
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination polymers and polygons using di-pyridyl-thiadiazole spacers and substituted phosphorodithioato NiII complexes: potential and limitations for inorganic crystal engineering†
M. Carla
Aragoni
*a,
Massimiliano
Arca
a,
Simon J.
Coles
 b,
Miriam
Crespo Alonso
a,
Susanne L.
Coles (née Huth)
c,
Robert P.
Davies
*d,
Michael B.
Hursthouse
b,
Francesco
Isaia
a,
Romina
Lai
a and
Vito
Lippolis
a
b,
Miriam
Crespo Alonso
a,
Susanne L.
Coles (née Huth)
c,
Robert P.
Davies
*d,
Michael B.
Hursthouse
b,
Francesco
Isaia
a,
Romina
Lai
a and
Vito
Lippolis
a
aDipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Cagliari, S.S. 554 bivio per Sestu, 09042 Monserrato-Cagliari, Italy. E-mail: aragoni@unica.it
bEPSRC National Crystallography Service, School of Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK
cDepartment of Chemistry, Faculty of Natural and Environmental Sciences, University of Southampton, Highfield, Southampton SO17 1BJ, UK
dDepartment of Chemistry, Imperial College London, South Kensington, London, SW7 2AZ, UK. E-mail: r.davies@imperial.ac.uk
First published on 6th June 2016
Abstract
Coordinatively unsaturated P-substituted dithiophosphonato, dithiophosphato, and dithiophosphito complexes {[Ni((MeO)2PS2)2] (1), [Ni((EtO)2PS2)2] (2), [Ni(MeOdtp)2] (3), and [Ni((Ph)2PS2)2] (4)} were reacted with the bis-functional ligands 3,5-di-(4-pyridyl)-1,2,4-thiadiazole (L1) and 3,5-di-(3-pyridyl)-1,2,4-thiadiazole (L2) to give the coordination polymers (1–4·L1)∞, (3·L2)∞, and (4·L2·2C7H8)∞ and the discrete dimers (1–2·L2)2, all characterised by single crystal X-ray diffraction. A comparison of the structures shows that L1 can be exploited for the predictable assembly of undulating chains independent of the nature of the NiII complex, while L2 allows for the existence of different supramolecular constructs ensuing from different ligand conformations deriving from the rotation of the pyridyl rings.
Introduction
The past decades experienced an impressive number of novel multidentate bridging ligands designed to construct a variety of discrete metal–organic polygons and polyhedra (MOPs) or metal–organic frameworks (MOFs) with high dimensionality. These supramolecular assemblies are of great appeal due to a combination of their intrinsic beauty and promising applications in various fields such as gas storage, ion exchange, chemical sensing, catalysis, energy transfer, and separation.1,‡The properties of these crystalline materials are critically dependent on their network structures, and the deliberate creation of a crystalline network, planned using properly designed building blocks, remains, nowadays, a challenge. Several aspects need to be evaluated in developing a network based on coordination polymers: the building blocks, i.e. metal nodes and organic spacers, metal coordination environments, formation conditions, and weak secondary interactions. Multi-topic organic molecules are commonly used as spacers, and an opportune choice of the number and position of donor atoms can be used to direct the network assembly, although factors such as flexibility of the ligand and different accessible conformations need to be examined and taken into account. The metal coordination environment is exceptionally difficult to control when “naked” metal ions are used as nodes, and in view of that, the use of neutral coordination complexes held together by additional donor molecules or secondary bonding interactions has gained striking importance.2 In fact, by reducing the degrees of freedom of the system, for instance by using cis-protected metal blocks in place of the naked metal ions or by using a neutral, coordinatively unsaturated metal complex, less uncertainty can be expected. Moreover, the use of neutral synthons leads to self-reliant supramolecular assemblies which do not require the presence of a counterion and thus reduce the occurrence of isomerism.
In this respect, we have been developing a synthetic program based on the ability of neutral dithiophosphonato NiII complexes [Ni(ROdtp)2] [ROdtp = (RO)(4-MeOC6H4)PS2−; R = alkyl substituent]3 to assemble coordination polymers in combination with a variety of polypyridyl donors, in particular 4,4′-bipyridine and its analogues.§,4 This assembly process is based on the capability of the coordinatively unsaturated NiII ion of these square-planar complexes to axially bind monodentate ligands, such as pyridine, to yield discrete octahedral complexes.5,6 In addition, suitable N–L–N bidentate bipyridyl-based spacers yield 1D coordination polymers of the type [Ni(ROdtp)2(N–L–N)]∞.7,8 The primary structural motif of these polymers mainly depends on the features of the spacers such as length, rigidity and orientation of the donor atoms, as recently confirmed by the deliberate stereospecific generation of homochiral polymeric helices built from a designed enantiopure binaphthyl-based ligand.9
The substituents on the phosphorus atoms are responsible for the connection of the polymers through hydrogen bonds and face-to-face or edge-to-face π–π interactions, thus influencing the final 3D architecture.7 As a consequence, coordination polymers and 3D assemblies with different structures and architectures can be built up by varying either the bridging ligands or the substituents on the P atom of the initial Ni complexes. In order to better understand the process of molecular recognition between components and how the steric information contained in the P-substituents and in the orientation of ligand binding sites combine to give the final structure, differently P-substituted dithiophosphonato, dithiophosphato, and dithiophosphito complexes {[Ni((MeO)2PS2)2] (1), [Ni((EtO)2PS2)2] (2), [Ni(MeOdtp)2] (3), and [Ni((Ph)2PS2)2] (4)} were reacted with the bis-functional ligands 3,5-di-(4-pyridyl)-1,2,4-thiadiazole (L1)10 and 3,5-di-(3-pyridyl)-1,2,4-thiadiazole (L2)10 (Scheme 1).
