibility, multi-shot experiments are possible. But, care must be taken to ensure that no permanent build-up of photoproducts is occurring that may interfere with the transient signal of interest. Jean-Ruel et al.14,15 exploited the bi-directional switching in photochromic systems to present a different approach that made possible ultrafast studies of photochromism in diarylethene crystals. The authors used a 633 nm continuous wave (CW) helium–neon (HeNe) laser to convert the photoproducts back to their initial condition, returning the system to its original state.
Here we extend this approach and use the ring opening reaction of a spirooxazine (SO) to show that reversibility can be improved by implementation of a non-collinear optical parametric amplifier (NOPA).16 The role of the NOPA is to revert the system back to its original state (photoreversion). Compared to CW lasers, the use of a NOPA offers several advantages such as broad bandwidths, higher flux, ultrashort pulse durations and the possibility of precisely synchronising the time delay with respect to the pump and probe pulses. The protocols that will be mentioned have been explicitly developed to resolve the structural dynamics of the ring opening reaction using femtosecond electron diffraction17 and will be reported separately. Information obtained from these experiments would be indispensable in understanding the reaction mechanism in the solid state and would also offer new insights for the design and fabrication of new materials.
Spirooxazines (SOs) and spiropyrans (SPs) have received much attention due to their ability to reversibly switch conformations, and hence their properties, upon absorption of a photon of appropriate wavelength.18–20 Such photochromic compounds have potential applications as photoswitches,21,22 memory devices23,24 and logic gates.25 A common use of SOs is in ophthalmic lenses for protection against ultraviolet (UV) radiation from the sun.26 SOs consist of an indoline ring joined to an orthogonal oxazine ring by a sp3 hybridised spiro carbon. A well accepted mechanism of SO photochromism is that upon UV irradiation, the bond between the spiro carbon and the oxygen in the oxazine ring is broken to produce a ring-opened isomer. Isomerisation involves rotation of the oxazine unit by almost 90 degrees relative to its position in the closed-ring form. The resulting planar structure is known as merocyanine (MC). Due to an extended π system and hence electron delocalisation, MC has characteristic absorption in the visible region of the spectrum. MC returns to the SO state either thermally or photochemically (Fig. 1).
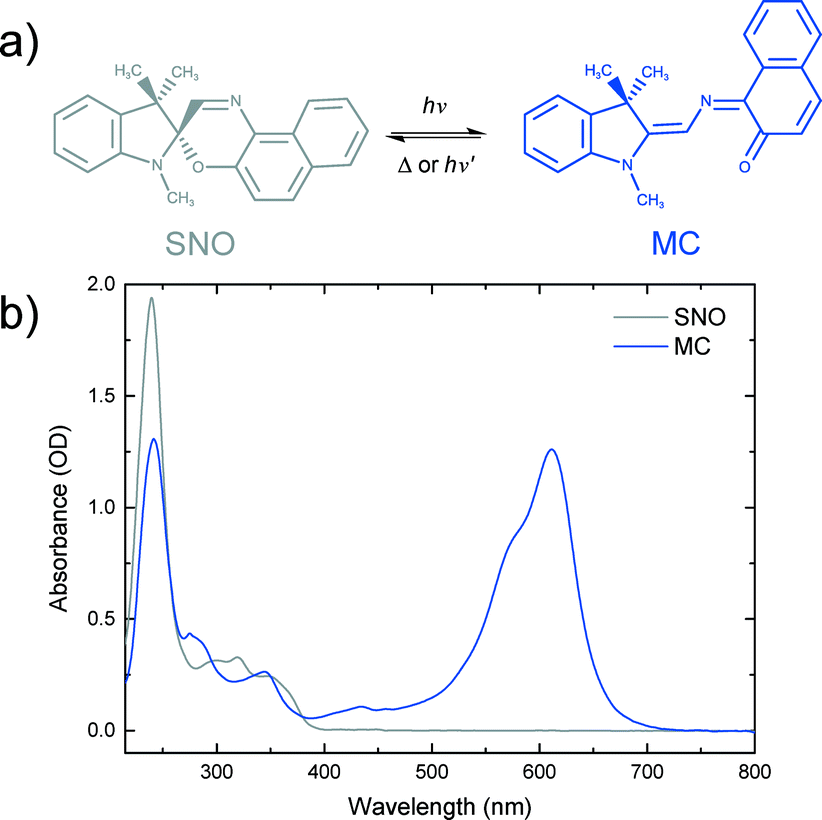 | ||
| Fig. 1 a) Photochromic reaction of spironaphthooxazine (SNO). b) Steady-state spectra of a 0.5 mM SNO solution in ethanol and merocyanine (MC) produced under UV excitation of the SNO solution. | ||
