
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tunable interfaces on tetracene and pentacene thin-films via monolayers†
Selma
Piranej
a,
David A.
Turner
a,
Shawn M.
Dalke
a,
Haejun
Park
a,
Brittni A.
Qualizza
a,
Juvinch
Vicente
b,
Jixin
Chen
b and
Jacob W.
Ciszek
*a
aDepartment of Chemistry and Biochemistry, Loyola University Chicago, Chicago, Illinois 60660, USA. E-mail: jciszek@luc.edu
bDepartment of Chemistry and Biochemistry, Ohio University, Athens, Ohio 45701, USA
First published on 17th June 2016
Abstract
To eliminate many of the traditional weaknesses of thin-film organic semiconductor materials, chemistry has been developed which reacts with the surface of these materials in a manner reminiscent of monolayers on traditional substrates. In the described approach, vapor phase small molecules react with the surface of tetracene and pentacene substrates to form an adlayer via classical Diels–Alder chemistry. The bonding is confirmed via measurement of several coupled vibrations via polarization modulation infrared reflection absorption spectroscopy, which importantly allows for differentiation from physisorbed materials. These films are then used to tune the materials' interaction with overlayers, as measured via a change in the contact angle the surface generates with water.
Introduction
The adoption of organic semiconductors within light emitting diode devices continues unabated. Currently, these materials dominate the small display market, where they can appear within both the active lighting element and the thin-film transistor which drives the LEDs within each pixel.1 However, despite the materials' successes, progress in controlling interfacial properties has lagged.2 The crux of the problems is the inability to generate a thin covalent coating on the surface. If the chemical identity of such a layer is tunable, the layer could address many of the problems pressing scientists and engineers including improving adhesion/stiction of overlayers,2 reducing contact resistance,3 and eliminating interfacial trap states.4 In fact, monolayers on inorganic substrates have long been shown to eliminate these same problems.5–8 The goal, thus, is to develop a viable means to generate monolayer like coatings on the surface of organic semiconductors (Fig. 1a). Specifically, we seek to generate controllable surface coatings on pentacene, which is relevant for thin-film transistors, and on tetracene, which is simpler in terms of both chemistry and stability. | ||
| Fig. 1 (a) Schematic of the vapor based reaction of small molecules onto a sublimed tetracene or pentacene substrate. (b) Top: General mechanism for the Diels–Alder reaction ([4+2] cycloaddition). Bottom: Representative examples of the adsorbates that have been shown to successfully react with a tetracene single crystals in ref. 9. Bolded bonds indicate those involved in the reaction. EWG = electron withdrawing group. | ||
The challenge in developing chemistry for surfaces comprised of acenes is unique. In contrast to more traditional inorganic surfaces, these organic semiconductors are made of building blocks that are held together by weak interactions.10–15 Thin-films of pentacene and tetracene are thus susceptible to solvent, high temperatures, and even extended periods under ultrahigh vacuum.16,17 Less obvious is the fact the orientation of the molecules within the surface and their tight packing can prevent the necessary transition states for a successful surface reaction.9 Perhaps in light of these challenges, only a limited number of successful demonstrations exist.
The first reported reaction of these materials was via the exposure of rubrene and tetracene single crystals to alkyl triethoxy- or trichlorosilane vapors which polymerize off of oxygenated defects on the surface.18,19 While the chemistry may prove to be effective (indeed, it is the standard means to functionalize glass slides),20 we thought it appropriate to develop a chemistry more germane to the chemical structure of the organic semiconductor surface. In doing so, one can design a system that reacts one adsorbate per surface site with chemistry that terminates at single layer thickness. If properly chosen, it would be able to install a variety of functional groups for interacting with overlayers,21 tuning interfacial dipoles,3 or other relevant effects. With an eye on these benchmarks, we turned to a classical chemistry that seems ideal for the conjugated π systems common to organic semiconductors: the Diels–Alder reaction. Here, a [4+2] cycloaddition occurs between an electron rich diene (common to the acene family) and a double bond within the adsorbate (Fig. 1b, bold).22
Our initial demonstrations were of vapor phase small molecules that reacted on an idealized test surface: tetracene single crystal substrates.9 Surface functionalization was facile, maintained the bulk substrate integrity, and worked for a range of adsorbates. This work aims to demonstrate that the Diels–Alder reaction is equally applicable to the relatively disordered microstructure of thin-films, and to adapt the work for pentacene. In particular, we use polarization modulation infrared reflection absorption spectroscopy (PM-IRRAS) to demonstrate the species is both covalently attached and that the associated vibrations match the spectra of a Diels–Alder adduct. As a first step towards adding new functionality to these materials, we also demonstrate the adlayer's ability to effect surface energy, as measured by a change in contact angle between a water droplet and the surface.