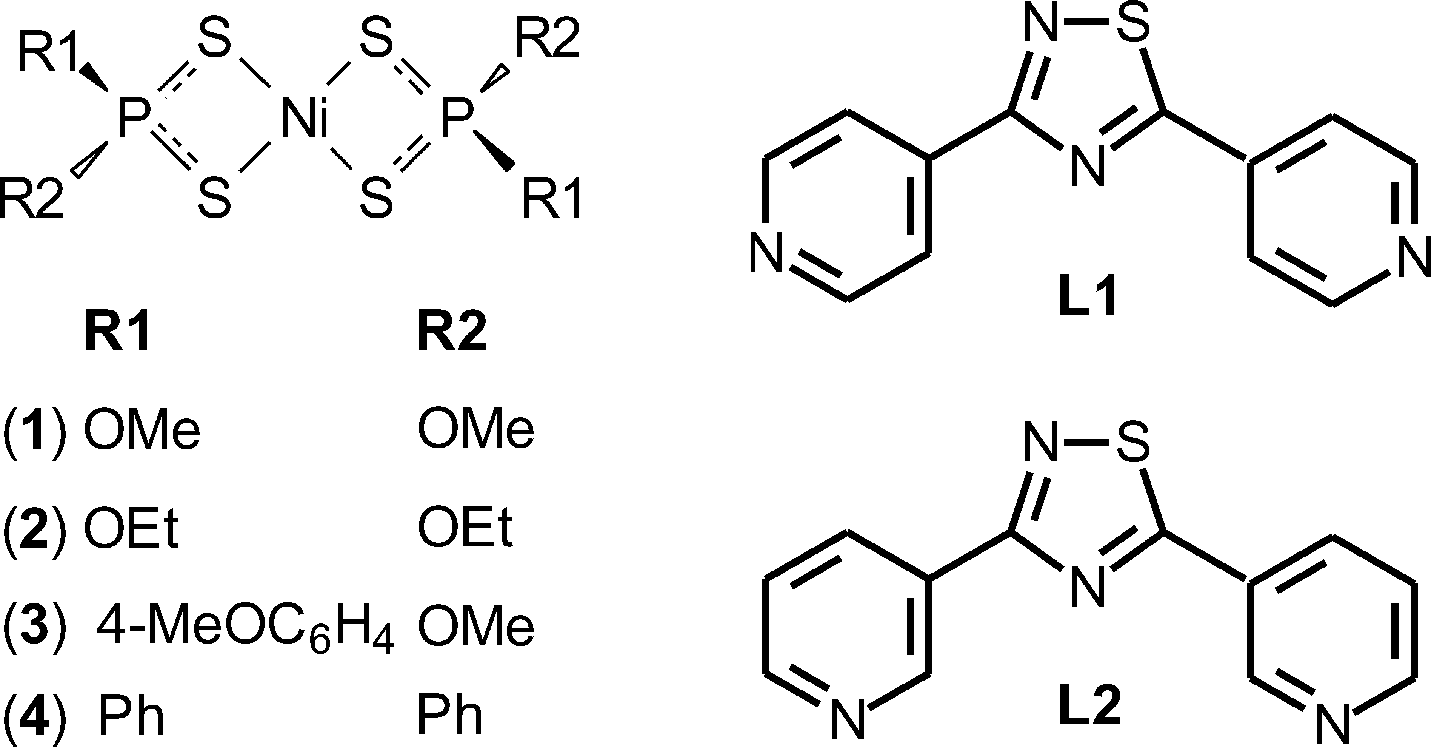 | ||
| Scheme 1 General scheme of complexes 1–4 (left) and ligands L1 and L2 (right). | ||
We have recently investigated the reactivity of L1 and L2 with I2 and IBr, in both polar and apolar media, thereby elucidating the role of specific directional interactions, namely, NH+⋯N and N⋯I, combined with geometrical features of the molecules, in the formation of different supramolecular constructs.10 The investigation of these ligands is extended here to the formation of coordination polymers.
Experimental
Materials
All commercially available compounds were used as received. Bis[O-alkyl-dithiophosphato]Ni complexes {[Ni((RO)2PS2)2], R = Me (1), Et (2)},5 the dithiophosphonato NiII complex [Ni(MeOdtp)2] [MeOdtp = (MeO)(4-MeOC6H4)PS2−],3 and bis-functional ligands 3,5-di-(4-pyridyl)-1,2,4-thiadiazole (L1) and 3,5-di-(3-pyridyl)-1,2,4-thiadiazole (L2)10 were synthesised according to previously reported procedures. The solvents used were freshly distilled over the appropriate drying agent and used directly from the stills.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 215 m, 1019 vs, 945 vs, 848 w, 830 m, 805 m, 773 s, 713 m, 673 s, 657 s, 644 m, 620 w, 546 w, 410 w cm−1. FT-Raman (3500–50 cm−1, 600 mW, solid state, relative intensities between parentheses related to the highest peak taken equal to 10.0): 3075 (4.8), 3027 (4.2), 2933 (5.1), 2888 (5), 1981 (4.1), 1611 (10), 1513 (5.7), 1413 (8.5), 1338(5.9), 1229 (5.3), 1215 (5.5), 1095 (7.7), 1015 (7.7), 999 (5.7), 729 (4.2), 650 (6.1), 548 (8.8), 376 (7.5) cm−1.
215 m, 1019 vs, 945 vs, 848 w, 830 m, 805 m, 773 s, 713 m, 673 s, 657 s, 644 m, 620 w, 546 w, 410 w cm−1. FT-Raman (3500–50 cm−1, 600 mW, solid state, relative intensities between parentheses related to the highest peak taken equal to 10.0): 3075 (4.8), 3027 (4.2), 2933 (5.1), 2888 (5), 1981 (4.1), 1611 (10), 1513 (5.7), 1413 (8.5), 1338(5.9), 1229 (5.3), 1215 (5.5), 1095 (7.7), 1015 (7.7), 999 (5.7), 729 (4.2), 650 (6.1), 548 (8.8), 376 (7.5) cm−1.
Characterisation
1H- and 31P-NMR spectra were recorded at 25 °C in D6-DMSO on a Bruker DPX400 NMR spectrometer with internal standards. Elemental analyses were performed using an EA1108 CHNS-O Fisons instrument. FT-infrared spectra were recorded on a Thermo Nicolet 5700 spectrometer at room temperature using a flow of dry air. Middle IR spectra (resolution 4 cm−1) were recorded as KBr pellets, with a KBr beam-splitter and KBr windows. X-ray structure determinations and crystallographic data for compounds (1·L1)∞, (2·L1)∞, (3·L1)∞, (1·L2)2, and (3·L2)∞ were collected at 120(2) K by means of combined phi and omega scans using a Bruker-Nonius Kappa CCD area detector, situated at the window of an FR591 rotating anode (graphite Mo-Kα radiation, λ = 0.71073 Å). The data for compound (2·L2)2 were collected at 120(2) K by means of fine-slice/omega scans using a Bruker SMART APEX2 CCD diffractometer with Daresbury SRS station 9.8 synchrotron source (silicon 111, λ = 0.6893 Å). The data for compound (4·L1)∞ were collected at 173(2) K by means of combined phi and omega scans using a diffractometer with enhanced X-ray source (graphite Mo-Kα radiation). The data for compound (4·L2·2C7H8)∞ were collected at 173(2) K by means of omega scans using an Oxford Diffraction Xcalibur PX Ultra diffractometer with enhanced ultra (Cu) X-ray source (graphite Cu-Kα radiation, λ = 1.54184 Å). The structures were solved by direct methods and SHELXS-97 and refined on F2 using SHELXL-97.12,13 Anisotropic displacement parameters were assigned to all non-hydrogen atoms. Hydrogen atoms were included in the refinement, but thermal parameters and geometry were constrained to ride on the atom to which they are bonded. The data of (1·L1)∞, (2·L1)∞ and (3·L1)∞ were corrected for absorption effects using SADABS V2.10.14 The data for (4·L1)∞ and (4·L2)∞ were refined using CrysAlis RED,15 implemented in SCALE3 ABSPACK scaling algorithm for empirical absorption correction using spherical harmonics. The structures have been deposited with the Cambridge Crystallographic Data Centre: deposition numbers CCDC 1474147, 1474143, 1474148, 1474145, 1474150, 1474144, 1474146, and 1474149 for (1·L1)∞, (2·L1)∞, (3·L1)∞, (4·L1)∞, (1·L2)2, (2·L2)2, (3·L2)∞, and (4·L2·2C7H8)∞, respectively. Theoretical calculations based on Density Functional Theory (DFT)16 were carried out on L1 and L2 using the Gaussian 0917 commercial suite of software by adopting the mPW1PW18 functional and Schäfer, Horn, and Ahlrichs double-zeta plus polarisation (pVDZ) all-electron basis sets (BSs) for all atomic species.19 A potential energy surface was carried out by rotating by an angle τ one of the pyridine rings (−180.0° ≤ τ ≤ 180.0°; Δτ = 5.0°). The programmes GaussView 5.0.8 and Molden 5.2 were used to investigate the charge distributions and MO shapes.20Results