Photochemistry of spironaphthooxazine (SNO) has been investigated experimentally in different media27–31 and also theoretically.32–34 From solution phase studies, it is known that the photochromic reaction of SNO occurs via the excited singlet state manifold35 leading to a very rapid process in which the so-called intermediate X, having the orthogonal parent geometry, is formed and isomerisation to the trans-merocyanine is completed in less than 10 ps.36 While the photochromic reaction always takes place in solution and amorphous films, the reaction may not be possible in crystalline form due to spatial confinement and the large structural change associated with the reaction. To explore this, Suzuki and coworkers37–39 carried out detailed studies of the photochromic reaction of SNO in microcrystalline state using ultrafast diffuse reflectance spectroscopy. They performed the experiments exciting the sample with 390 nm light and changing the sample position after every 50 pump shots to avoid accumulating artefactual signals from damaged sample areas. Under low excitation conditions (fluence <1 mJ cm−2), they reported rapid C–O bond breaking, but isomerisation leading to the MC form was not observed. Instead, the ring-opened transient returned to the original spiro-form on a nanosecond time scale. However, at higher fluences, they noted the formation of the MC photoproduct.37 They later proposed a cooperative model39 to explain this observation in which a high concentration of the ring-opened products was attained by intense femtosecond laser excitation and the interaction between the open form with the excited state consequently created local free volumes within the crystalline environment making it possible for the molecules to undergo isomerisation to MC.
We carried out transient absorption spectroscopy experiments on crystals of SNO with a thickness of 500 nm, in transmission mode, using a home-built spectrometer (see the ESI† file for preparation methods and details of the experimental setup). A fraction of the fundamental output of a Ti:Sapphire laser (800 nm, 200 μJ, 40 fs) was frequency-tripled (266 nm, 1.4 mJ cm−2) and used as the pump while a single stable filament of white light supercontinuum (380–900 nm) generated by focussing 2 μJ of the 800 nm light in water was used as the probe. The optical path length of the probe with respect to the pump pulse was varied using an optical delay line. For all of the experiments discussed here, the repetition rate of the pump was fixed to 31.25 Hz and 250 Hz for the probe. The pump repetition frequency was chosen to ensure no damage to the sample occurred (thermal or other) while maintaining relatively fast data collection. The spectra obtained with each probe pulse were recorded by a low-noise charge-coupled device (CCD). We performed two separate experiments in order to compare the approach that we introduce with the more conventional one. The following acquisition scheme was adapted for the experiments. For each time delay, 100 pump-probe cycles were acquired. For each pump pulse, 8 spectra were recorded and labelled according to their distance to the immediately preceding pump pulse, as shown in Fig. 2. For instance, pulse number 6 corresponded to the spectrum measured 24 ms after the pump. All the pulses with the same label were averaged and the transient absorption spectra were calculated as:
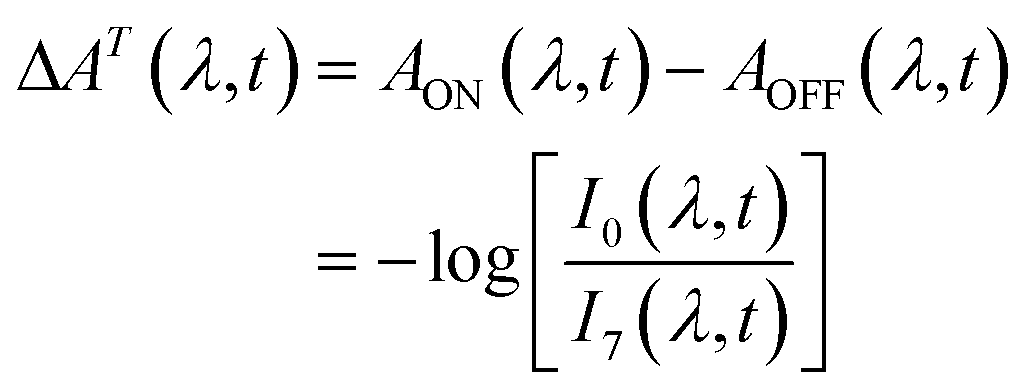 | (1) |
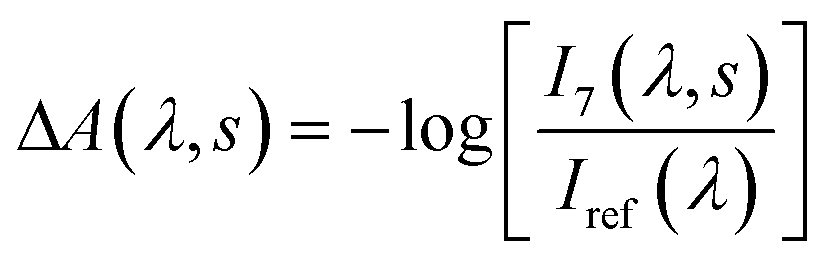 | (2) |
Plots corresponding to ΔA(λ,s) for the experiment using only the pump and probe beams are shown in Fig. 3a. It is obvious from these plots that the absorption in the whole probe region grows with the number of excitation shots hitting the sample. The absorption profile after 4000 shots resembles that of the MC absorption in solution (Fig. 1) with the peak slightly red shifted, typical of species in the solid state. This suggests that MC is being formed in the crystal and its concentration increases reaching a steady-state after about 8000 pump pulses. The number of shots leading to a steady-state concentration of species was heavily dependent on the experimental conditions, with the rate of product formation increasing with higher fluences or repetition rates. Indeed, with repetition rates of the pump laser as low as 2.5 Hz, the build-up of MC was still observed albeit with much slower rates. The build-up manifested itself as a bleach band with the maximum at about 600 nm in the transient absorption spectra (see the ESI† file) obtained under these conditions. We interpreted the bleach as the excitation of the trapped MC caused by the 266 nm pump. After the experiment, colouration of the probed region was detectable by eye (see the ESI† file).
 | ||
| Fig. 2 Timing diagram for the acquisition scheme used in the transient absorption experiments. | ||
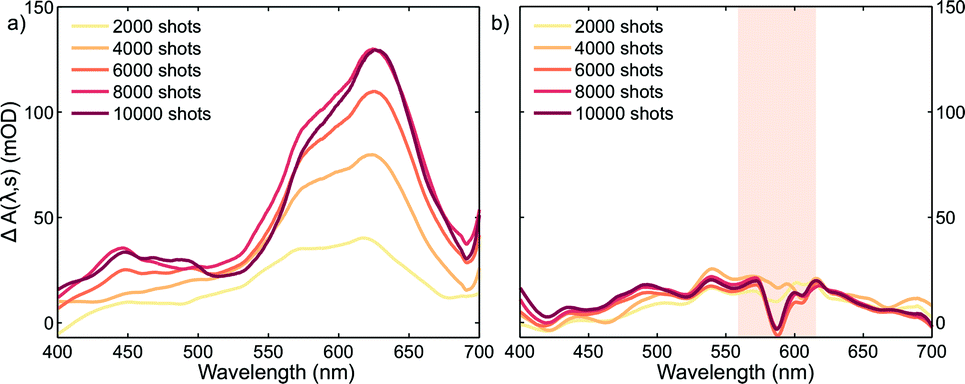 | ||
| Fig. 3 Change in the absorption spectra of SNO thin crystals measured 28 ms after excitation for different number of 266 nm laser shots (1.4 mJ cm−2) a) without using the NOPA photoreversion pulse and b) with the photoreversion pulse (2.8 mJ cm−2). The spectral region covered by the NOPA is shaded. In this region, the scattered light coming from the NOPA beam reaching the detector produced fluctuations increasing the noise significantly. | ||
It is evident that the accumulation of the photoproducts was preventing us from performing experiments under reversible conditions and a new strategy was needed. Therefore, we carried out a three-beam experiment where we used the output of a NOPA with the wavelength tuned to the maximum of the photoproduct absorption (see Fig. 3a), operating at 1 kHz and delayed by ∼10 ns with respect to the probe pulse as our third beam to switch the MC produced back to the spiro form and thus halt the build-up of the photoproduct. For this purpose, 32 NOPA pulses (λ = 600 ± 25 nm, 2.8 mJ cm−2) hit the sample after each UV excitation pulse as shown in Fig. 2. The results of ΔA(λ,s) for this experiment are presented in Fig. 3b showing clearly that no significant amount of MC remained even after 104 UV laser shots. This result indicates that the sample was returning to the original state before each pump-probe sequence. It is also noted that NOPA excitation did not have any adverse effect on SNO by the virtue that the sample is transparent in the region of the NOPA spectrum and the peak intensity used (<10 GW cm−2) was below the multiphoton damage threshold.