Experimental section
General experimental methods
Tetracene and pentacene (sublimed grade, 99.99% trace metal basis), N-methylmaleimide, maleic anhydride, and N-hydroxymaleimide, were purchased from Sigma-Aldrich and used without additional purification. Metal evaporations were performed in a Kurt J. Lesker NANO 38 thermal evaporator. Infrared spectra of standard powder samples were taken using a Thermo Nicolet Nexus 470 FT-IR with a Thermo Scientific Smart Endurance ATR attachment. Surface infrared spectra were taken on a Bruker Optics Tensor 37 FT-IR with a liquid nitrogen cooled MCT detector with a polarization modulation accessory (PMA 50) under nitrogen. PM-IRRAS spectra were converted to absorbance as described elsewhere.23 Organic semiconductors were deposited as thin-films using a home built sublimation chamber with a source to sample distance of 16–17.5 cm. Also attached is an Inficon SQM-160 thin-film deposition monitor which reports the growth of the film. Water contact angles were measured on a home built system at room temperature using a Veho VMS-004 Deluxe USB Microscope and Microcapture software.General organic semiconductor sublimation procedure
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2SO4/H2O2) for 30 min at room temperature then washed four times using 18 MΩ water and dried using nitrogen. Uncoated glass microscope slides were used for contact angle experiments while slides to be utilized for the spectroscopies were coated in gold. Following cleaning, 5 nm of chromium were thermally deposited followed by 100 nm of gold at a base pressure of <1 × 10−6 Torr. Both metals were evaporated at a rate of 1 Å s−1.
:1 H2SO4/H2O2) for 30 min at room temperature then washed four times using 18 MΩ water and dried using nitrogen. Uncoated glass microscope slides were used for contact angle experiments while slides to be utilized for the spectroscopies were coated in gold. Following cleaning, 5 nm of chromium were thermally deposited followed by 100 nm of gold at a base pressure of <1 × 10−6 Torr. Both metals were evaporated at a rate of 1 Å s−1.
Contact angle
A thin layer of tetracene was prepared following the standard sublimation procedure. The sample, reacted or unreacted, was placed in front of the camera and approximately 10 μL of 18 MΩ water was dropped onto the surface. Tangent lines were manually drawn on the drop image and the contact angle was measured using a protractor.AFM measurement
The sample, reacted or unreacted, was measured using an MFP 3D microscope (Asylum Research) in AC mode under ambient conditions. Diamond-like-carbon coated nano probes with a resonance frequency of ∼190 KHz and a spring constant of ∼48 N m−1 (TAP190DLC, Budget Sensors) were used.Results and discussion
Background on reaction conditions
Several comments about the experimental setup and subsequent reaction are necessary. First, reactions were initially conducted within the sublimation chamber, which has a separate isolatable dosing source. This setup allowed reactions to be performed without breaking vacuum, something presumed desirable as oxygen or other volatile adsorbates might render the surface unreactive. Later experiments on both tetracene and pentacene indicated that any hindrance of the Diels–Alder reaction from brief air exposure (10 min) was small. Second, the Diels–Alder on acene substrates is remarkably temperature sensitive. This unusual behavior will be detailed in a separate work. Third, the substrates of interest become volatile at modest temperatures, with a loss of material observed above 40 °C for tetracene in ultra-high vacuum.16 In order to ensure that the sublimed film was constant, we used a temperature where acene vaporization is negligible (40 °C for tetracene, 50 °C for pentacene). Surfaces were reacted for 48 h for tetracene, and 24 h for pentacene.The reaction itself was accomplished by placing the tetracene or pentacene thin-film into a ∼50 cm3 chamber containing approximately 5 mg of solid source material. Vapor from the source (N-methylmaleimide, maleic anhydride, and N-hydroxymaleimide) diffused to the thin-film and reacted while the system was kept sealed under vacuum. After the reaction, one end of the chamber was cooled with liquid nitrogen for 1 min in order to condense any residual vapors. The sample was then moved to high vacuum for 40 min (≤1 × 10−4 Torr) to remove any physisorbed species on the tetracene/pentacene. The reacted thin-film was then removed from vacuum and analyzed.