The bis-functional ligands L1 and L2 (Scheme 1) were first isolated by Meltzer et al. in 1955,21 but their complexing ability towards metal ions, or as Lewis donors, has not been investigated to date. In fact, a search in the Cambridge Crystallographic Database revealed only a recent work by Ondrejkovicova et al.22 Both L1 and L2 contain two pyridyl groups linked by a 1,2,4-thiadiazole ring acting as a rigid, non-reactive spacer. Compared with the popular 4,4′-bipyridine linker, the pyridyl rings in L1 and L2 feature different geometries and separation lengths (9.95 and 9.60 Å, respectively). Moreover, due to the different positions of the nitrogen atoms in L1 and L2 and to the different rotational conformations possible for the pyridyl rings, several orientations of the binding sites can be expected. A potential energy surface (PES) analysis carried out on L1 and L2 at the DFT level in the gas phase by rotating one pyridyl substituent by an angle τ ranging between −180 and 180° clearly shows that both donors display an energy minimum when the central 1,2,4-thiadiazole ring is coplanar with the pyridine moieties (Fig. 1). In the case of L2, two planar isomers, cisoid and transoid, are possible, differing in energy by less than 1 kcal mol−1 and showing similar metric parameters. As previously described, L1 and L2 feature the highest Kohn–Sham molecular orbitals localized on the negatively charged nitrogen atoms of the two pyridine moieties, which are therefore available to behave as donor sites towards Lewis unsaturated metal complexes.10 The reactions of L1 with nickel dithiophosphato {[Ni((RO)2PS2)2] [R = Me (1), Et (2); Scheme 1]}, dithiophosphonato {[Ni(MeOdtp)2] (3) [MeOdtp = (CH3O)(4-MeOC6H4)PS2−, Scheme 1]}, and dithiophosphito {[Ni(Ph2PS2)2] (4)} complexes under solvothermal conditions afforded solid, crystalline compounds, which were isolated and identified by means of single crystal X-ray diffraction as coordination polymers of formulae (1·L1)∞, (2·L1)∞, (3·L1)∞, and (4·L1)∞, respectively. Crystallographic data and selected bond lengths and angles are reported in Tables S1† and 1, respectively. With the exception of compound (4·L1)∞, the L1 molecules are found in two different but essentially superimposable orientations, arising from a 180° rotation of the molecule about the direction passing through the midpoint of the N–S bond and through the remaining nitrogen atom of the penta-atomic ring (Fig. S1–S3†). As a consequence, the nitrogen and the adjacent sulfur atoms show fractional occupancies refined at values of about 60% and 40%, respectively, and only the major orientation of the molecule is illustrated in the figures. In the case of compound (2·L1)∞, the L1 molecule is located about a crystallographic two-fold axis, and therefore, the resulting disorder is modelled with occupancies of 50% for each orientation.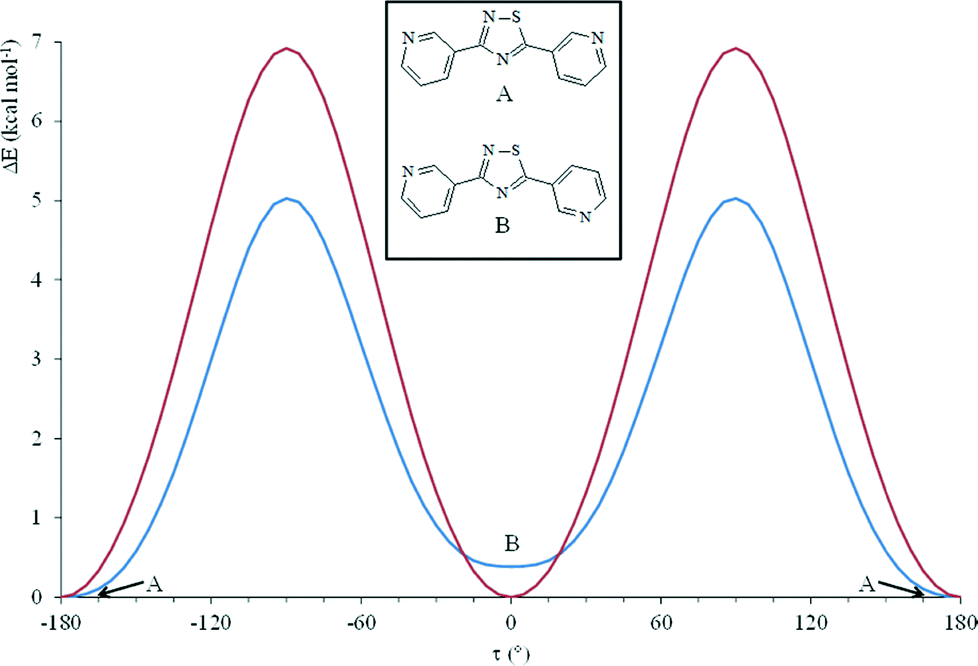 | ||
| Fig. 1 Relative electronic energy variation ΔE as a function of the rotation angle τ of one pyridine ring (rotation step 5.0°) calculated at the DFT level for L1 (red) and L2 (blue). In the inset, the two possible cisoid (A) and transoid (B) L2 conformers are depicted. ΔEB − ΔEA = 0.39 kcal mol−1. | ||
| (1·L1)∞ | (2·L1)∞ | (3·L1)∞ | (4·L1)∞ | |
|---|---|---|---|---|
| a In order to compare structural parameters of the polymers which reside in different space groups a common artificial numbering scheme has been adopted as shown in Fig. S1. b Average of the bond parameters for the two symmetry independent fragments (P1S1S2Ni1) and (P2S3S4Ni2). | ||||
| Ni–N1 | 2.095(3) | 2.1042(16) | 2.1343(18) | 2.1376(16) |
| Ni–N4 | 2.100(3) | 2.1042(16) | 2.137(2) | 2.1052(16) |
| Ni–S1 | 2.5041(12)b | 2.4846(5) | 2.4876(6)b | 2.4836(5)b |
| Ni–S2 | 2.4585(11)b | 2.4683(5) | 2.4662(6)b | 2.4940(6)b |
| P1–S1 | 1.9735(15)b | 1.9693(8) | 1.9993(9)b | 2.0001(8)b |
| P1–S2 | 1.9814(15)b | 1.9884(8) | 1.9974(9)b | 2.0025(7)b |
| N1–Ni–S1 | 91.02(8) | 89.72(5) | 90.94(5) | 91.22(4) |
| N1–Ni–S2 | 89.58(9) | 90.39(5) | 89.12(5) | 88.76(5) |
| S1–Ni–S2 | 82.04(4)b | 82.010(17) | 82.55(2)b | 82.42(2)b |
| S1–P1–S2 | 110.95(7)b | 110.39(3) | 109.70(4)b | 110.2(4)b |
| C2–C3–C6–N2 | 19(3) | 31.9(3) | 34.2(3) | 4.7(3) |
| C9–C8–C7–N2 | 17(3) | 31.9(3) | 20.3(3) | 11.0(3) |
| Py(N1)^Py(N4) | 25.8 | 63.7 | 31.9 | 16.1 |
| Py(N1)^Tdz | 20.1 | 32.9 | 36.5 | 4.9 |
| Py(N4)^Tdz | 17.3 | 32.9 | 20.0 | 11.4 |