Thereafter, we performed transient absorption experiments with the NOPA beam and compared them with the solution-phase photochromic reaction, measured without using the NOPA but by flowing the sample in order to renew it after every pump-probe cycle. For the case of crystals, transient spectra were measured for three different regions on the sample and then averaged. The results are shown in Fig. 4a and b as 2D maps. Spectral traces corresponding to selected time delays are also plotted (Fig. 4c and d). Clearly, MC was formed in the solution, as evidenced by the intense growing absorption band at 550 nm, but the signature for its production was absent from the crystal data for the delay range covered. Instead, a strong decaying absorption feature centered at about 500 nm was observed. These results are similar to the ones from Suzuki et al. with 390 nm pump,38 indicating that excitation with 266 nm light also produces a ring-opened transient species. The ability to overcome the sample damage by exciting with broadband 600 nm light also supports their model for the damage mechanism in SO crystals, whereby a large local free volume in crystal – necessary for spatial reorganisation during isomerisation to planar MC – is created due to the build-up of intermediate species which, once removed via photoreversion, significantly minimises sample degradation.
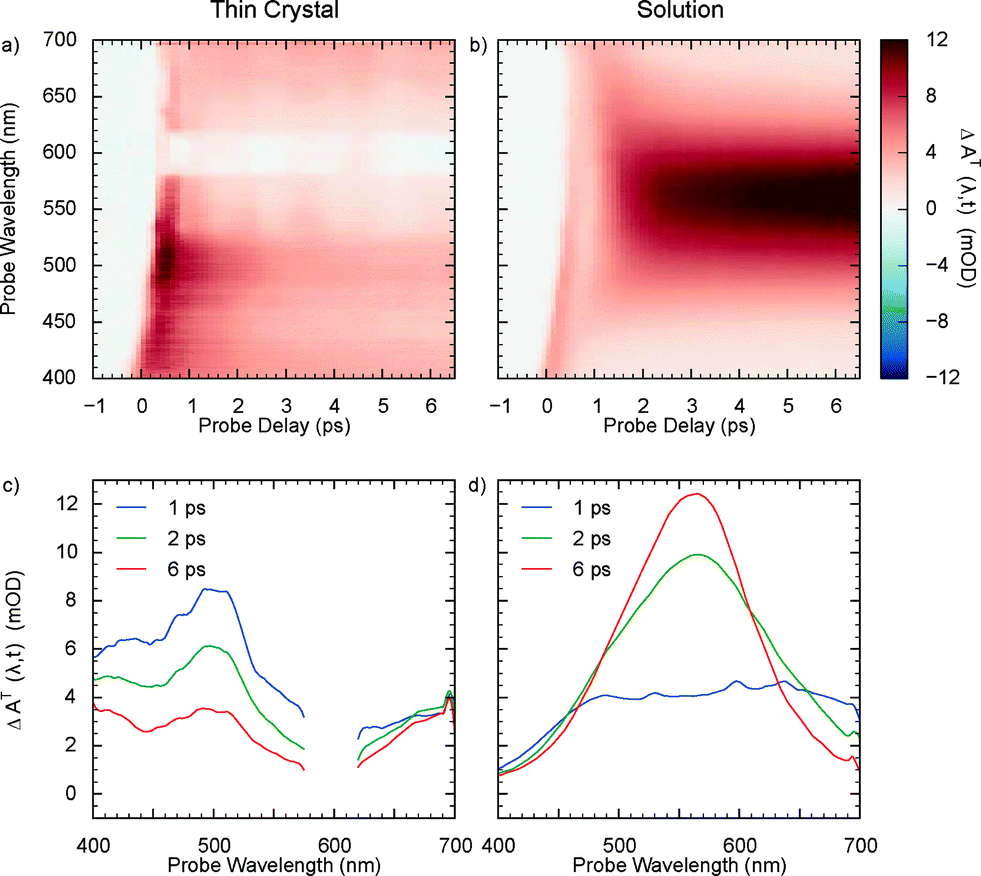 | ||
| Fig. 4 Transient absorption spectra of a) a SNO thin crystal using the photoreversion approach and b) 20 mM SNO solution in hexane using only pump (0.46 mJ cm−2) and probe beams and flowing the sample. The plots were not corrected for probe dispersion. c) and d) Traces along the wavelength for different probe delays for thin crystal and solution, respectively. The region between 575 and 625 nm in the thin crystal is affected by fluctuations in the power of the NOPA beam and is masked. | ||
In summary, we improved the reversibility of SNO photoswitching in the crystalline state by using a NOPA to revert the photoproducts back to the initial state. Due to the ultrashort pulse duration of the NOPA and the possibility to synchronise it to the pump pulse, this process can be driven faster than the deformation of the crystal which requires an accumulated propagation of the stress to strain, that takes considerably longer than nanoseconds to manifest itself. This relatively simple approach is expected to be quite generic for photochromic systems and solves the reversibility problem and the need for using one shot data collection methods to follow reaction dynamics in the solid state. It is