Analysis of functionalized tetracene surfaces
Polarization modulation infrared reflection absorption spectroscopy (PM-IRRAS) is a technique exquisitely suited for monitoring the reaction of tetracene and pentacene surfaces, provided these species are on a sufficiently thick metal backing (≥100 nm). On metal substrates, s-polarized light destructively interferes near the surface rendering the field near zero. PM-IRRAS dynamically removes the s-polarized signal and amplifies signal through the use of a lock in amplifier.24 These effects, in concert, yield a measurement that is sensitive enough to rapidly acquire the spectra of species that are present at well below a monolayer coverage, and eliminates the need for background measurements of the substrate. Just as important, the penetration of the infrared light means information about the subsurface tetracene or pentacene can be obtained at the same time as the reacted surface. From the spectra, it is possibly to both determine the extent of reaction and to comment on whether the reaction only occurs at the surface or throughout the thin-film. Infrared spectroscopy is also well suited to provide direct evidence of bond formation between the adsorbate and surface.Our initial analysis began with the many Diels–Alder adducts generated in the lab via solution phase synthesis, all of which have been fully characterized via1H NMR.9,25 These materials act as standards for our surface work. IR spectra of the powder adducts were collected via attenuated total reflectance (ATR) and the spectra were compared to the two starting materials to identify new stretches indicative of the newly formed adduct. Of the compounds listed in ref. 9 and 25, the adduct formed between N-methylmaleimide and tetracene has the most distinct vibrations (≥15 cm−1 difference from either starting material). It contains 6 prominent and distinct peaks that are located at 792, 803, 845, 1143, 1478 and 1499 cm−1; all of these are in spectral regions devoid of any peaks (major or minor) in either starting materials. These six peaks are still apparent in a 10:1 mixture of tetracene to standard adduct and thus were deemed likely to be visible when the adduct is but a ∼0.5 nm coating on a ∼40 nm tetracene thin-film. The spectra of the tetracene, N-methylmaleimide, and standard powder adduct, as well as the 10:1 mixture can be found in the ESI† (Fig. S1–S4).
Once measured, ATR data for the standard compounds were compared to the layer formed by the reaction of N-methylmaleimide and a tetracene thin-film. It was found that of the six characteristic stretches observed in the standard powder samples, all are present. Representative regions of the infrared spectra can be seen in Fig. 2, along with the data for the standard powder adduct. Additional regions can be found in the ESI† (1350–1200 cm−1 and 1225–1075 cm−1, Fig. S5 and S6).
 | ||
| Fig. 2 (a–c) Infrared spectra of selected regions of N-methylmaleimide (dashed line), tetracene (dotted line), and the Diels–Alder adduct formed during reaction of these two (solid line). Spectra labeled with transmittance are from pure powder samples generated via standard techniques9,25 (or commercially available) and were acquired via ATR-IR. Spectra labeled with absorbance are from thin-films deposited on gold substrates and were acquired via PM-IRRAS. Grey bands indicate strong adsorption bands present in the adduct but absent in the starting materials which can be used to identify the Diels–Alder adduct. | ||
As can be seen, the fidelity between the standard powder adduct and the surface reaction is excellent in the region spanning 1400 to 1550 cm−1, and the only feature not matching between the standard powder (solid line top) and the surface adduct (solid line, bottom) is a feature at 1539 cm−1 which is from the bulk tetracene (dotted line, bottom). From comparing the surface species and the standard N-methylmaleimide spectrum (dashed line, top), it is apparent that physisorption is not an appropriate explanation for the observed stretches. The same correlation between the standard sample and the “monolayer” can also be seen in the region from 775 to 925 cm−1, though analysis here is complicated by strong vibrations from the tetracene substrate at 904 cm−1 which obscures many of the useful features. What can be gleaned is that the features at 792, 802 and 840 cm−1 offer further proof for the successful formation of an adsorbed layer.
Considering the extent to with the standard adduct and the reacted thin-film match spectroscopically, the major species is clearly covalently bonded to the surface. However, the system should also be examined to comment on the absence of physisorbed material. From our experiments, we believe that little to no physisorbed material is present. This statement is justified in four ways. First, if present, the species should be apparent spectroscopically via bands at 1053, 1252 and 1587 cm−1. The latter two are completely absent, while the former is a minor feature. Second, N-methylmaleimide is rather volatile, with a vapor pressure of 50 mTorr at 7 °C.26 As such it is removed from the surface easily. Fig. S7 (ESI†) shows a tetracene thin-film on a salt plate, where N-methylmaleimide was sublimed onto the thin-film, while the substrate temperature was kept below 0 °C. When this same sample is exposed to a gentle stream of nitrogen for 5 minutes, the N-methylmaleimide peak disappears leaving only the signal for tetracene. Third, application of high vacuum conditions do not change the spectra significantly in regions associated with N-methylmaleimide. Neither the small feature at 1053 cm−1 nor any other peaks change noticeably in intensity, something that is extremely unlikely if the material were not covalently bonded to the surface. Fourth, the surface texture changes when exposed to the adsorbates, as measured by AFM (Fig. S8, ESI†). These surface changes have often been used by solid phase chemists as an indication of reaction.27 Based on these four arguments, we presume that no significant amount of physisorbed material is present, though we continued to treat each sample with high vacuum as a precautionary measure.
Analysis of functionalized pentacene surfaces
Similar experiments can be performed on thin-films of pentacene. For this system, the vibrational spectra of the pentacene layer, standard powder adduct, and N-methylmaleimide contain substantially more overlap reducing the number of vibrations that can be used diagnostically. In fact, only two frequencies (corresponding to 1775 cm−1 and 1126 cm−1) are in regions free from interference from the starting materials. Both are observed in reacted surface (Fig. 3a and S9, ESI†), and again, the fidelity is excellent. While the two regions are the most useful, others (containing overlapping signals) can be analyzed if care is taken in preparing a reference pentacene thin-film (ideally the same substrate before reaction, or another from the same sublimation). For example, in Fig. 3b, the adduct peaks can be clearly observed as added intensity at 1299 and 1280 cm−1, despite the presence of significant pentacene vibrations at those frequencies. This approach will become necessary if other adsorbates are to be analyzed.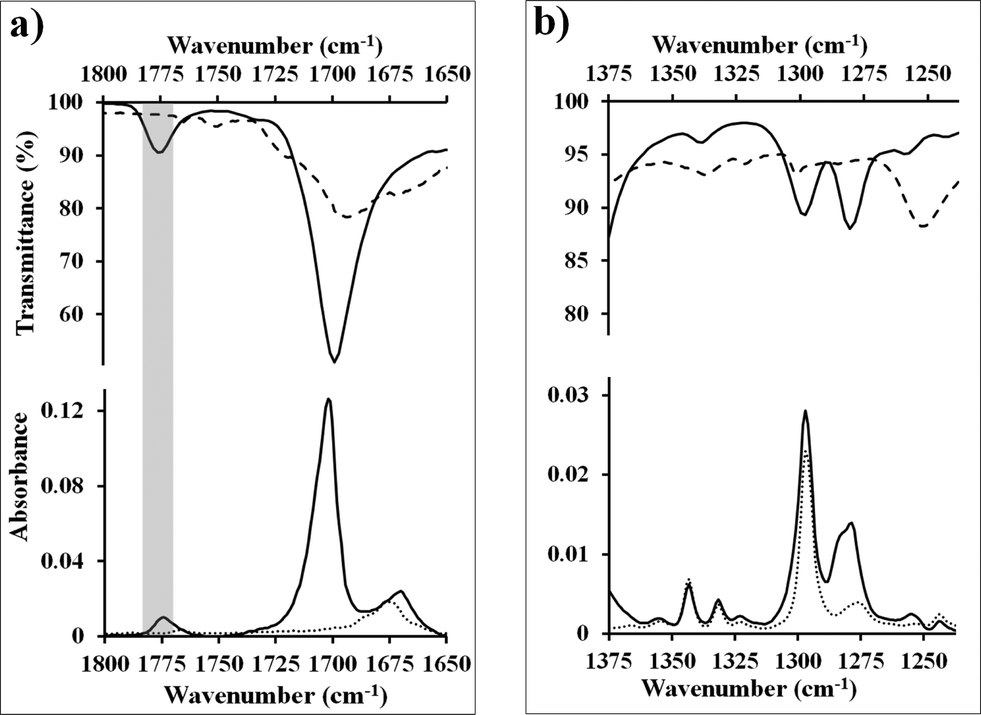 | ||
| Fig. 3 (a, b) Infrared spectra of selected regions of N-methylmaleimide (dashed line), pentacene (dotted line), and the Diels–Alder adduct formed during reaction of these two (solid line). Spectra labeled with transmittance are from pure powder samples generated via standard techniques9,25 (or commercially available) and were acquired on an ATR. Spectra labeled with absorbance are from substrates generated on gold surfaces and were acquired via PM-IRRAS. Fidelity between the powder adduct (top) and PM-IRRAS spectra (bottom) demonstrate a Diels–Alder adduct has been formed. | ||
Comparison of the reacted surfaces to other unreacted samples from the same lot also allows us to comment on the state of the subsurface material. For thin-films of pentacene generated during the same sublimation run, the measured intensity of the vibrations typically varies by less than 10%. Thus, PM-IRRAS can determine whether the subsurface pentacene remains unperturbed, or whether it has been lost to sublimation or consumed by the reaction. The reacted sample shown in Fig. 3 was reanalyzed, with a particular focus on the prominent signals at 907 and 731 cm−1 which correspond to out-of-plane vibrations of pentacene.28 These peaks are appropriate for subsurface analysis as the adduct has no significant vibration at 731, and only a weak vibration at 904 cm−1.28 As can be seen in Fig. 4, all the vibrations corresponding to the pentacene thin-film remain, and are only minimally diminished. In fact the most notable change corresponds to an increase in intensity at 760 cm−1 which is the formation of adduct at the surface. It is also important to note that none of the features associated with a change in the microstructure can be seen. Reorientation of the pentacene film would be expected to display strong stretches at 1145 and 1162 cm−1.28 The latter region is easily analyzed and, as can be seen in Fig. 4, no signal is present. The relative lack of change in the features of the pentacene thin-film seem to suggest that the electronic properties of pentacene should remain unchanged; this is of great importance when these materials are used in devices.
 | ||
| Fig. 4 Infrared spectra of substantial features associated with the pentacene thin-film both before (dotted line) and after reaction with N-methylmaleimide (solid line). Outside of the formation of the surface adduct, the remainder of the thin-film is unperturbed and the absence of a feature at 1160 cm−1 (red arrow) indicates that the film has not changed orientation. Data has not been baseline corrected or converted to absorbance. The offset and slight curve is due to the Bessel function that is part of normal data acquisition with a PEM controller.23 | ||
Contact angle
Adlayers are remarkably effective at tuning a materials' interaction with overlayers. When the interaction is highly optimized (e.g. oxygenated terminal groups for adhesion to Ni, Cu, or Cr overlayers) the increase in adhesion strength can be several orders of magnitudes.21 To demonstrate the ability of a thin organic monolayer to affect similar change, tetracene was analyzed via contact angle measurements showing the wettability of the surface. Though contact angle is a remarkably simple technique, it can also provide a wealth of information about the energy of interaction between the layer and its surrounding media, and is often used as a simple surrogate for surface energy (γs) and adhesion/stiction.8,10Before these tests were attempted, an acceptable initial value for tetracene's contact angle was required (no prior reports exist). Measurements of progressively thicker surfaces find that, initially, tetracene thin-films have a low contact angle (likely because of the discontinuous nature of the initial layers), and that by 100 nm, the surface reaches a consistent value above which further deposition does not affect the contact angle (Fig. S10, ESI†). The average contact angle for a 100 nm thick tetracene film was 74°.
In order to affect a useful change, the goal was to lower the contact angle via a monolayer, and in turn raise the surface energy and adhesion of the surface. The first attempts via simple adsorbates (maleic anhydride, N-hydroxymaleimide, Fig. 5a–c) had a modest effect that were in line with literature precedence on classical surfaces,29 and demonstrate the potential effects of the monolayers. For maleic anhydride, the average contact angle across multiple maleic anhydride treated substrates was 63°. For N-hydroxymaleimide it was 60°. These samples were also useful in discerning the variance and reproducibility of the contact angle. In the case of the maleic anhydride treated substrates the standard deviation was 13°. Contact angle values for individual substrates showed little deviation across the sample (the mean deviation was 3°), suggesting consistent coverage at the macroscopic scale.