Coordination polymers (1–4·L1)∞ show similar coordination environments: the nickel ions display distorted octahedral geometries with the equatorial plane occupied in iso-bidentate bonds with two dithiophosphoric ligands and the pyridine rings belonging to spacer L1 axially bridging adjacent NiII ions to form infinite polymeric chains. The relevant bond lengths and angles (Table 1) are similar to those found in analogous coordination polymers.7–9 The structures of the coordination polymers share the presence of neutral undulated polymeric chains with very similar pitches: Ni–Ni distances, through coordinate bonds, of 13.70, 14.15, 13.97, and 14.02 Å for (1·L1)∞, (2·L1)∞, (3·L1)∞, and (4·L1)∞, respectively (Fig. 2). These chains are closely analogous to the C11(12) chains of type [⋯(Py∩PyH+⋯Py∩PyH+)n⋯] built up through the moderate NH+⋯N bonds derived by the protonation of L1 molecules, reported for comparison in Fig. 2e.10 This confirms that the orientation of the nitrogen atoms para-positioned in the outwards pyridyl rings of L1 self-governs the geometry of the resulting supramolecular aggregates, leading to similar shapes independent of the nature of the interacting Lewis acid. The asymmetric unit of compound (1·L1)∞ (Fig. S2† and 2a) contains two independent NiII ions, namely, Ni1 and Ni2, lying on crystallographic inversion centres with coordination sites differing in the orientation of the methoxy substituents at the phosphorous atoms. The two coordination sites are bridged by L1 molecules, thus forming 1D parallel chains propagating along the 101 direction. These chains interact with each other through C–H⋯S short contacts, involving the pyridine rings and the coordinated sulphur atoms, leading to the 2D layers shown in Fig. 3a. The polymeric chains pack in two different orientations (coloured blue and yellow in Fig. 3b and S3†) generated by inversion along the screw axes parallel to the 010 direction. As a consequence, symmetry related 2D layers formed by differently oriented chains alternate when packed and interact through synergic C–H⋯O bonds (“d” and “e” in Fig. 3b and S3a†) involving the pyridine rings and the methoxy P-substituents belonging to the Ni1b coordination sphere in an R22(7) motif. This packing arrangement enables the formation of “rippled” void channels of about 60 Å3 corresponding to 2.2% of the unit cell volume (Fig. S3b†). The presence of OEt P-substituents in place of the OMe ones in compound (2·L1)∞ (Fig. 2b and S4†) does not result in significant changes in either the NiII coordination environment or the primary motif of the polymer, and no relevant intramolecular interactions are worthy of note. The polymeric chains assume four symmetry related orientations as illustrated in Fig. 4. Pairs of symmetry related chains propagating in the [10(1/2)] (yellow and green in Fig. 4) and [−10(1/2)] (blue and grey in Fig. 4) directions, respectively, interact through weak H bonds involving the P-substituents and the central thiadiazole ring of L1 (“a” in Fig. 4) and form 2D layers which stack along the [010] direction leading to a very dense packing with no voids.
 | ||
| Fig. 2 Polymeric and protonated chains involving ligand L1 in compounds (1·L1)∞ (a), (2·L1)∞ (b), (3·L1)∞ (c), (4·L1)∞ (d), and (L1H+)∞ (e; ref. 10). | ||
 | ||
| Fig. 3 Packing view of (1·L1)∞ showing parallel chains interacting through C–H⋯S interactions (a; H⋯S distances and C–H⋯S angles: a, C9i–H9i⋯S2 2.89(3), 120(2); b, C8i–H8i⋯S5 2.85(3), 122(2); c, C15i–H15i⋯S3 2.91(4) Å, 126(2)°. (i) x, −1 + y, z); (b) view along the 010 direction with polymeric chains coloured in blue and yellow according to their orientation. The NiII ions and the O and H atoms involved in the described H bonds (d, C6ii–H6ii⋯O3 2.37(4), 3.288(5), 155(3); e, C5ii–H5ii⋯O4 2.60(4) Å, 3.419(5) Å, 148(3)°; (ii) x, 1.5 − y, 0.5 + z) are displayed with conventional colours. | ||
 | ||
| Fig. 4 Packing view along the 001 direction of (2·L1)∞ showing polymeric chains differently coloured according to their orientation. The NiII ions and the H atoms involved in the described H bonds [a, C2–H2a⋯N3i/S3i 2.54(4)/2.73(3), 3.426(10)/3.617(5), 147(2)/148(2); b, C5–H5⋯S2ii 2.85(2) Å, 3.364(2) Å, 115(2)°; (i) x, −y, 0.5 + z; (ii) −0.5 + x, 0.5 − y, 1 − z] are displayed with conventional colours. H atoms not involved in the shown interactions have been omitted for clarity. The labels used for the described interactions refer to the original numbering scheme (Fig. S2†). | ||
The coordination polymers (3·L1)∞ and (4·L1)∞ (Fig. 2c and d, S5 and S6†) are characterised by the presence of aryl substituents at the phosphorous atoms which are expected to engender additional intra- and inter-molecular aromatic interactions. Similarly to those described previously herein, the asymmetric units of compounds (3·L1)∞ and (4·L1)∞ contain two independent NiII ions located on crystallographic inversion centres and differing only in the orientation of the aryl P-substituents. In the coordination polymer (3·L1)∞, the interactions involving the aromatic rings are intramolecular in nature (edge-to-face interaction “a” in Fig. 5). Weak interactions between the methoxy groups (interactions “b” and “c” in Fig. 5) connect the chains in layers which pack parallel to each other, forming a three-dimensional network through C–H⋯S interactions mainly involving the coordinated sulphur atoms and the methoxy substituents (interactions “d”–“g” in Fig. S5b†).
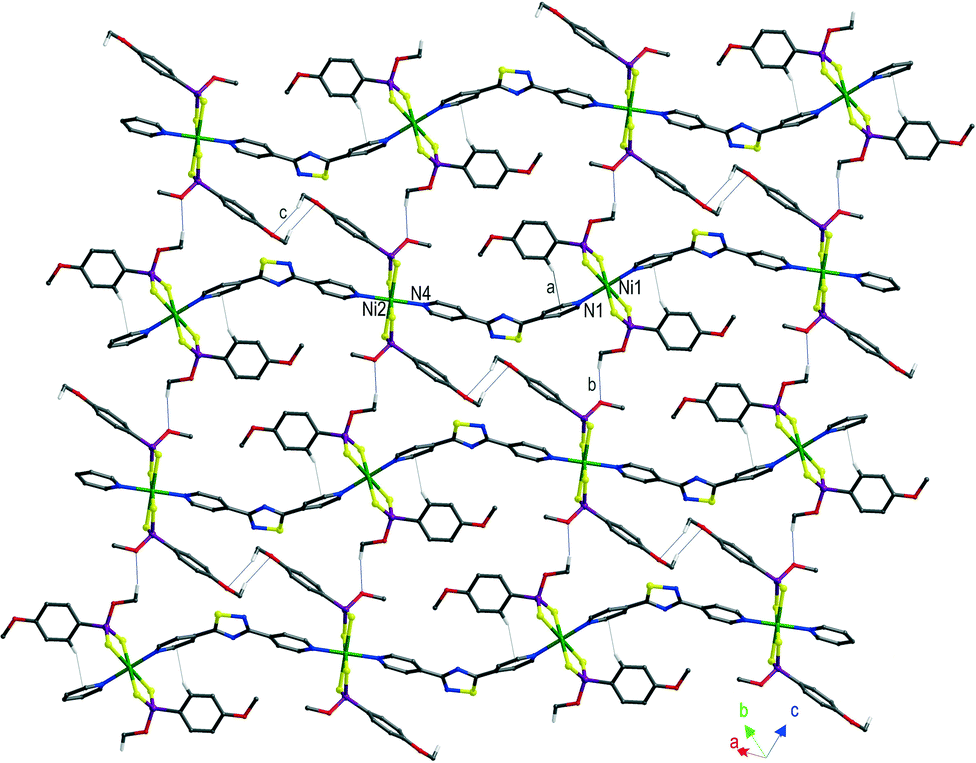 | ||
| Fig. 5 Packing views of (3·L1)∞ showing layers of undulated chains; a, C2–H2⋯CntPy(N1) 2.62 Å; b, C8–H8c⋯O3i 2.24, 3.171(3), 158.0; c, C15–H15a⋯O4ii 2.53 Å, 3.237(3) Å, 129.0°; (i) −1 + x, −1 + y, z; (ii) 2 − x, 2 − y, −z. H atoms not involved in the shown interactions have been omitted for clarity. The labels used for the described interactions refer to the original numbering scheme (Fig. S5†). | ||
Aromatic interactions become prevalent in polymer (4·L1)∞ due to the presence of phenyl substituents at the phosphorous atoms, so that the polymeric chains pack in layers built up by face-to-face and edge-to-face interactions only (“a”–“g” in Fig. 6). The layers pack parallel to the a axis through weak C–H⋯S interactions (“h”–“j” in Fig. S6b†). Despite the fact that all the coordination polymers featuring L1 as a spacer exhibit the same primary motif found in the supramolecular aggregates of L1H+ (Fig. 2e), the presence and orientation of the P-substituents influence the final architecture of the polymers via aromatic or C–H⋯S intramolecular interactions. The main consequence is the loss of planarity of the bridging L1 ligands (Table 1), probably ascribable to the conformational arrangement the pyridine rings adopt to optimize these interactions. It is interesting to note that in the H-bonded chains built up by protonated L1 molecules, where such interactions are not present, the ligands retain planarity.10
 | ||
| Fig. 6 Packing view of (4·L1)∞ showing layers built up by aromatic interactions a–g: a, CntPh(C24–C29)⋯CntPh(C24–C29)’ 4.16, 0; b, CntPy(N1)⋯H26i–C26i 3.32, 49; c, CntPy(N15)⋯H21ai–C21i 3.08, 133; d, CntPy(N15)⋯CntiiPh(C36–C41) 3.67, 11; e, CntPh(C30–C35)⋯CntiPh(C18–C23) 3.90, 19; f, C34–H34a⋯CntiiiPy(N1) 3.58, 109; g, C19iv–H19aiv⋯CntPy(N1) 3.90 Å, 129°. Symmetry codes: (i) −x, 1 − y, 1 − z; (ii) 1 − x, 1 − y, 2 − z; (iii) x, −1 + y, z; (iv) −x, 2 − y, −z. H atoms not involved in the shown interactions have been omitted for clarity. The labels used for the described interactions refer to the original numbering scheme (Fig. S6†). | ||
The reactions of L2 with nickel complexes 1–4 under solvothermal conditions afforded crystalline compounds recognised by means of single crystal X-ray diffraction as the dimers (1·L2)2 and (2·L2)2 and the coordination polymers (3·L2)∞ and (4·L2)∞ in compound (4·L2·2C7H8)∞, respectively (Fig. 7–10 and S8–S11†). The two different constructs reflect the different conformations of L2 behaving as a convergent linker in the dimeric structures and as a divergent linker in the polymers. Crystallographic data and selected bond lengths and angles are reported in Tables S2† and 2. Similarly to the previously discussed cases of polymers (1–3·L1)∞, in the crystal structures of the dimer (1·L2)2 and the polymers (3–4·L2)∞, the spacer L2 is located in two essentially superimposable orientations. As a consequence, the nitrogen and the adjacent sulfur atoms show fractional occupancies refined at values of 68.8 and 31.2% for (1·L2)2 and 61.5 and 31.5% for (4·L2)∞ (the figures represent the molecules in the major orientation only).
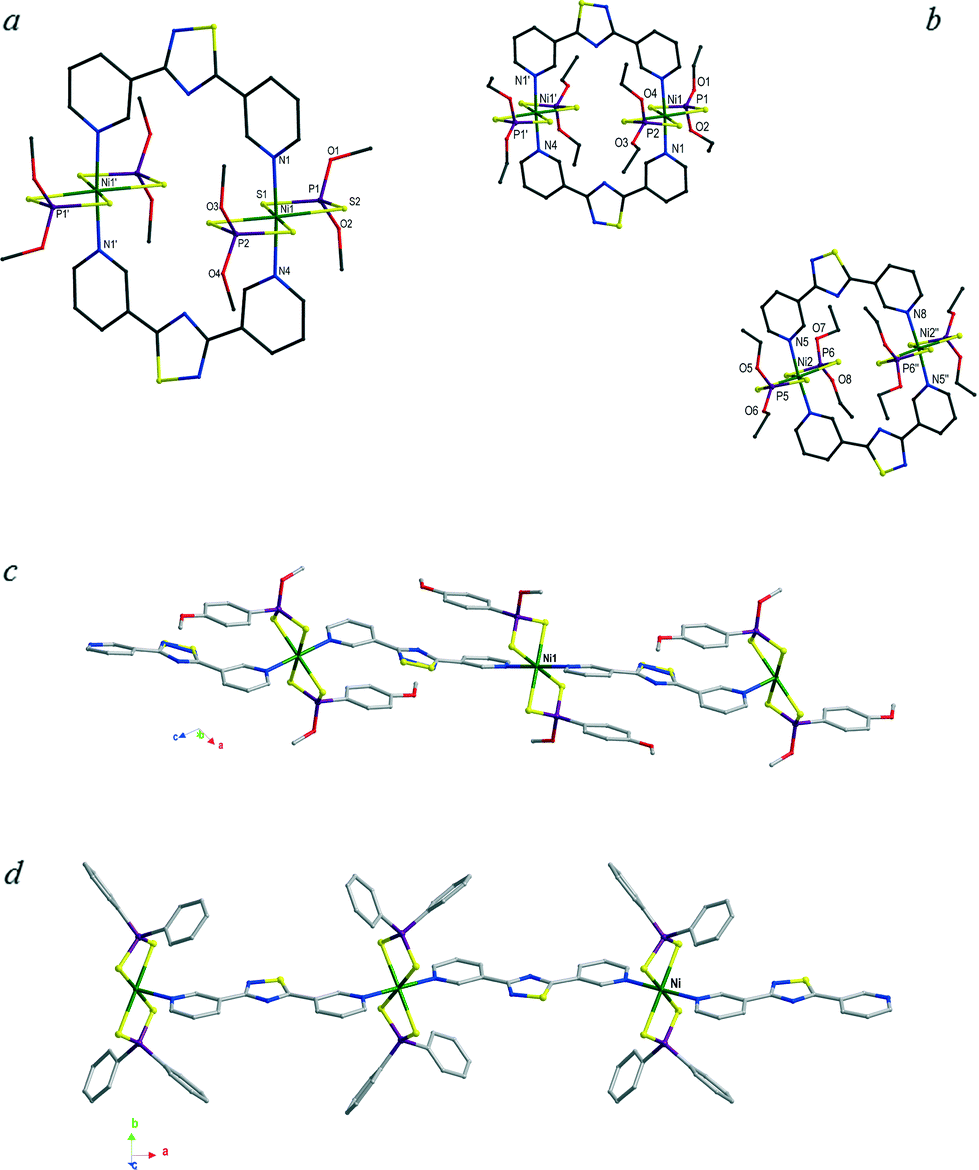 | ||
| Fig. 7 Dimeric units and polymeric chains for (1·L2)2 (a), (2·L2)2 (b), (3·L2)∞ (c), and (4·L2)∞ in compound (4·L2·2C7H8)∞ (d). The labels reported refer to the original numbering schemes (Fig. S8–S12†). H atoms have been omitted for clarity. Only the labels used in the discussion are reported here. | ||
| (1·L2)2 | (2·L2)2 | (3·L2)∞ | (4·L2·2C7H8)∞ | |
|---|---|---|---|---|
| a Average of the bond parameters for the two fragments (P1S1S2Ni) and (P2S3S4Ni). b Average of the bond parameters for the symmetry independent coordination environment around Ni1 and Ni2. | ||||
| Ni–N1 | 2.093(5) | 2.103(5)b | 2.160(2) | 2.129(6) |
| Ni–N4 | 2.099(5) | 2.100(5)b | 2.160(2) | 2.129(6) |
| Ni–S1 | 2.4928(18)a | 2.500(2)b | 2.4915(6) | 2.4809(15) |
| Ni–S2 | 2.4846(18)a | 2.487(2)b | 2.4871(6) | 2.4995(17) |
| P1–S1 | 1.979(2)a | 1.969(3)b | 1.9974(8) | 2.0059(19) |
| P1–S2 | 1.981(2)a | 1.976(3)b | 1.9915(9) | 1.997(2) |
| N1–Ni–S1 | 90.66(15) | 89.93(19)b | 88.91(5) | 90.82(11) |
| N1–Ni–S2 | 88.99(15) | 89.76(18)b | 90.52(5) | 89.35(11) |
| S1–Ni–S2 | 81.89(10)a | 81.98(8)b | 81.86(2) | 83.00(5) |
| S1–P1–S2 | 110.93(10)a | 112.0(2)b | 109.71(4) | 111.08(8) |
| N1–Ni–N4 | 175.7(2) | 176.3(2)b | 180.00 | 180.00 |
| N2–C6–C3–C2 | 9.6(3) | 1.4(11)b | 11.7(3) | 0.0 |
| N2–C7–C8–C9 | 17.9(3) | 4.4(11)b | 11.7(3) | 0.0 |
| Py(N1)^Py(N4) | 12.3 | 5.0b | 19.2 | 0.0 |
| Py(N1)^Tdz | 4.3 | 3.0b | 14.7 | 0.0 |
| Py(N4)^Tdz | 20.3 | 7.2b | 14.7 | 0.0 |
In the case of compound (3·L2)∞, the L2 molecule is located about a crystallographic two-fold axis, and therefore, the resulting disorder is modelled with occupancies of 50% for each orientation. The asymmetric unit of the dimer (2·L2)2 contains two independent units featuring differently oriented ligands L2 (Fig. 7b). Both dimers (1–2·L2)2 and polymers (3–4·L2)∞ display octahedral NiII coordination environments similar to those previously discussed, with two iso-bidentate dithiophosphoric ligands in the equatorial plane and the axial positions occupied by the pyridine rings of L2 bridging spacers.
The bond lengths and angles (Table 2) are also similar to those found in the analogous coordination polymers (1–4·L1)∞. Notwithstanding similarities with the results obtained with L1, the use of L2 as a linker leads to different constructs. In fact, as evidenced by DFT calculations (see above), L2 can exist either as a transoid or as a cisoid isomer, and a convergent or a divergent conformation can be distinguished depending on the orientation of the N atoms of the pyridyl rings that can point inwards or outwards with respect to the pinch angle. In (1·L2)2 and (2·L2)2, the pyridyl rings of ligand L2 are oriented in a convergent fashion leading to closed rings rather than polymeric chains (Fig. 7a and b). It is interesting to note that this construct is far from predictable in its behaviour since a convergent fashion does not necessarily lead to closed rings. This is demonstrated by the formation of the mono-dimensional C11(10) spirals found in the two structures of (HL2)I5 and (HL2)IBr2 assembled through NH+⋯N bonds between protonated L2 molecules exhibiting a convergent cisoid conformation.10 In dimers (1·L2)2 and (2·L2)2, two spacers bridge two dithiophosphato nickel complexes through axial coordination generating eicos-atomic planar wheels with openings of about 8 × 8 Å2 and inner Ni⋯Ni distances of 7.73, 7.77, and 7.58 Å for (1·L2)2 and the two independent units of (2·L2)2, respectively.
Despite the analogies in their structures, the dimers (1·L2)2 and (2·L2)2 show different packing arrangements. The dimers (1·L2)2 are arrayed in regular perforated layers assembled by N⋯H and S⋯H interactions (Fig. 8a). Symmetry related parallel layers pack in an off-set compact arrangement along the b direction, as shown in Fig. 8.
 | ||
| Fig. 8 Packing views showing the (a) layers formed by interacting (1·L2)2 dimers and (b) packing of intercalating layers evidenced in different colours; in the inset, the intercalating layers packing along the 010 direction are presented. All the hydrogen atoms with the exception of those involved in the shown interactions have been omitted. Interactions: a: C14i–H14i⋯N3, 2.70(9), 3.331(13), 124; b: C15ii–H15ii⋯S2, 2.93(9), 3.618(8), 127; c: C1iii–H1biii⋯N2, 2.57, 3.365(19), 138; d: C2iii–H2aiii⋯S5, 2.88, 3.542(8), 126; e: C8iv–H8iv⋯O3, 2.59 Å, 3.504(9) Å, 161°. Symmetry codes: (i) −1 + x, y, −1 + z; (ii) 1 − x, 1 − y, 1 − z; (iii) −x, 0.5 + y, 0.5 − z; (iv) x, 1.5 − y, 0.5 + z. | ||
In the crystal packing of (2·L2)2, the two symmetrical independent units (containing Ni1 and Ni2 ions) that, as previously observed differ in the orientation of the thiadiazole ring (Tdz), interact with each other through π–π interactions involving the pyridine and Tdz rings forming puckered ribbons (Fig. 9a). A view along the a axis (Fig. 9b) shows the ribbons aligned in a parallel arrangement through H bonds and weaker interactions mainly involving the sulphur atoms and the pyridine hydrogens. A cisoid but divergent conformation of L2 leads to the coordination polymers (3·L2)∞ and (4·L2)∞ (Fig. 7c and d and S10–S12†). The asymmetric units of both compounds contain one independent NiII ion situated on a crystallographic inversion centre in the first case and on a mirror plane in the latter. The polymeric supramolecular constructs present close analogies with the C11(10) chains formed by head-to-tail NH+···N bonds between adjacent pyridine rings of protonated L2 molecules featuring a trans conformation.10 Both complexes 3 and 4 feature aromatic substituents that are involved in π-type interactions which govern the assembly of the relevant polymers. In (3·L2)∞, the aromatic P-substituents are engaged in intramolecular π–π interactions with the facing pyridine rings and intermolecular C–H···O interactions with the oxygen belonging to the MeO P-substituents which connect the chains in layers (interactions a and b in Fig. S12a†). The layers pack in parallel through C–H···S interactions involving the para-methoxy group of the Ar–P substituents and the coordinated sulphur atoms (interactions c–e in Fig. S12b†), thus forming an overall three-dimensional network. Solvent accessible voids of 69 Å3 (in blue in the inset of Fig. S12†) correspond to 4% of the unit cell volume. In polymer (4·L2)∞, the exclusive presence of phenyl substituents at the phosphorous atoms results in the aromatic interactions becoming prevalent such that the parallel polymeric chains pack through edge-to-face interactions only (Fig. 10a). The resulting network features empty channels suitable for the inclusion of toluene molecules which engage in π-interactions with the phenyl rings (Fig. 10b and c). It is interesting to note that, in contrast to the previously discussed structures (see also Tables 1 and 2), (4·L2)∞ contains perfectly planar L2 ligands.