equally notable that this method also permits the prospect of probing the dynamics of the back reactions thereby developing a complete picture of the bi-directional processes. With this approach, it is possible to get complete information from well-defined areas of the sample, circumventing the need for translation. Moreover, if the ensemble dynamics from different spots is required, many cycles can be run at each spot increasing the signal-to-noise ratio dramatically, compared to single-shot experiments. This progress in reducing the photodegradation in SO also opens up the possibility of applying ultrafast diffraction techniques that would enable direct probing of the transient intermediate structures and aid with the understanding of the degradation mechanism. We hope that this approach will enable a wider class of studies involving solid-state photoreversible systems on ultrafast time scales.
Acknowledgements
This work was funded by the Max Planck Society in collaboration with the Centre for Free Electron Laser Science and the Hamburg Centre for Ultrafast Imaging. K. M. S. acknowledges the University of Leeds for partial funding of studentship. G. C. thanks the Alexander von Humboldt Foundation for support. D. S. B. acknowledges support from the Natural Sciences and Engineering Research Council of Canada. We thank Michal A. Kochman for useful discussions.References
- L. E. Hatcher and P. R. Raithby, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1448–1456 CrossRef PubMed.
- F. Toda, Acc. Chem. Res., 1995, 28, 480–486 CrossRef.
- E. T. Nibbering, H. Fidder and E. Pines, Annu. Rev. Phys. Chem., 2005, 56, 337–367 CrossRef CAS PubMed.
- A. H. Zewail, J. Phys. Chem. A, 2000, 104, 5660–5694 CrossRef CAS.
- A. Rosspeintner, B. Lang and E. Vauthey, Annu. Rev. Phys. Chem., 2013, 64, 247–271 CrossRef CAS PubMed.
- T. Suzuki and B. J. Whitaker, in Imaging in Molecular Dynamics, ed. B. J. Whitaker, Cambridge University Press, 2003, ch. 7, pp. 165–186 Search PubMed.
- A. Stolow, A. E. Bragg and D. M. Neumark, Chem. Rev., 2004, 104, 1719–1758 CrossRef CAS PubMed.
- S. Koshihara, Y. Tokura, K. Takeda and T. Koda, Phys. Rev. Lett., 1992, 68, 1148–1151 CrossRef CAS PubMed.
- K. Onda, S. Ogihara, K. Yonemitsu, N. Maeshima, T. Ishikawa, Y. Okimoto, X. Shao, Y. Nakano, H. Yamochi, G. Saito and S.-y. Koshihara, Phys. Rev. Lett., 2008, 101, 067403 CrossRef PubMed.
- E. Collet, N. Moisan, C. Baldé, R. Bertoni, E. Trzop, C. Laulhé, M. Lorenc, M. Servol, H. Cailleau, A. Tissot, M.-L. Boillot, T. Graber, R. Henning, P. Coppens and M. B.-L. Cointe, Phys. Chem. Chem. Phys., 2012, 14, 6192 RSC.