 | ||
| Fig. 5 Representative images of the contact angle formed between water and sublimed films of tetracene (100 nm). (a) An unreacted tetracene film. (b) A film that had been reacted with maleic anhydride vapors. (c) A film that had been reacted with N-hydroxymaleimide vapors. (d) A maleic anhydride reacted film that had been exposed to water vapors for 48 h. The associated change in the carbonyl stretch indicates consumption of the anhydride moiety. The new broad peak at ∼1575 cm−1 also suggest the potential presence of anionic charge in the adduct.30,31 | ||
Creation of a high energy surface requires functional groups such as carboxylic acids. However, dienophiles of this structure would have much lower volatility and slower reactivity.32 As such, a more practical approach was to take the maleic anhydride layer and convert it to the diacid. Such an approach is also advantageous as a single anhydride is converted to two carboxylic acids, effectively doubling the density of high energy groups. Conversion was reasonably facile, and was accomplished by exposing the sample to water vapor at 70 °C for 48 h (under a nitrogen atmosphere).
The results were remarkable: the contact angle of the tetracene substrate was reduced to 16° (Fig. 5d). Control experiments show that though humidity itself does not appreciably change the tetracene sample (Fig. S11†). Furthermore, when the sample shown in Fig. 5d is dried in a desiccator, no change in the contact angle occurs, again demonstrating the ring opening is the dominant cause of this change. Though it is difficult to make a sweeping generalization about how this change in contact angle would impact adhesive forces (as these are highly dependent on the adhering material and the nature of the induced interaction), such a change should generically increase the adhesion force by a factor of two.33 These terminal groups are also ideal for minimizing the penetration of metal overlayers into the bulk semiconductor.34 Additionally, there is no reason to believe that this approach is incompatible with other organic semiconductors such as phenacenes, triphenodioxazines, or other polyaromatics.35–38 As such this approach portends well to utilization in complex electronic devices.
Conclusion
In summary, we have demonstrated the ability to alter the surface layer of thin-film organic semiconductors via small molecular adsorbates. Infrared results serve to confirm the formation of a covalently bonded Diels–Alder adduct at the surface and that the bulk of the thin-film acene (non-surface) remains unaltered. This surface coverage will allow scientists to begin engineering the properties of these materials which we have demonstrated by changing the wettability of the surface. The versatility of the approach and its relative simplicity suggest that this chemistry can proliferate much in the same way monolayers revolutionized classical surfaces two decades ago.Acknowledgements
JWC thanks NSF CAREER award #1056400 for supporting this work and Dr. Dan Batzel for some preliminary studies. BAQ thanks the Arthur J. Schmitt foundation for a graduate fellowship.References
- L. Zhou, S. Park, B. Bai, J. Sun, S.-C. Wu, T. N. Jackson, S. Nelson, D. Freeman and Y. Hong, IEEE Electron Device Lett., 2005, 26, 640–642 CrossRef CAS.
- Y. G. Seol, N.-E. Lee, S. H. Park and J. Y. Bae, Org. Electron., 2008, 9, 413–417 CrossRef CAS.
- B. H. Hamadani, D. A. Corley, J. W. Ciszek, J. M. Tour and D. Natelson, Nano Lett., 2006, 6, 1303–1306 CrossRef CAS PubMed.
- W. L. Kalb, S. Haas, C. Krellner, T. Mathis and B. Batlogg, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 155315 CrossRef.
- P. E. Laibinis and G. M. Whitesides, J. Am. Chem. Soc., 1992, 114, 9022–9028 CrossRef CAS.
- J. Adams, G. Tizazu, S. Janusz, S. R. J. Brueck, G. P. Lopez and G. J. Leggett, Langmuir, 2010, 26, 13600–13606 CrossRef CAS PubMed.
- H. Ma, H.-L. Yip, F. Huang and A. K.-Y. Jen, Adv. Funct. Mater., 2010, 20, 1371–1388 CrossRef CAS.
- J. F. Kang, A. Ulman, S. Liao, R. Jordan, G. Yang and G. Liu, Langmuir, 2001, 17, 95–106 CrossRef CAS.
- B. A. Qualizza, S. Prasad, M. P. Chiarelli and J. W. Ciszek, Chem. Commun., 2013, 49, 4495–4497 RSC.
- J. N. Israelachvili, Intermolecular and surface forces, Academic Press, Amsterdam; Boston, 2nd edn, 1991 Search PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430–3434 CrossRef CAS PubMed.
- H. M. Duong, M. Bendikov, D. Steiger, Q. Zhang, G. Sonmez, J. Yamada and F. Wudl, Org. Lett., 2003, 5, 4433–4436 CrossRef CAS PubMed.
- J. Xiao, H. M. Duong, Y. Liu, W. Shi, L. Ji, G. Li, S. Li, X.-W. Liu, J. Ma, F. Wudl and Q. Zhang, Angew. Chem., Int. Ed., 2012, 51, 6094–6098 CrossRef CAS PubMed.
- C. Wang, J. Zhang, G. Long, N. Aratani, H. Yamada, Y. Zhao and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 6292–6296 CrossRef CAS PubMed.
- A. Langner, A. Hauschild, S. Fahrenholz and M. Sokolowski, Surf. Sci., 2005, 574, 153–165 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, 83rd edn, 2002 Search PubMed.
- D. J. Ellison, B. Lee, V. Podzorov and C. D. Frisbie, Adv. Mater., 2011, 23, 502–507 CrossRef CAS PubMed.
- M. F. Calhoun, J. Sanchez, D. Olaya, M. E. Gershenson and V. Podzorov, Nat. Mater., 2008, 7, 84–89 CrossRef CAS PubMed.
- D. L. Angst and G. W. Simmons, Langmuir, 1991, 7, 2236–2242 CrossRef CAS.
- J. M. Burkstrand, J. Appl. Phys., 1981, 52, 4795–4800 CrossRef CAS.
- I. Fleming, Pericyclic reactions, Oxford University Press, Oxford, New York, 1999 Search PubMed.
- Handbook of Vibrational Spectroscopy, ed. J. M. Chalmers and Peter R. Griffiths, John Wiley & Sons Ltd., 2002, vol. 2 Search PubMed.
- P. R. Griffiths, Fourier transform infrared spectrometry, Wiley-Interscience, Hoboken, N. J., 2nd edn, 2007 Search PubMed.
- B. A. Qualizza and J. W. Ciszek, J. Phys. Org. Chem., 2015, 28, 629–634 CrossRef CAS.
- M. V. Roux, P. Jiménez, M. Á. Martín-Luengo, J. Z. Dávalos, Z. Sun, R. S. Hosmane and J. F. Liebman, J. Org. Chem., 1997, 62, 2732–2737 CrossRef CAS PubMed.
- G. Kaupp, J. Schmeyers and J. Boy, Tetrahedron, 2000, 56, 6899–6911 CrossRef CAS.
- W. S. Hu, Y. T. Tao, Y. J. Hsu, D. H. Wei and Y. S. Wu, Langmuir, 2005, 21, 2260–2266 CrossRef CAS PubMed.
- C. D. Bain, E. B. Troughton, Y. T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321–335 CrossRef CAS.
- M. Ilczyszyn, D. Godzisz and M. M. Ilczyszyn, Spectrochim. Acta, Part A, 2003, 59, 1815–1828 CrossRef CAS.
- L. G. Kaake, Y. Zou, M. J. Panzer, C. D. Frisbie and X.-Y. Zhu, J. Am. Chem. Soc., 2007, 129, 7824–7830 CrossRef CAS PubMed.
- W. E. Bachmann and L. B. Scott, J. Am. Chem. Soc., 1948, 70, 1458–1461 CrossRef CAS PubMed.
- B. D. Beake and G. J. Leggett, Phys. Chem. Chem. Phys., 1999, 1, 3345–3350 RSC.
- G. C. Herdt, D. R. Jung and A. W. Czanderna, Prog. Surf. Sci., 1995, 50, 103–129 CrossRef CAS.
- D. Biermann and W. Schmidt, J. Am. Chem. Soc., 1980, 102, 3163–3173 CrossRef CAS.
- D. Biermann and W. Schmidt, J. Am. Chem. Soc., 1980, 102, 3173–3181 CrossRef CAS.
- G. Gruntz, H. Lee, L. Hirsch, F. Castet, T. Toupance, A. L. Briseno and Y. Nicolas, Adv. Electron. Mater., 2015, 1, 1500072 CrossRef.
- Y. Nicolas, F. Castet, M. Devynck, P. Tardy, L. Hirsch, C. Labrugère, H. Allouchi and T. Toupance, Org. Electron., 2012, 13, 1392–1400 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data of standard powder adducts, prestine surfaces, and reacted surfaces (ATR-IR, PM-IRRAS, contact angles). See DOI: 10.1039/c6ce00728g |
| This journal is © The Royal Society of Chemistry 2016 |