 | ||
| Fig. 9 Packing views showing (a) the ribbons formed by π–π interacting (2·L2)2 dimers and (b) packing view along the 100 direction of interacting ribbons. All the hydrogen atoms with the exception of those involved in the shown interactions have been omitted. Shown π–π interactions (a and b), weak contacts (c–e, g, and h), and the H bond (f): a, CntPy(N8)⋯CntPy(N1)iii 3.58, Py(N5)^Py(N4)iii 1; b, CntTdz(S15)⋯CntTdz(S5)iii 3.44 Å, Tdz(S15)∧Tdz(S5)iii 1°; c, C38–H38⋯O3iii 2.40 Å; d, C6iii–H6ciii⋯C38, 2.78; e, C12ii–H12ii⋯S2, 2.89; g, C1ix–H1aix⋯S12, 2.99; h, C2ix–H2aix⋯S13, 3.04 Å; f, C32viii–H32viii⋯S12, 2.77 Å, 3.596(9) Å, 146°. Symmetry codes: (i) 2 − x, −y, 1 − z; (ii) 1 − x, 1 − y, −z; (iii) x, −1 + y, z; (iv) −1 + x, y, −1 + z; (v) 1 − x, −y, −z; (vi) −x, 1 − y, −1 − z; (vii) −1 + x, −1 + y, −1 + z; (viii) 2 − x, 1 − y, 1 − z; (ix) 1 + x, y, 1 + z. | ||
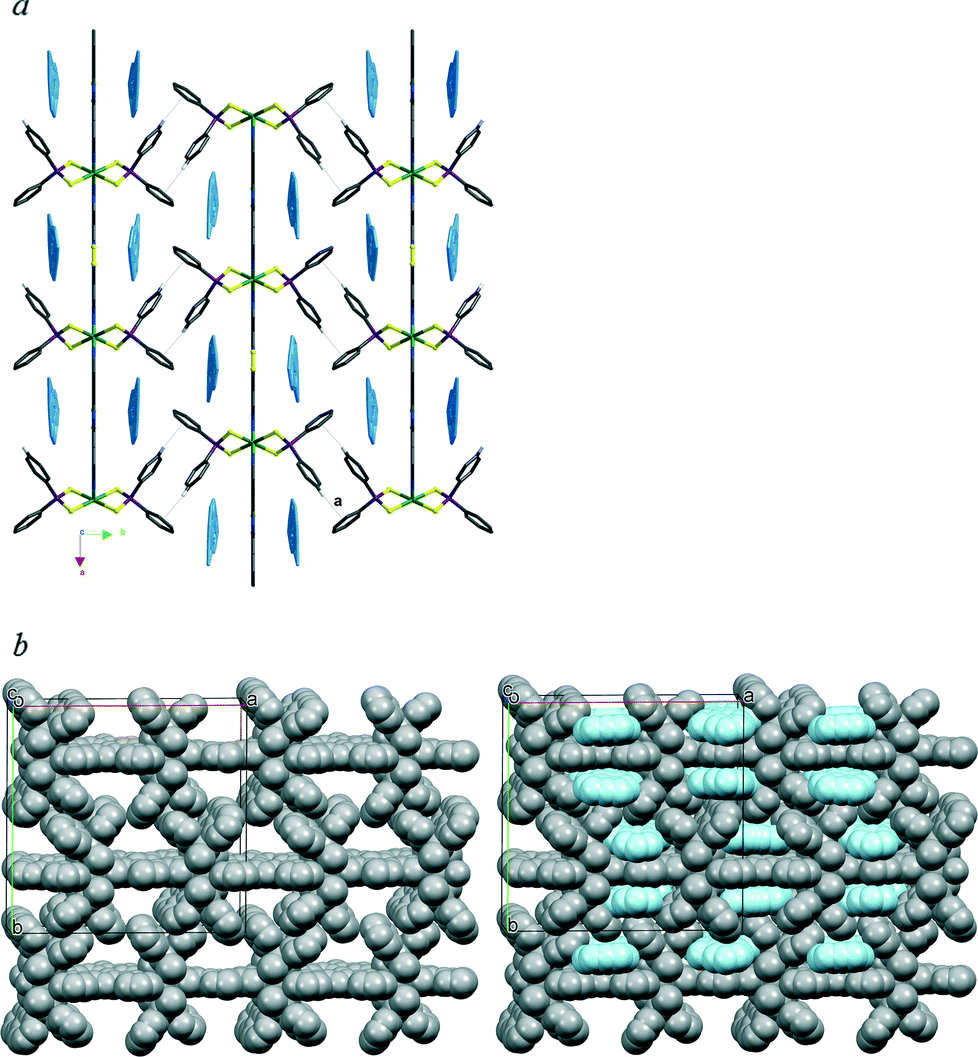 | ||
| Fig. 10 (a) Packing view along the 001 direction of the parallel chains of (4·L2)∞ with the toluene molecules included in the crystal evidenced in light blue colour. All the hydrogen atoms have been omitted with the exception of those involved in the shown interaction a, C27–H27a⋯Cnt(Ph(C18–C23))i 2.84 Å, (PhC24–C29)∧(PhC18–C23)i 91.4°. Symmetry code: (i) 1.5 − x, 1 − y, 0.5 + z. (b) Space-fill view of the network with (right) and without (left) the solvent molecules. | ||
Conclusions
The reaction of L1 with the differently P-substituted dithiophosphonato, dithiophosphato, and dithiophosphito complexes 1–4 yielded the corresponding coordination polymers (1–4·L1)∞ featuring polymeric [⋯(Py∩Py⋯M⋯Py∩Py)⋯] chains built from coordination to axial vacant sites on the nickel complexes. These results indicate that L1 can be used as a spacer for the predictable assembly of smoothly undulating chains independent of the nature of the interacting Lewis acid, since the orientation of the nitrogen atoms para-positioned in the outwards pyridyl rings of L1 self-governs the geometry of the resulting supramolecular construct. On the contrary, L2 allows for the existence of different supramolecular constructs ensuing from different ligand conformations deriving from the rotation of the pyridyl rings.In particular, the results suggest that aromatic P-substituents capable of π-interacting with the aromatic rings of the ligand tend to favour divergent constructs. The influence of secondary interactions involving the P-substituents is confirmed by the loss of planarity of L1 and L2 in order to enhance inter-molecular packing interactions.
Acknowledgements
MCA, MA, FI and VL kindly acknowledge the Dipartimento di Scienze Chimiche e Geologiche of the Università degli Studi di Cagliari and Fondazione di Sardegna for financial support (PRID 2015). MCA and MA kindly acknowledge RAS for the financial support coming from L.R. 7, CRP-78365.Notes and references
- Y. Wang, B. Yuan, Y.-Y. Xu, X.-G. Wang, B. Ding and X.-J. Zhao, Chem. – Eur. J., 2015, 21, 2107–2116 CrossRef CAS PubMed; X. Zhang, W. Wang, Z. Hu, G. Wang and K. Uvdal, Coord. Chem. Rev., 2015, 284, 206–235 CrossRef; P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC; A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gándara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863–8866 CrossRef PubMed; J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC; V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994–6010 RSC; P. R. Ashton, V. Balzani, A. Credi, O. Kocian, D. Pasini, L. Prodi, N. Spencer, J. F. Stoddart, M. S. Tolley, M. Venturi, A. J. P. White and D. J. Williams, Chem. – Eur. J., 1998, 4, 590–607 CrossRef.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC; M. C. Aragoni, M. Arca, F. A. Devillanova, M. B. Hursthouse, S. L. Huth, F. Isaia, V. Lippolis, A. Mancini, S. Soddu and G. Verani, Dalton Trans., 2007, 2127–2134 RSC; A.-L. Cheng, N. L. Yan-Feng Yue, Y.-W. Jiang, E.-Q. Gao, C.-H. Yan and M.-Y. He, Chem. Commun., 2007, 407–409 RSC; Z.-F. Chen, S.-F. Zhang, H.-S. Luo, B. F. Abrahams and H. Liang, CrystEngComm, 2007, 9, 27–29 RSC; K. Biradha, M. Sarkar and L. Rajput, Chem. Commun., 2006, 4169–4179 RSC; I. Goldberg, Chem. Commun., 2005, 1243–1254 RSC; D. Braga, L. Brammer and N. R. Champness, CrystEngComm, 2005, 7, 1–19 RSC; L. Brammer, Chem. Soc. Rev., 2004, 33, 476–489 RSC; S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed; C. B. Aakeröy, J. Desper and J. Valdés-Martínez, CrystEngComm, 2004, 6, 413–418 RSC; B. Rather and M. J. Zaworotko, Chem. Commun., 2003, 830–831 RSC; E.-Q. Gao, S.-Q. Bai, Z.-M. Wanga and C.-H. Yan, Dalton Trans., 2003, 1759–1764 RSC; S. Sain, T. K. Maji, G. Mostafa, T.-H. Lub and N. R. Chaudhuri, New J. Chem., 2003, 27, 185–187 RSC; N. L. Rosi, M. Eddaoudi, J. Kim, M. O'Keeffe and O. M. Yaghi, CrystEngComm, 2002, 4, 401–404 RSC; F. A. Cotton, C. Lin and C. A. Murillo, J. Chem. Soc., Dalton Trans., 2001, 499–501 RSC; M. E. Braun, C. D. Steffek, J. Kim, P. G. Rasmussen and O. M. Yaghi, Chem. Commun., 2001, 2532–2533 RSC; M. O'Keeffe, M. Eddaoudi, H. Li, T. Reineke and O. M. Yaghi, J. Solid State Chem., 2000, 152, 3–20 CrossRef.
- M. Arca, A. Cornia, F. A. Devillanova, A. C. Fabretti, F. Isaia, V. Lippolis and G. Verani, Inorg. Chim. Acta, 1997, 262, 81–84 CrossRef CAS.
- K. Biradha, M. Sarkar and L. Rajput, Chem. Commun., 2006, 4169–4179 RSC.
- M. C. Aragoni, M. Arca, F. Demartin, F. A. Devillanova, C. Graiff, F. Isaia, V. Lippolis, A. Tiripicchio and G. Verani, J. Chem. Soc., Dalton Trans., 2001, 2671–2677 RSC.
- I. Haiduc, Handbook of Chalcogen Chemistry, ed. F. A. Devillanova, Royal Society of Chemistry, 2006, pp. 593–643 Search PubMed.
- M. C. Aragoni, M. Arca, N. R. Champness, A. V. Chernikov, F. A. Devillanova, F. Isaia, V. Lippolis, N. S. Oxtoby, G. Verani, S. Z. Vatsadze and C. Wilson, Eur. J. Inorg. Chem., 2004, 10, 2008–2012 CrossRef.
- M. C. Aragoni, M. Arca, N. R. Champness, M. De Pasquale, F. A. Devillanova, F. Isaia, V. Lippolis, N. S. Oxtoby and C. Wilson, CrystEngComm, 2005, 7, 363–369 RSC.
- M. Crespo Alonso, M. Arca, F. Isaia, R. Lai, V. Lippolis, S. K. Callear, M. Caricato, D. Pasini, S. J. Coles and M. C. Aragoni, CrystEngComm, 2014, 16, 8582–8590 RSC.
- M. C. Aragoni, M. Arca, C. Caltagirone, C. Castellano, F. Demartin, A. Garau, F. Isaia, V. Lippolis, R. Montis and A. Pintus, CrystEngComm, 2012, 14, 5809–5823 RSC.
- P. Porta, A. Sgamellotti and N. Vinciguerra, Inorg. Chem., 1968, 7, 2625–2629 CrossRef CAS.
- G. M. Sheldrick, SHELX suite of programs for crystal structure solution and refinement, Univ. of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS V2.10, University of Göttingen, 2003 Search PubMed.
- Oxford Diffraction Ltd., Version 1.171.32.5 Search PubMed (release 08-05-2007 CrysAlis171 .NET).
- C. J. Cramer, in Essentials of Computational Chemistry, Wiley, Chichester, England, 2nd edn., 2004, ch. 8 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- (a) G. Schaftenaar and J. H. Noordik, J. Comput.-Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS PubMed; (b) R. Dennington, T. Keith and J. Millan, GaussView, Ver. 5.08, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- R. I. Meltzer, A. D. Lewis and J. A. King, J. Am. Chem. Soc., 1955, 77, 4062–4066 CrossRef CAS.
- I. Ondrejkovicova, R. Uhrecky, M. Koman, Z. Faberova, D. Lackova, J. Mrozinsky, B. Kalinska and Z. Padelkova, Inorg. Chim. Acta, 2014, 414, 33–38 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data and figures for (1–4·L1)∞, (1–2·L1)2, (3·L2)∞, and (4·L2·2C7H8)∞. CCDC 1474143–1474150. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce00991c |
| ‡ The 112th issue of Chem. Rev. (2012) and the 16th issue of Chem. Soc. Rev. (2014) were entirely devoted to the synthesis and applications of MOFs, showing how rapidly this branch of solid state materials chemistry is evolving. |
| § Among the ligands most commonly employed as spacers, the choice of using 4,4′-bipyridine and its analogues is due to their versatility. In fact, by introducing different groups between the two pyridyl rings, a wide variety of either linear or bent, rigid or flexible spacers are available. See for example ref. 4. |
| This journal is © The Royal Society of Chemistry 2016 |