- T. Ishikawa, S. A. Hayes, S. Keskin, G. Corthey, M. Hada, K. Pichugin, A. Marx, J. Hirscht, K. Shionuma, K. Onda, Y. Okimoto, S. Y. Koshihara, T. Yamamoto, H. Cui, M. Nomura, Y. Oshima, M. Abdel-Jawad, R. Kato and R. J. D. Miller, Science, 2015, 350, 1501–1505 CrossRef CAS PubMed.
- T. Shin, J. W. Wolfson, S. W. Teitelbaum, M. Kandyla and K. A. Nelson, Rev. Sci. Instrum., 2014, 85, 083115 CrossRef PubMed.
- P. R. Poulin and K. A. Nelson, Science, 2006, 313, 1756–1760 CrossRef CAS PubMed.
- H. Jean-Ruel, R. R. Cooney, M. Gao, C. Lu, M. A. Kochman, C. A. Morrison and R. J. D. Miller, J. Phys. Chem. A, 2011, 115, 13158–13168 CrossRef CAS PubMed.
- H. Jean-Ruel, M. Gao, M. A. Kochman, C. Lu, L. C. Liu, R. R. Cooney, C. A. Morrison and R. J. D. Miller, J. Phys. Chem. B, 2013, 117, 15894–15902 CrossRef CAS PubMed.
- G. Cerullo and S. D. Silvestri, Rev. Sci. Instrum., 2003, 74, 1–18 CrossRef CAS.
- R. J. D. Miller, Science, 2014, 343, 1108–1116 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1683–1684 CrossRef CAS PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- Y. Prostota, P. J. Coelho, J. Pina and J. Seixas de Melo, Photochem. Photobiol., 2011, 10, 1346 RSC.
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2012, 25, 378–399 CrossRef PubMed.
- D. A. Parthenopoulos and P. M. Rentzepis, Science, 1989, 245, 843–845 CrossRef CAS PubMed.
- G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741–1754 CrossRef CAS PubMed.
- G. Jiang, Y. Song, X. Guo, D. Zhang and D. Zhu, Adv. Mater., 2008, 20, 2888–2898 CrossRef CAS.
- K. Tamura, S. Mitsuuchi, K. Wada and A. Takata, Photochromic lens, US Patent App. 13/667, 183, 2014 Search PubMed.
- S. Aramaki and G. H. Atkinson, Chem. Phys. Lett., 1990, 170, 181–186 CrossRef CAS.
- L. Poisson, K. D. Raffael, B. Soep, J.-M. Mestdagh and G. Buntinx, J. Am. Chem. Soc., 2006, 128, 3169–3178 CrossRef CAS PubMed.
- N. Tamai and H. Masuhara, Chem. Phys. Lett., 1992, 191, 189–194 CrossRef CAS.
- R. S. S. Kumar, L. Lüer, D. Polli, M. Garbugli and G. Lanzani, Opt. Mater. Express, 2011, 1, 293 CrossRef CAS.
- J. Harada, Y. Kawazoe and K. Ogawa, Chem. Commun., 2010, 46, 2593 RSC.
- I. Gómez, M. Reguero and M. A. Robb, J. Phys. Chem. A, 2006, 110, 3986–3991 CrossRef PubMed.
- F. Liu and K. Morokuma, J. Am. Chem. Soc., 2013, 135, 10693–10702 CrossRef CAS PubMed.
- C. Walter, S. Ruetzel, M. Diekmann, P. Nuernberger, T. Brixner and B. Engels, J. Chem. Phys., 2014, 140, 224311 CrossRef PubMed.
- C. Lenoble and R. S. Becker, J. Photochem., 1986, 34, 83–88 CrossRef CAS.
- N. P. Ernsting and T. Arthen-Engeland, J. Phys. Chem, 1991, 95, 5502–5509 CrossRef CAS.
- T. A. Mototsugu Suzuki and H. Masuhara, Phys. Chem. Chem. Phys., 2002, 4, 185–192 RSC.
- K. T. Mototsugu Suzuki, T. Asahi and H. Masuhara, Chem. Phys. Lett., 2003, 368, 384–392 CrossRef.
- M. Suzuki, T. Asahi and H. Masuhara, ChemPhysChem, 2005, 6, 2396–2403 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and complementary results are included. See DOI: 10.1039/c6ce01049k |
| ‡ K. M. Siddiqui and G. Corthey contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |