DOI:
10.1039/C6CE01039C
(Paper)
CrystEngComm, 2016,
18, 5333-5337
Anion hydrogen bonding from a ‘revealed’ urea ligand†
Received
3rd May 2016
, Accepted 28th May 2016
First published on 30th May 2016
Abstract
Hydrogen bonding from a urea group to hydrogen bond acceptor anions can adopt either R12(6) or R22(8) motifs depending on the proximity of hydrogen bond acceptor atoms. However, for the sterically bulky and weaker hydrogen bond acceptor triflate anion, hydrogen bond acceptor polymorphism is observed.
Introduction
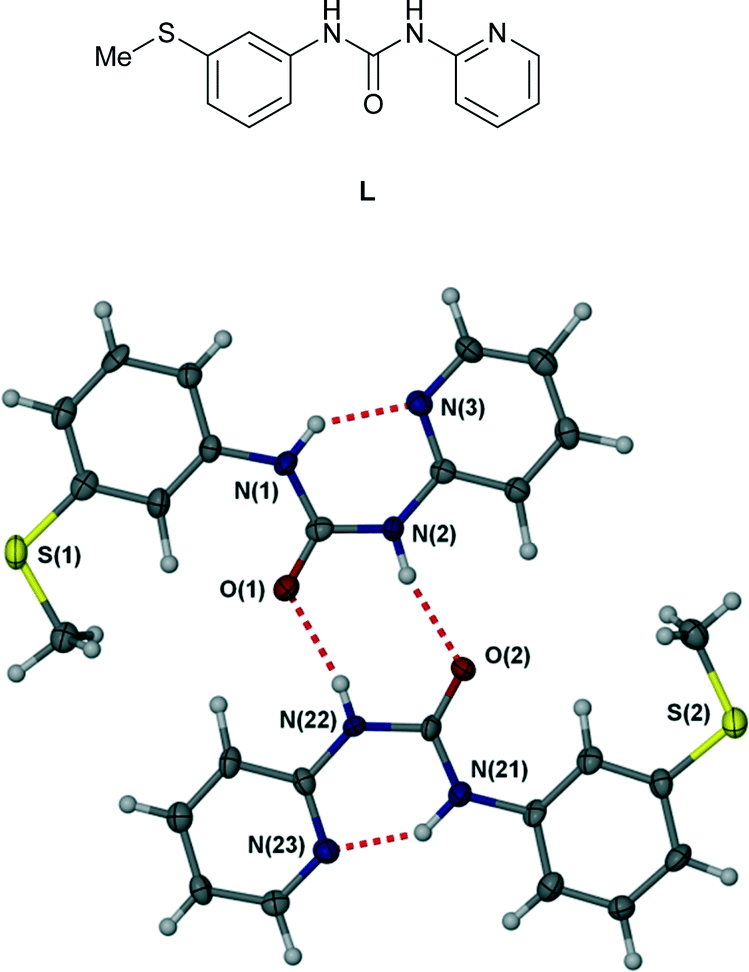
The scope, geometry, strength and directionality of hydrogen bonding interactions to metal-coordinated anions has generated considerable interest both on a fundamental level1–8 and in applications such as extraction of halometalate anions in mining processes.9–11 The directionality, steric constraints and polarization of the M–X bond (in comparison to a spherical halide anion, for example) places considerable constraints on complexed anion hydrogen bonded geometries and hence must be understood in order to inform the design of metal salt ion pair binding hosts. Seminal contributions in the area were the work of Gillon, Orpen and coworkers4,12,13 and of Brammer and coworkers2,5,14,15 who showed, for example, that hydrogen bonding from a single NH group of a pyridinium unit to a cis-MCl2 fragment occurs to the centroid of the two chloride ligands. Complexed anion binding by a double hydrogen bond donor group such as the urea functionality can invert this behaviour with both NH donors interacting with a single acceptor atom (the R12(6) motif in graph set nomenclature16,17) being particularly common.1 In this work we report a series of copper(II) complexes of ligand L exhibiting hydrogen bonding to anions. In the free state, ligand L, while highly polymorphic, consistently adopts an intramolecular R22(8) hydrogen bonded ring motif18 (Fig. 1) and hence the urea functionality is unavailable for anion hydrogen bonding. We have previously reported the formation of a range of Ag(I) complexes of ligand L in which the proximity of the pyridyl nitrogen atom and urea group results in the bonding of contact ion pairs.19 The coordination mode of copper(II) is very different however and results in significantly different anion binding behaviour.
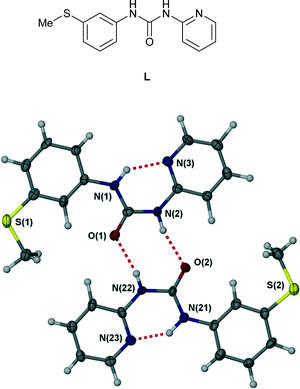 |
| | Fig. 1 Intermolecular R22(8) hydrogen bonded ring motif in free ligand L.18 | |
Results and discussion
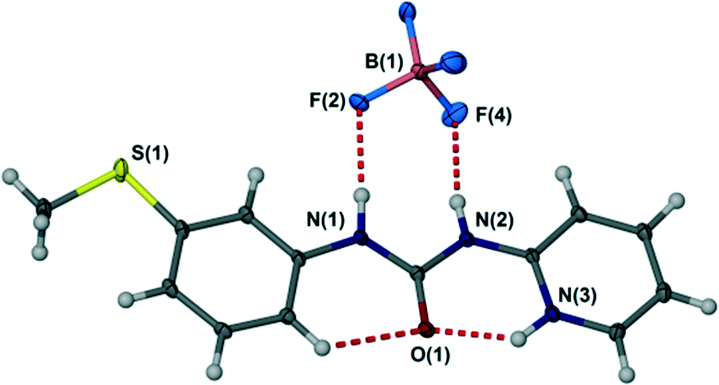
The crystal structure of the free ligand L is based on an R22(8) hydrogen bonded ring with an unusual syn, anti conformation of the aryl groups of the N,N′-diarylurea ligand relative to the urea carbonyl. This feature arises from the formation of a strong intramolecular hydrogen bond to the pyridyl nitrogen atom. Rendering this acceptor unavailable is expected to change the ligand conformation to reveal a more conventional syn, syn conformation predisposed to the formation of urea α-tape or R12(6) type hydrogen bonds to the urea NH groups in which both NH donors are aligned in the same direction.20 This behaviour is exemplified by the single crystal X-ray structure of the protonated ligand LH+BF4− (see Experimental section) in which the syn, syn ligand displays NH+⋯O and CH⋯O hydrogen bonding to the urea carbonyl group and hydrogen bonding from both urea NH functionalities to the BF4− anion giving an R22(8) anion hydrogen bonded motif, Fig. 2.
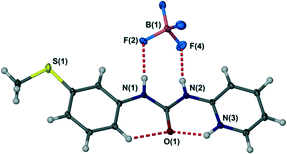 |
| | Fig. 2
Syn, syn conformation of protonated L in LH+BF4− and the R22(8) hydrogen bond motif to the BF4− anion. Selected hydrogen bonded distances (Å): N(1)⋯F(2) 2.870(2), N(2)⋯F(4) 2.824(2), N(3)⋯O(1) 2.645(2), C(5)⋯O(1) 2.940(2). | |
Reaction of ligand L with a range of copper(II) salts in polar solvent mixtures results in crystals of the following complexes that were analysed by X-ray crystallography: [{Cu(L)(μ-Cl)Cl}2] (1), [Cu(L)2Br2] (2), [Cu(L)2(NO3)2] (3) and [Cu(L)2(CF3SO3)2] (4). The triflate complex 4 exists in two polymorphic modifications, forms A and B, depending on the crystallization solvent. In each case 1–4 the copper(II) ion is chelated by the pyridyl nitrogen and urea carbonyl oxygen atoms to give a 6-membered chelate ring. Cu–O distances range from 1.95–1.98 Å and Cu–N 2.00–2.04 Å, consistent with related structures such as [Cu(N,N′-di-2-pyridylurea)2(NO3)2],21 [Cu(1-benzyl-3-(2-pyridinyl)urea)2Cl2] (ref. 22) and a series of dinuclear stacked analogues reported by us previously.23 Complexes 2–4 are all closely related (albeit not isomorphous) mononuclear 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 M:L complexes, exhibiting a Jahn–Teller distorted octahedral copper(II) centre with long axial bonds to the anionic ligands. In contrast the chloride complex 1 is a 1:1 complex exhibiting a chloride bridged dimeric structure, making the anionic ligands somewhat less accessible for hydrogen bonding. Since all complexes were formed from similar concentrations of mixtures of 1:1 stoichiometry the different stoichiometry of product in the case of 1 is likely to arise from the better ligating ability of chloride for copper(II).
:2 M:L complexes, exhibiting a Jahn–Teller distorted octahedral copper(II) centre with long axial bonds to the anionic ligands. In contrast the chloride complex 1 is a 1:1 complex exhibiting a chloride bridged dimeric structure, making the anionic ligands somewhat less accessible for hydrogen bonding. Since all complexes were formed from similar concentrations of mixtures of 1:1 stoichiometry the different stoichiometry of product in the case of 1 is likely to arise from the better ligating ability of chloride for copper(II).
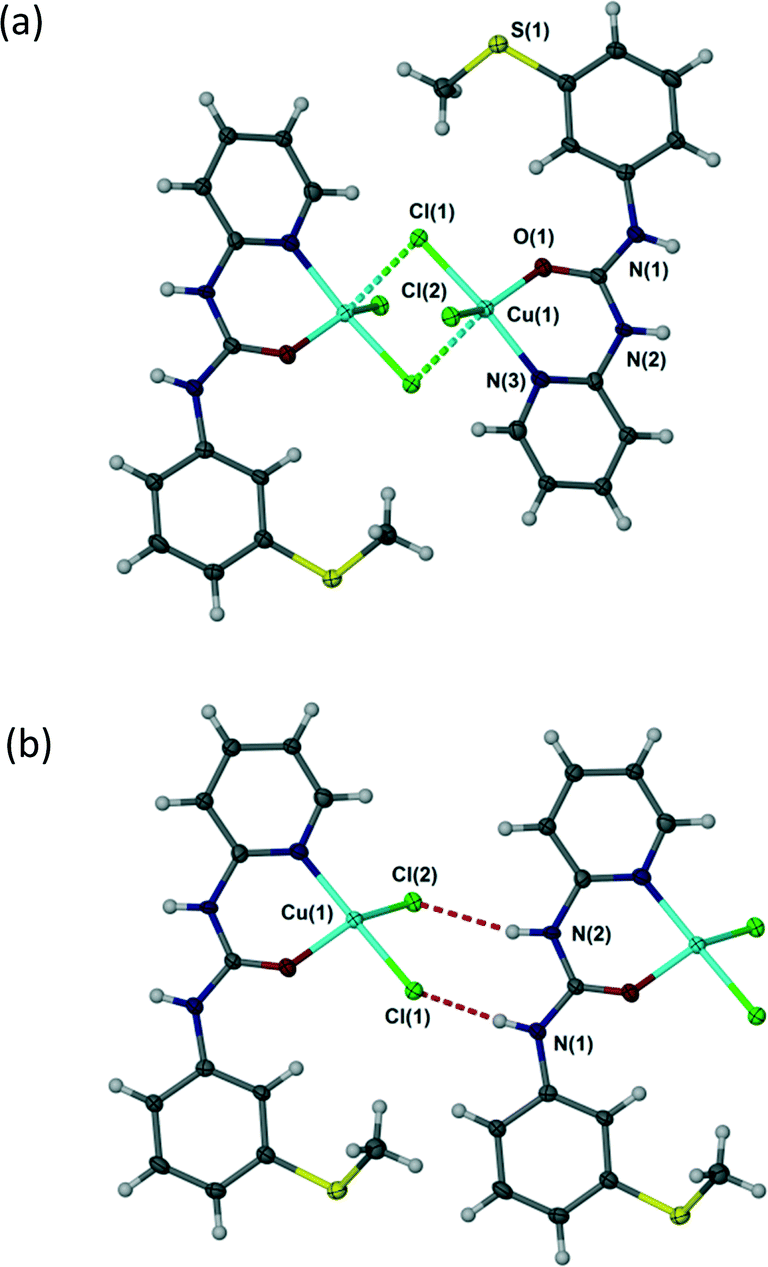
In all complexes 1–4 the urea aryl substituents adopt a syn, syn conformation and hence the urea NH groups point away from the metal centre and are co-aligned. Despite the bridged structure, complex 1 exhibits an R22(8) hydrogen bonding interaction with both chloride ligands acting as hydrogen bond acceptors, Fig. 3. This interaction mode contrasts with a range of trans dihalide complexes of the related ligand N,N′-p-tolyl-3-pyridylurea which tends to form R12(6) interactions as a result of the exposed nature of the terminal halide ligands. Complex 1 is similar, however, to the cis-dihalide complex [Zn(N,N′-p-tolyl-3-pyridylurea)2Cl2] and to LH+BF4− (Fig. 2).1
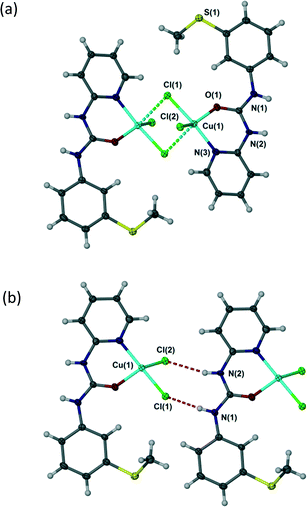 |
| | Fig. 3 (a) Dimeric structure of [{Cu(L)(μ-Cl)Cl}2] (1) – long axial Cu–Cl interactions shown as dotted lines. (b) R22(8) hydrogen bond motif in 1 (one half of the chloride-bridged dimer is shown for clarity). Selected distances (Å): N(1)⋯Cl(1) 3.156(6), N(2)⋯Cl(2) 3.371(6). | |
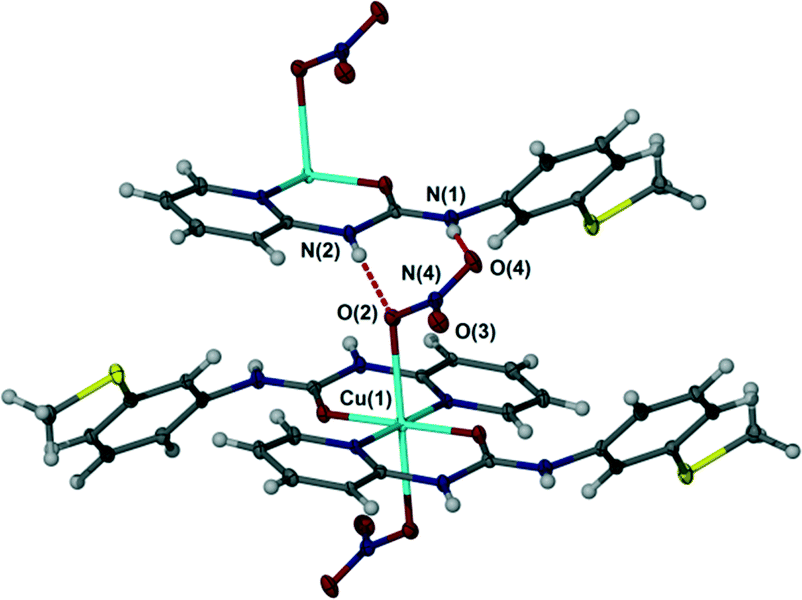
In contrast to the chloride complex, the bromide complex 2 exhibits long bonds to mutually trans axial bromide ligands and as a result hydrogen bonding is of the R12(6) type in which each bromide anion acts as an acceptor to two NH hydrogen bond donors of similar length, Fig. 4, in a way that is related to the N,N′-p-tolyl-3-pyridylurea analogues.1 The nitrate salt 3 also exhibits long, trans diaxial coordination of the anions however because the anion itself is polyatomic with adjacent pairs of oxygen atoms the hydrogen bonded geometry is R22(8) with a single syn, syn urea group interacting with an ‘NO2’ group comprising the coordinated oxygen atom O(2) and one uncoordinated atom O(4). The remaining nitrate oxygen atom O(3) does not form any short interactions and the closest contact is to a pyridyl CH atom (Fig. 5).
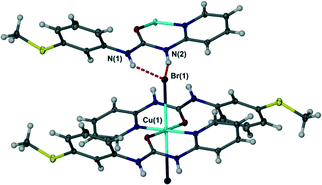 |
| | Fig. 4 R12(6) motif in bromide complex 2. Selected distances (Å): N(1)⋯Br(1) 3.3399(16), N(2)⋯Br(1) 3.2871(15). | |
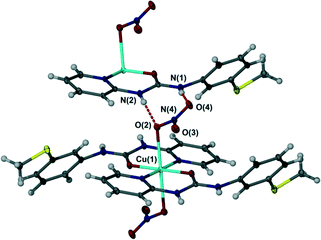 |
| | Fig. 5 R22(8) hydrogen bond motif in nitrate complex 3. Selected distances (Å): N(1)⋯O(2) 2.817(3), N(2)⋯O(4) 2.810(3). | |
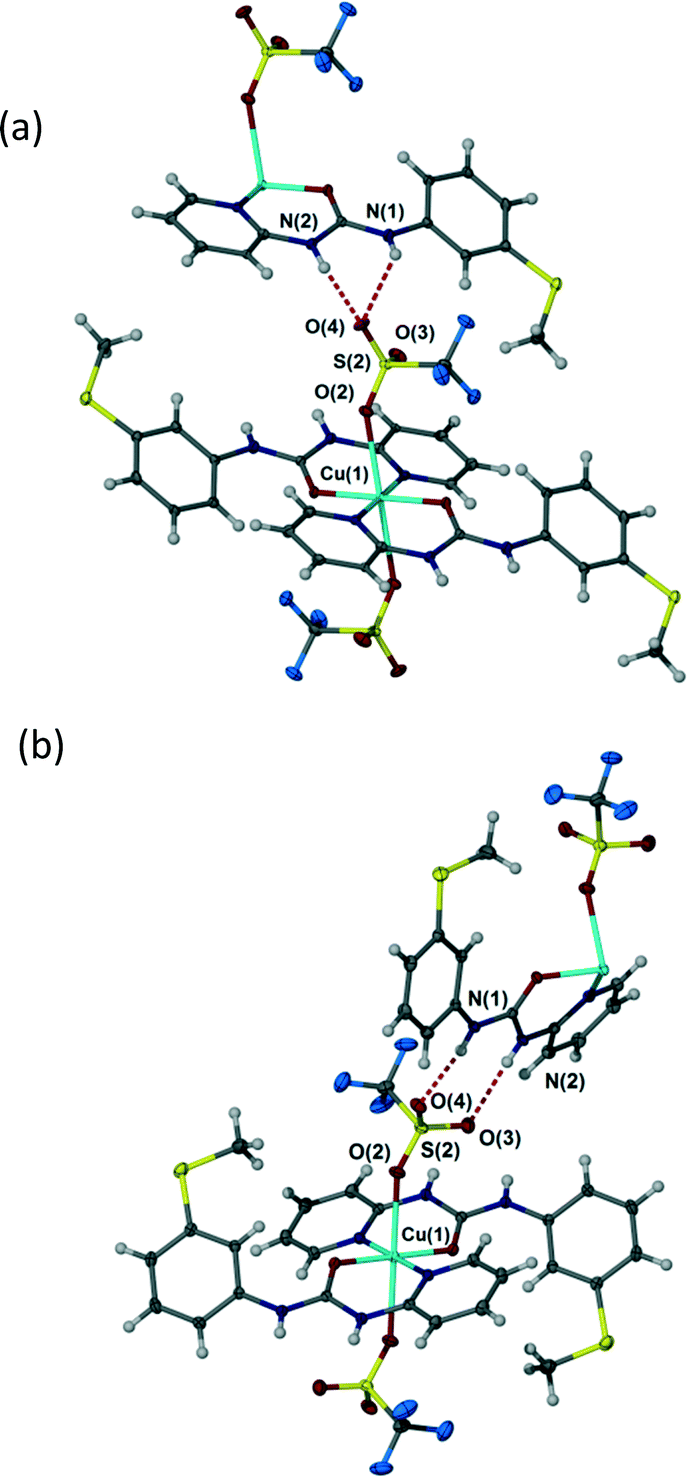
Depending on crystallization conditions (see Experimental section) the triflate complex 4 exists in two polymorphic modifications, A and B. Forms A and B differ in a very interesting way in the context of the preceding discussion. The coordination complex 4 itself in both polymorphs is a 1:2 M:L species with unidentate trans diaxial triflate anions. However, in form A the anion adopts a highly unsymmetrical R12(6) hydrogen bonding interaction with an adjacent urea group with NH⋯O distances of 2.89 and 3.23 Å (Fig. 6a) while in Form B the polyatomic anion forms an R22(8) interaction involving symmetrical hydrogen bonding to the two uncoordinated sulfonyl oxygen atoms (Fig. 6b), hydrogen bonded distances 2.83 and 2.98 Å. The fact that the triflate salt thus does not seem to exhibit a strong preference for a particular hydrogen bonding geometry may be attributed to the relatively diffuse negative charge of the –SO3− group and the steric constraints of the –CF3 group which are absent in the nitrate analogue.
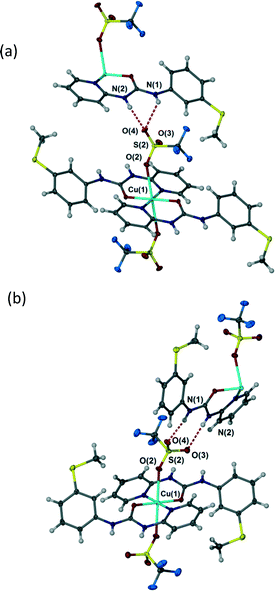 |
| | Fig. 6 Hydrogen bonding in triflate complex 4 (a) highly unsymmetrical R12(6) interaction in Form A, selected distances (Å): N(1)⋯O(4) 3.255(5), N(2)⋯O(4) 2.892(5). (b) R22(8) interaction in Form B. Selected distances (Å): N(1)⋯O(4) 2.825(2), N(2)⋯O(3) 2.982(3). | |
Conclusions
Coordination to copper(II) forces a syn, syn conformation for the N,N′-diaryl urea ligand and allows the mode of interaction to coordinated anions to be explored. In the case of strong hydrogen bond acceptors in which two acceptor atoms are adjacent to one another as in the “CuCL2” unit of complex 1 and in nitrate complex 3 the R22(8) motif is formed. Mononuclear anions result in an R12(6) arrangement as in the bromide complex 2. For the weaker acceptor, sterically bulky triflate anion both arrangements appear to be almost equienergetic resulting in anion hydrogen bond acceptor polymorphism in complex 4.
Experimental
X-ray crystallography
Suitable single crystals were grown by slow evaporation. Crystallographic measurements were carried out on a Rigaku R-AXIS Spider IP diffractometer (compounds 1 and 2) and on a Bruker SMART CCD 6000 diffractometer (all other compounds) using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) at the temperature of 120(2) K, maintained by open flow N2 Cryostream (Oxford Cryosystems) cryostates. Structures were solved using direct methods with and refined by full-matrix least squares on F2 for all data using SHELXTL and OLEX2 software.24,25 All non-hydrogen atoms were refined with anisotropic displacement parameters; H-atoms were located on the difference map and refined isotropically. Molecular graphics were produced using the programs X-Seed26 and POV-Ray.27 Crystal data and parameters of refinement are listed in Table 1. Crystallographic data for the structures has been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC-1476704–1476709.
Table 1 Crystal data and structure refinement data
| Compound |
LHBF4 |
1
|
2
|
3
|
4
A
|
4
B
|
| Empirical formula |
C13H14SON3BF4 |
C13H13N3OCuCl2S |
C26H26N6O2CuBr2S2 |
C26H26N8CuS2O8 |
Cu(C13H13N3OS)2(CF3SO3)2 |
| Formula weight |
347.14 |
393.76 |
742.01 |
706.21 |
880.33 |
| Temperature |
120(2) |
120(2) |
120(2) |
120(2) |
120(2) |
120(2) |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
Monoclinic |
Triclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P21/c |
P |
P21/n |
|
a/Å |
6.6282(3) |
7.7680(16) |
7.8394(16) |
8.9556(3) |
8.2365(3) |
12.6428(3) |
|
b/Å |
10.3741(5) |
9.1578(18) |
8.1920(16) |
21.6859(9) |
9.6360(3) |
9.0531(2) |
|
c/Å |
11.1178(5) |
10.466(2) |
12.372(3) |
7.1433(4) |
11.5943(4) |
15.2733(4) |
|
α/° |
96.700(10) |
90.77(3) |
104.40(3) |
90.00 |
110.59(2) |
90.00 |
|
β/° |
92.305(10) |
98.75(3) |
92.42(3) |
98.90(1) |
97.00(2) |
99.138(10) |
|
γ/° |
99.927(10) |
95.52(3) |
116.28(3) |
90.00 |
103.85(2) |
90.00 |
| Volume/Å3 |
746.47(6) |
732.1(3) |
679.3(2) |
1370.6(1) |
814.57(5) |
1725.94(7) |
|
Z
|
2 |
2 |
1 |
2 |
1 |
2 |
|
ρ
calc mg mm−3 |
1.544 |
1.786 |
1.814 |
1.711 |
1.795 |
1.694 |
|
μ, m mm−1 |
0.266 |
1.999 |
3.937 |
1.018 |
1.023 |
0.966 |
|
F(000) |
356 |
398 |
371 |
726 |
447 |
894 |
| Reflections collected |
9369 |
9240 |
17441 |
13846 |
10725 |
23373 |
| Independent reflections/Rint |
3948/0.0420 |
2846/0.0990 |
4319/0.0336 |
3301/0.0534 |
4645/0.0374 |
5028/0.0524 |
| Data/restrains/parameters |
3948/0/264 |
2846/0/192 |
4319/0/179 |
3301/0/253 |
4645/0/242 |
5028/0/293 |
| Goodness-of-fit on F2 |
1.099 |
1.036 |
1.021 |
1.063 |
0.874 |
1.020 |
|
R
1 indexes [I > 2σ(I)] |
0.0479 |
0.0864 |
0.0268 |
0.0405 |
0.0641 |
0.0391 |
| wR2 indexes [all data] |
0.1324 |
0.2519 |
0.0627 |
0.1069 |
0.1662 |
0.1084 |
Synthesis
The Ligand 1-(3-methylsulfanyl-phenyl)-3-pyridin-2-yl-urea (L) was prepared as previously described.19
Fluoroboric acid salt HL+BF4−.
Ligand L (0.30 g, 0.116 mmol) was dissolved in THF (4 ml) and added to a solution of copper(II) tetraflouoroborate hexahydrate (0.40 g, 0.155 mmol) in THF (4 ml). The mixture was refluxed under nitrogen for 19 h, filtered and left to evaporate slowly over several days resulting in the formation of crystals suitable for X-ray diffraction analysis. IR (ν/cm−1) 711 (br), 1054 (br), 1619 (s), 1673 (s), 2925 (w), 3081 (m), 3206 (br).
[{Cu(L)(μ-Cl)Cl}2] (1).
Ligand L (0.30 g, 0.116 mmol) was dissolved in THF (2 ml) and added to a solution of copper(II) chloride (0.20 g, 0.118 mmol) in MeOH (2 ml). The mixture was left to slowly evaporate for few days resulting in a formation of green crystals suitable for X-ray analysis. IR (ν/cm−1) 1387 (s), 1535 (s), 1578 (s), 1608 (s), 1709 (s), 3129 (w, br), 3278 (w, br).
[Cu(L)Br2] (2).
Ligand L (0.30 g, 0.116 mmol) was dissolved in THF (2 ml) and added to a solution of copper(II) bromide (0.19 g, 0.116 mmol) in distilled water (1 ml). The mixture was left to slowly evaporate resulting in a formation of green crystals suitable for X-ray analysis. IR (ν/cm−1) 1478 (s), 1535 (s), 1613 (m), 1660 (s), 3026 (w) and 3325 (vw).
[Cu(L)(NO3)2] (3).
Ligand L (0.30 g, 0.116 mmol) was dissolved in THF (2 ml) and added to a solution of copper nitrate (0.22 g, 0.116 mmol) in THF:H2O (1:1 v/v, 2 ml). The mixture was left to slowly evaporate resulting in a formation of green crystals suitable for X-ray analysis. IR (ν/cm−1) 1030 (m, NO3), 1231 (vs), 1414, (m) 1478, (m) 1529 (m), 1668 (m), 3027 (br).
[Cu(L)(CF3SO3)2] (4) (Form A).
Ligand L (0.12 g, 0.046 mmol) in THF (2 ml) was mixed with copper trifluromethanesulfonate (0.168 g, 0.046 mmol) in methanol:H2O (1:1 v/v, 2 ml) and the mixture allowed to evaporate slowly resulting in a formation of green crystals suitable for X-ray diffraction analysis. IR (ν/cm−1) 1227 (s), 1438 (s), 1548 (s), 1585 (s), 1651 (s), 3282 (br).
[Cu(L)(CF3SO3)2] (4) (Form B).
Ligand L (0.30 g, 0.116 mmol) was dissolved in THF (2 ml) and mixed with copper(II) trifluromethanesulfonate (0.19 g, 0.116 mmol) in methanol:acetonitrile (1:1 v/v, 2 ml) and the mixture was left to evaporate slowly. This resulted in the formation of crystals suitable for X-ray diffraction analysis. IR (ν/cm−1) 1411 (s), 1467 (s), 1531 (s), 1579 (s), 1612 (s), 1718 (s), 3199 (m), 3279 (m).
Acknowledgements
We thank Durham University, the Higher Education Commission of Pakistan, Charles Wallace Trust Pakistan and Dr. Wali Mohammad Trust for partial funding.
References
- D. R. Turner, B. Smith, A. E. Goeta, I. R. Evans, D. A. Tocher, J. A. K. Howard and J. W. Steed, CrystEngComm, 2004, 6, 633 RSC.
- L. Brammer, E. A. Bruton and P. Sherwood, Cryst. Growth Des., 2001, 1, 277 CAS.
- F. Zordan, G. M. Espallargas and L. Brammer, CrystEngComm, 2006, 8, 425 RSC.
- A. L. Gillon, G. R. Lewis, A. G. Orpen, S. Rotter, J. Starbuck, X.-M. Wang, Y. Rodríguez-Martín and C. Ruiz-Pérez, J. Chem. Soc., Dalton Trans., 2000, 3897 RSC.
- L. Brammer, J. K. Swearingen, E. A. Bruton and P. Sherwood, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4956 CrossRef CAS PubMed.
- A. Bencini, A. Bianchi, P. Dapporto, E. Garcia-España, M. Micheloni, J. A. Ramirez, P. Paoletti and P. Paoli, J. Chem. Soc., Chem. Commun., 1990, 753 RSC.
- U. Suksangpanya, A. J. Blake, P. Hubberstey and C. Wilson, CrystEngComm, 2002, 4, 552 RSC.
- U. Suksangpanya, A. J. Blake, E. L. Cade, P. Hubberstey, D. J. Parker and C. Wilson, CrystEngComm, 2004, 6, 159 RSC.
- K. J. Bell, A. N. Westra, R. J. Warr, J. Chartres, R. Ellis, C. C. Tong, A. J. Blake, P. A. Tasker and M. Schröder, Angew. Chem., Int. Ed., 2008, 47, 1745 CrossRef CAS PubMed.
- I. Carson, K. J. MacRuary, E. D. Doidge, R. J. Ellis, R. A. Grant, R. J. Gordon, J. B. Love, C. A. Morrison, G. S. Nichol, P. A. Tasker and A. M. Wilson, Inorg. Chem., 2015, 54, 8685 CrossRef CAS PubMed.
- M. R. Healy, J. W. Roebuck, E. D. Doidge, L. C. Emeleus, P. J. Bailey, J. Campbell, A. J. Fischmann, J. B. Love, C. A. Morrison, T. Sassi, D. J. White and P. A. Tasker, Dalton Trans., 2016, 45, 3055 RSC.
- A. L. Gillon, A. G. Orpen, J. Starbuck, X.-M. Wang, Y. Rodríguez-Martín and C. Ruiz-Pérez, Chem. Commun., 1999, 2287 RSC.
- B. Dolling, A. L. Gillon, A. G. Orpen, J. Starbuck and X. M. Wang, Chem. Commun., 2001, 567 RSC.
- L. Brammer, Dalton Trans., 2003, 3145 RSC.
- G. Aullón, D. Bellamy, L. Brammer, E. A. Bruton and A. G. Orpen, Chem. Commun., 1998, 653 RSC.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120 CrossRef CAS.
- K. Fucke, N. Qureshi, D. S. Yufit, J. A. K. Howard and J. W. Steed, Cryst. Growth Des., 2010, 10, 880 CAS.
- N. Qureshi, D. S. Yufit, J. A. K. Howard and J. W. Steed, Dalton Trans., 2009, 5708 RSC.
- L. S. Reddy, S. Basavoju, V. R. Vangala and A. Nangia, Cryst. Growth Des., 2005, 6, 161 Search PubMed.
- M. Tiliakos, P. Cordopatis, A. Terzis, C. P. Raptopoulou, S. P. Perlepes and E. Manessi-Zoupa, Polyhedron, 2001, 20, 2203 CrossRef CAS.
- L. He, X.-L. Luo and W.-Q. Zhang, Chin. J. Struct. Chem., 2008, 27, 1384 CAS.
- M.-O. M. Piepenbrock, K. M. Anderson, B. C. R. Sansam, N. Clarke and J. W. Steed, CrystEngComm, 2009, 11, 118 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 CrossRef CAS.
-
C. J. Cason, Persistence of Vision Raytracer Pty Ltd, v3.7, Williamstown, Australia, 2002–2013 Search PubMed.
Footnote |
| † CCDC Single crystal X-ray structural details in CIF format have been deposited with the CCDC deposition numbers 1476704–1476709. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce01039c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a