
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymorphic transformations in glycine co-crystals at low temperature and high pressure: two new examples as a follow-up to a glycine–glutaric acid study†
E. A.
Losev
*ab,
B. A.
Zakharov
ab and
E. V.
Boldyreva
a
aInstitute of Solid State Chemistry and Mechanochemistry SB RAS, Kutateladze 18, Novosibirsk, 630128, Russia. E-mail: losev.88@mail.ru; b.zakharov@yahoo.com; eboldyreva@yahoo.com; Fax: +7 8 383 363 41 32; Tel: +7 8 383 363 42 06
bREC-008, Novosibirsk State University, Pirogova 2, Novosibirsk, 630090, Russia
First published on 20th June 2016
Abstract
The effects of temperature and pressure on the co-crystals of glycine with DL-tartaric and phthalic acids (GTa and GPh, respectively) have been studied by X-ray diffraction and Raman spectroscopy in comparison with those on glycine–glutaric acid (GGa). On cooling, no phase transitions were observed in GTa or GPh, in contrast to the situation with GGa. On hydrostatic compression, both GTa and GPh underwent reversible phase transformations, accompanied by fracture. In the high-pressure phases, the main structural framework was preserved, and the number of crystallographically independent molecules in the unit cell increased. In GTa, dimers are squeezed together so that some hydrogen bonds get a three-centered character, and the interactions of one of the two glycine molecules change dramatically.
Introduction
Interest in studying and understanding the effects of temperature and pressure on multicomponent molecular crystals has grown considerably in recent years.1 Their properties differ from those of the corresponding single-component crystals and often cannot be predicted due to unique lattice structures. This has rendered the prediction of the properties of multi-component materials very challenging. Of particular note are the difficulties in predicting structural strain anisotropy, the possibility of structural phase transitions,2–6 or the potential for co-crystals to form salts via proton transfer.7–12Analysis of the Cambridge Structural Database (version 5.36 from November 2015)13 reveals ten two-component crystals of glycine with carboxylic acids (excluding hydrates and solvates), namely, three salts with oxalic acid,14–16 salts with malonic,17 maleic,18 3-nitrophthalic19 and 2-hydroxy-5-sulfobenzoic20 acids, as well as co-crystals with glutaric,21,22 phthalic23 and DL-tartaric24 acids. The ΔpKa (ΔpKa = pKa(base)- pKa(acid)) values (measured in aqueous solution) for glycine and other listed carboxylic acids fall in the range where it is impossible to predict if the two-component crystal will be a co-crystal or a molecular salt (Table S1 and Fig. S1 in the ESI†).25–29 Therefore, one might expect that the hydrogen atom of a carboxylic group of the acid in the co-crystals can be transferred from an acidic species to a basic one under pressure, similar to what has been observed for multi-component crystals of other compounds, e.g. for the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct formed between squaric acid and 4,4-bipyridine12 or for oxalic acid dihydrate with increasing pressure.30–32
:1 adduct formed between squaric acid and 4,4-bipyridine12 or for oxalic acid dihydrate with increasing pressure.30–32
Three glycine co-crystals with carboxylic acids have been reported to date, namely, those with glutaric,21,22DL-tartaric24 and phthalic23 acids (GGa, GTa, and GPh, respectively) (Fig. 1). Topologically, the crystal structures of GGa and GTa are similar, and have 3D hydrogen bond networks (Fig. 2). The crystal structure of GPh contains layers of glycine molecules, with phthalic acid molecules connected to these layers by hydrogen bonds. Weak hydrophobic stacking interactions exist between phthalic acid molecules (Fig. 2). The nature of the hydrogen bond networks is quite different in each case: GGa has only heteromolecular hydrogen bonds, GPh contains two types of glycine–glycine interactions (Fig. S2 in the ESI†) and GTa co-crystals have both glycine–glycine and acid–acid hydrogen bonds (Fig. S2 in the ESI†).
 | ||
| Fig. 1 Glutaric (a), DL-tartaric (b) and phthalic (c) acids. | ||
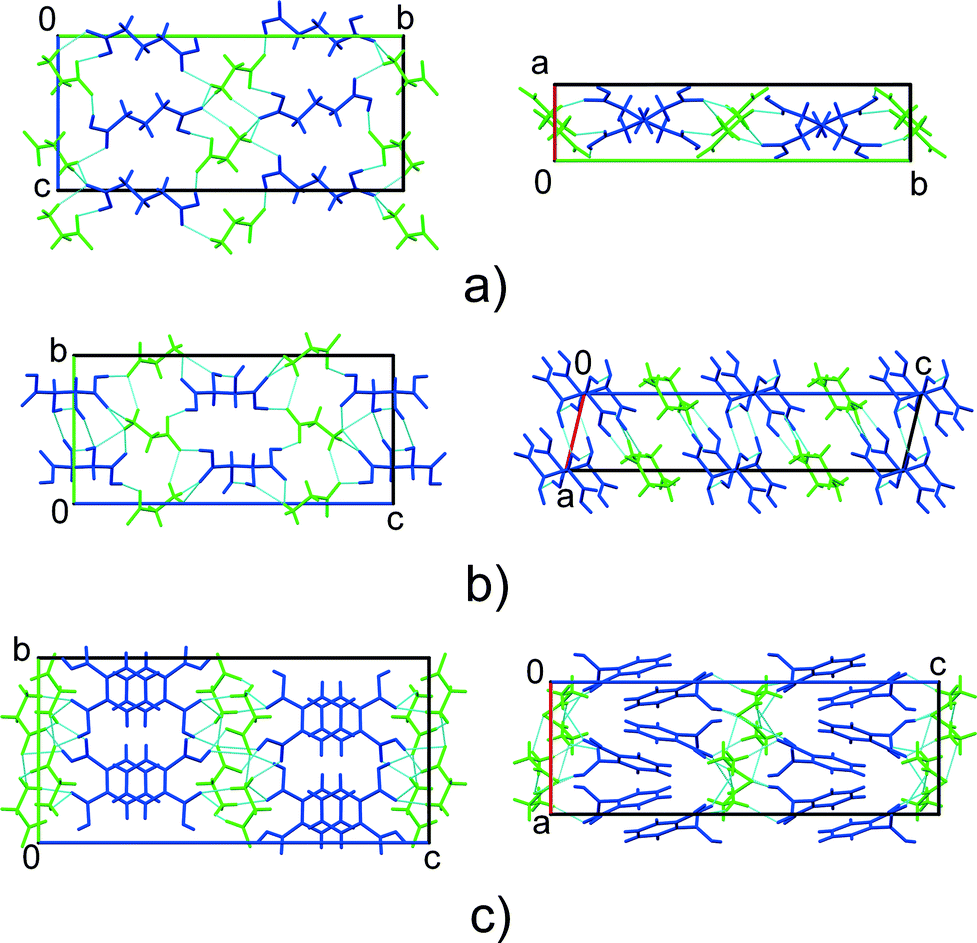 | ||
| Fig. 2 Comparison of the crystal structures of GGa (a), GTa (b) and GPh (c) co-crystals. Glycine – green, co-former – blue. | ||
In our previous work, GGa has been shown to undergo the same structural phase transition both on cooling33 and with increasing pressure.34 This transition is related to a change in the glutaric acid conformation and occurs at about 0.1 GPa (ambient temperature), which is considerably lower than the pressure at which proton transfer could be expected, judging from the data obtained for oxalic acid dihydrate (5–6 GPa) – the only multicomponent crystal constructed from neutral molecules – for which the fact of proton transfer on compression has been supported by high-quality single-crystal X-ray and neutron diffraction data, spectroscopic data, and computations.30,35 Another multicomponent glycine-containing crystal explored at high pressure was glycinium oxalate. Spectroscopic and computational studies revealed the symmetrisation of the position of hydrogen between adjacent semi-oxalate molecules at about 8 GPa.36 During the phase transition in GGa, the crystal symmetry changed from monoclinic to triclinic, with every second glutaric acid molecule changing its conformation.33 The aim of the present study was to investigate co-crystals of glycine with two other co-formers, DL-tartaric and phthalic acids (GTa and GPh, respectively), and to compare their response to variations in temperature and pressure with that of GGa. We also compared the response of GGa, GTa and GPh to pressure with that of selected amino acids salts.
Experimental section
Materials
All reagents were commercially available and were used without further purification. The following chemicals were used: α-glycine (Reactiv, Russia, pur. >95%), DL-tartaric acid (Reactiv, Russia, pur. >95%), phthalic acid (Sigma-Aldrich, puriss ≥99.5%), and distilled water.Crystallisation
GTa and GPh single crystals were grown from equimolar aqueous solutions of the starting components (total solid mass ∼0.6 g). Particular attention was required for the successful preparation of GPh crystals, and a three-step process was developed: 1. preparation of a saturated solution of α-glycine at room temperature; 2. addition of phthalic acid up to a saturation limit at approximately 70–80 °C; 3. slow cooling to room conditions, upon which GPh crystallised.Single crystal X-ray diffraction
X-AREA37 and X-RED32 (ref. 37) were used for cell refinement, data collection and data reduction. The crystal structures were solved by direct methods using SHELXS97 (ref. 38) and refined on F2 using X-STEP,37 SHELXL2014 (ref. 38) and OLEX2 Version 1.2.7.39 Mercury40 was used for the visualisation and analysis of structure motifs. Win_Strain41 was used to calculate the linear strain along the principal axes of the strain ellipsoid. All structures were deposited with the Cambridge Crystallographic Data Centre (CCDC numbers 1435507, 1437082–1437089 and 1437185–1437193 for GTa and GPh, respectively) and are available at http://www.ccdc.cam.ac.uk/.
The data were collected (pressure range for GTa: 0.1–5.9 GPa, for GPh: 0.2–6.0 GPa) using an Oxford Diffraction Gemini R Ultra X-ray diffractometer with a CCD area detector and Mo Kα radiation. The following software packages were used: CrysAlisPro45 (data collection, cell refinement and data reduction), SHELXS 97 (structure solution),38 SHELXL 2014,38 X-STEP 32 (ref. 37) (structure refinement) and OLEX2 Version 1.2.7 (structure refinement).39The absorption of X-rays by the diamonds was accounted for using ABSORB (Gaussian absorption correction).46 Reflections from the sample that overlapped with those from the diamond and gasket were excluded manually. Mercury40 was used for the structure visualisation and analysis.
The crystal structure of the GTa high-pressure phase was solved by direct methods implemented in SIR2014 (ref. 47) software. All atoms in the structure of GTa in the DAC experiments were refined isotropically since not enough reflections were obtained from the dataset for adequate anisotropic refinement. The hydrogen atom parameters for all DAC experiments were constrained and refined using a riding model (AFIX 33 with Uiso(H) = 1.2Ueq(N) and NH distances equal to 0.890 Å for NH3 groups; AFIX 83 with Uiso(H) = 1.2Ueq(O) and OH distances equal to 0.820 Å for O–H groups, AFIX 13 with Uiso(H) = 1.2Ueq(C) and CH distances equal to 0.980 Å for CH groups, AFIX 23 with Uiso(H) = 1.2Ueq(C) and CH distances equal to 0.970 Å for CH2 groups, AFIX 43 with Uiso(H) = 1.2Ueq(C) and CH distances equal to 0.930 Å for aromatic CH groups). All structures were deposited with the Cambridge Crystallographic Data Centre (CCDC numbers 1451443–1451454 and 1450716–1450717 for GTa and GPh, respectively) and are available at http://www.ccdc.cam.ac.uk/.
Raman spectroscopy
Raman experiments were performed in parallel with X-ray diffraction to obtain additional information on high-pressure structural transformations. For each pressure point, Raman spectra were recorded immediately after collection of X-ray diffraction data. Single-crystal Raman spectra were collected using a LabRAM Horiba single spectrometer with a CCD Symphony (Jobin Yvon) detector. The 488 nm line of an Ar+ laser with a beam size of 1 mm and 8 mW power was used for spectral excitation. The spectra at all pressures were measured in 180° backscattering collection geometry using a Raman microscope. While the spectral resolution was 2 cm−1, the observed broadening was significantly higher due to an increase in anharmonicity with increasing pressure.Optical microscopy
Microphotography was performed using a Nikon AZ100 optical microscope and the above-mentioned Raman microscope.Results and discussion
The parameters characterising the data collection and refinement, as well as the crystal data at different temperatures and pressures are summarised in Tables ST1–ST8 in the ESI.† In contrast with GGa, GTa and GPh were stable on cooling from 300 K to 100 K, during which monotonic changes to the volume and cell parameters were observed. There was no evidence of phase transitions over this temperature range (Fig. S3 and S4 in the ESI†). The volume decrease from 293 K to 100 K was slightly higher for GPh (2.6%) compared with that for GTa (2.2%). For comparison, a low-temperature reversible first-order phase transition, which was previously observed in GGa between 225 and 200 K, was associated with a volume change of approximately 1% compared to the volume at 300 K. Most probably, the conformational flexibility of glutaric acid is a reason for the phase transition in GGa at low temperature. In the case of GTa, a skeleton of DL-tartaric acid is stabilised due to hydrogen bonds between hydroxyl groups, whereas in the structure of GPh there are also multiple interactions between the aromatic rings of the phthalic acids and glycine layers that allow more compression compared with GGa.Judging from the almost equal values of the two C–O distances in the carboxylic group of glycine, the group remained non-protonated, i.e. no proton migration from the co-former – a carboxylic acid – to glycine occurred on cooling over the whole studied temperature range. The main structural changes were related to compression or extension of hydrogen bonds. The data on the geometries of the hydrogen bonds are summarised in Tables ST5–ST8 in the ESI.† Most of the hydrogen bonds in GTa and GPh shorten on cooling, whereas only one of them (O3–H3⋯O2 [x − 1, y, z] in GTa) extends slightly (by 0.4%, Fig. S5 and S6 in the ESI†). Interestingly, the O3–H3⋯O2 [x − 1, y, z] hydrogen bond is one of the shortest (and, supposedly, the strongest) intermolecular interactions in GTa (Fig. S5 and S6 in the ESI†). Evaluation of all torsion angles in the structure of GTa reveals a maximal change of about 2°, which could be sufficient to make the hydrogen bond longer. Alternatively, the bond extension can be related to a slight change of the molecular coordinates (shifting the molecule as a whole) with temperature, or to the cooperative effect of the compression of other hydrogen bonds in the 3D network, which accounts for the lowering of the overall lattice energy. Analysis of the linear strain in the two glycine co-crystals on cooling shows two different types of behavior of GTa and GPh crystal structures: GTa has one preferential direction of compression, whereas the compression of GPh along two principal axes is approximately equivalent. The maximum compression in the GTa structure is close to the direction of the mixed glycine–DL-tartaric acid hydrogen bonded chains. The maximum linear compression in the GPh structure lies along the stacks of the phthalic acid molecules, where weak hydrophobic interactions dominate. Another direction of the significant compression in GPh lies along the c-axis perpendicular to glycine layers and almost parallel with respect to the plane to which the benzene rings of phthalic acid are slightly inclined. The presence of non-hydrogen bonded layers of phthalic acid results in the appearance of two almost equivalent directions of compression (Fig. S7–S9 in the ESI†).
With increasing pressure, both GTa and GPh underwent reversible first-order phase transitions accompanied by fracture (Fig. 3). These transitions occurred at very low pressures (about 0.5 GPa and 1 GPa, respectively). For GTa, changes in the cell parameters and volume were tracked (Fig. 4), and the structure of the high-pressure phase was solved. The change from a monoclinic space group (P21/c) to a triclinic one (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) was accompanied by the doubling of the number of formula units per asymmetric unit cell (Z′) (Fig. 5), similar to what has been observed in the case of GGa (Fig. S10 in the ESI†). The increase in Z′ in the denser phases formed on cooling or at high pressures has been reported earlier also for single-component molecular crystals,48e.g. for chlorpropamide,49,50 although examples of the opposite are also known.51 Anisotropy of the linear strain and the directions of the principal axes of the strain ellipsoid for the GTa triclinic phase have been calculated in the range from 0.4 to 5.91 GPa (Table S2 and Fig. S11a in the ESI†). The triclinic phase of GTa contains two symmetrically independent dimeric chains of DL-tartaric acid that are stabilised by O–H⋯O hydrogen bonds between side OH-groups. An increase in the pressure causes the sliding of the tartaric acids relative to each other. The distance between O13 and O16 [2 − x, 2 − y, 2 − z] becomes shorter and at about 3 GPa reaches a value, which is usually assumed to be the threshold distance, below which an interaction can be classified as a hydrogen bond (about 2.8 Å at 5.9 GPa).52 Though the coordinates of the H-atoms cannot be considered as reliable, based on the O–O distances one can suppose that the O–H⋯O hydrogen bond becomes three-centered (Fig. S12 in the ESI†). Similar changes in the hydrogen bonds in the crystals of amino acids and their salts have been reported earlier for DL-alaninium semi-oxalate monohydrate,3L-serine,53 and DL-serine.54 Another major structural change is associated with the coordination of one of the glycine molecules: as pressure increases, the N11–O11 [2 − x, 1 − y, 2 − z] distance shortens, which can be related to the strengthening of the interaction between glycine and the corresponding tartaric acid molecules (Fig. S13 in the ESI†).
) was accompanied by the doubling of the number of formula units per asymmetric unit cell (Z′) (Fig. 5), similar to what has been observed in the case of GGa (Fig. S10 in the ESI†). The increase in Z′ in the denser phases formed on cooling or at high pressures has been reported earlier also for single-component molecular crystals,48e.g. for chlorpropamide,49,50 although examples of the opposite are also known.51 Anisotropy of the linear strain and the directions of the principal axes of the strain ellipsoid for the GTa triclinic phase have been calculated in the range from 0.4 to 5.91 GPa (Table S2 and Fig. S11a in the ESI†). The triclinic phase of GTa contains two symmetrically independent dimeric chains of DL-tartaric acid that are stabilised by O–H⋯O hydrogen bonds between side OH-groups. An increase in the pressure causes the sliding of the tartaric acids relative to each other. The distance between O13 and O16 [2 − x, 2 − y, 2 − z] becomes shorter and at about 3 GPa reaches a value, which is usually assumed to be the threshold distance, below which an interaction can be classified as a hydrogen bond (about 2.8 Å at 5.9 GPa).52 Though the coordinates of the H-atoms cannot be considered as reliable, based on the O–O distances one can suppose that the O–H⋯O hydrogen bond becomes three-centered (Fig. S12 in the ESI†). Similar changes in the hydrogen bonds in the crystals of amino acids and their salts have been reported earlier for DL-alaninium semi-oxalate monohydrate,3L-serine,53 and DL-serine.54 Another major structural change is associated with the coordination of one of the glycine molecules: as pressure increases, the N11–O11 [2 − x, 1 − y, 2 − z] distance shortens, which can be related to the strengthening of the interaction between glycine and the corresponding tartaric acid molecules (Fig. S13 in the ESI†).
 | ||
| Fig. 3 Images of co-crystals in the DAC. (a) Fracture of the glycine–DL-tartaric acid (GTa) co-crystals and (b) glycine–phthalic acid (GPh) co-crystals across the high-pressure phase transition. | ||
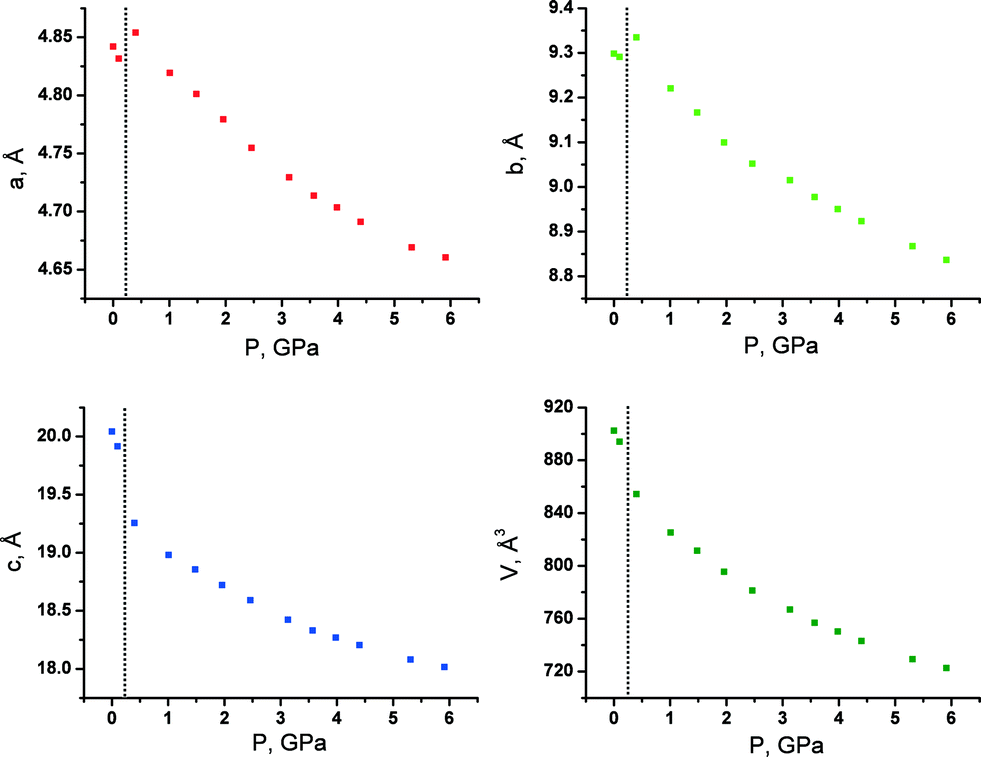 | ||
| Fig. 4 Unit cell parameters and volume of GTa at different pressures. The phase transition takes place between 0.1 and 0.4 GPa. | ||
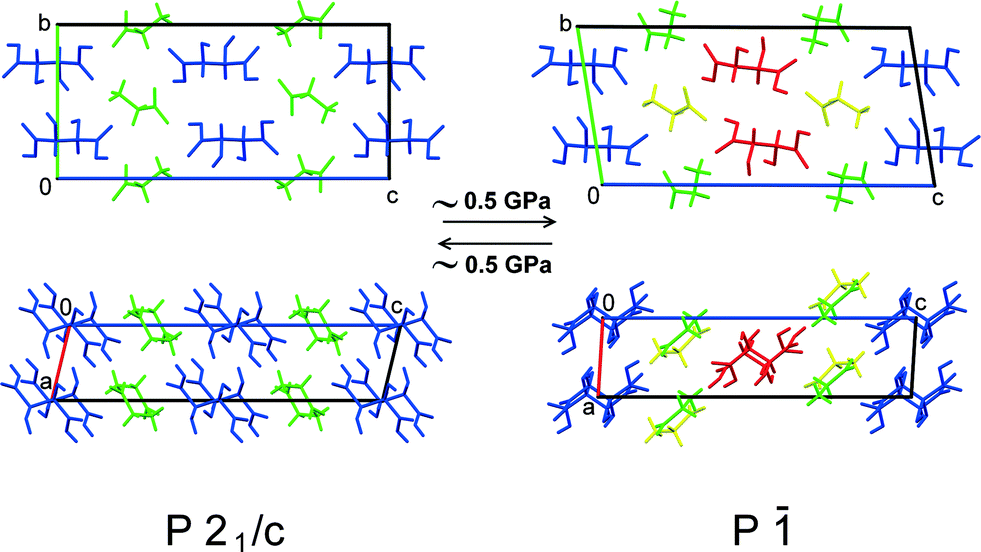 | ||
| Fig. 5 Structural changes in GTa due to the phase transition at high pressure. Different colours mark symmetry-independent molecules. | ||
This pressure-induced phase transition is similar to the ones observed earlier in GGa, both on cooling33 and with increasing pressure (Fig. S10 in the ESI†).34 As for the intriguing question regarding high-pressure proton transfer and an associated co-crystal–salt transition, experimental limitations did not offer sufficient precision to determine the proton positions within experimental error. That said, the pressure at which such a transfer might be expected (based on data for oxalic acid dihydrate) has not been reached, and future work can address this question more deeply. An attempt to record the Raman spectra of the single crystal in the DAC was not successful, with the laser beam decomposing the crystal despite the use of attenuating filters (Fig. S14 in the ESI†). Thus, information regarding proton transfer at high pressure was not obtainable.
The pressure-induced phase transition in GPh had an induction period of a few hours, so that the low-pressure phase could be maintained for a period of time at high pressure (over-pressurised ambient-pressure phase of GPh), after which the crystal was suddenly completely destroyed as a result of the phase transition into the high-pressure phase (Fig. 3b). The fragmentation of GPh rendered the collection of X-ray diffraction data suitable for structure solution impossible. It also should be mentioned that the initial crystal of GPh didn’t undergo destruction and phase transition at 0.19 and 0.50 GPa. At each of these two points, the total time during which the crystal was kept at a constant pressure was about 42 hours (spectroscopic and X-ray diffraction experiments) and no phase transitions were detected. Tentative conclusions on the structural changes across the phase transition were made based on the analysis of the original ambient-pressure crystal structure (Fig. 2c) and on the Raman spectra (which, in contrast to GPh, could be collected as the crystals did not burn) (Fig. 6). The rationale for the destructive nature of the phase transition rests on a dramatic change in the molecular packing. The change in the unit cell parameters before phase transition shows that the maximum compression occurs along the a- and c-axes (Fig. S11b in the ESI†). In this case, one might suggest that a steric conflict arises on compression due to shortening of the distances either between the aromatic rings of the phthalic acid and the glycine layers (compression along c), or between the aromatic rings (compression along a). One can suggest that these steric conflicts may result in shifts of the aromatic rings with respect to each other, such that a double layer is substituted for a single one. The Raman spectra changed across the phase transition, both in the low frequency range and in the range of vibrations of hydrogen bonded groups. The shift in band frequencies corresponding to the O–H and N–H stretching vibrations is not significant, supporting the idea that changes in the strengths of the hydrogen bonds across the phase transition are only minor (Fig. 6). This supports the hypothesis that major structural changes are related to stacking interactions of the aromatic rings, and not of the hydrogen bond network. The long induction period, the over-pressurising of the low-pressure phase prior to the phase transition and the spontaneous rapid propagation of the latter suggest a cooperative mechanism of the transformation, with slow nucleation but rapid nuclei growth, similar to the ones reported earlier for pressure-induced γ → δ glycine,55L-serine-I → L-serine-II,56 or β-alanine-II → β-alanine-V57 phase transitions at high pressure.
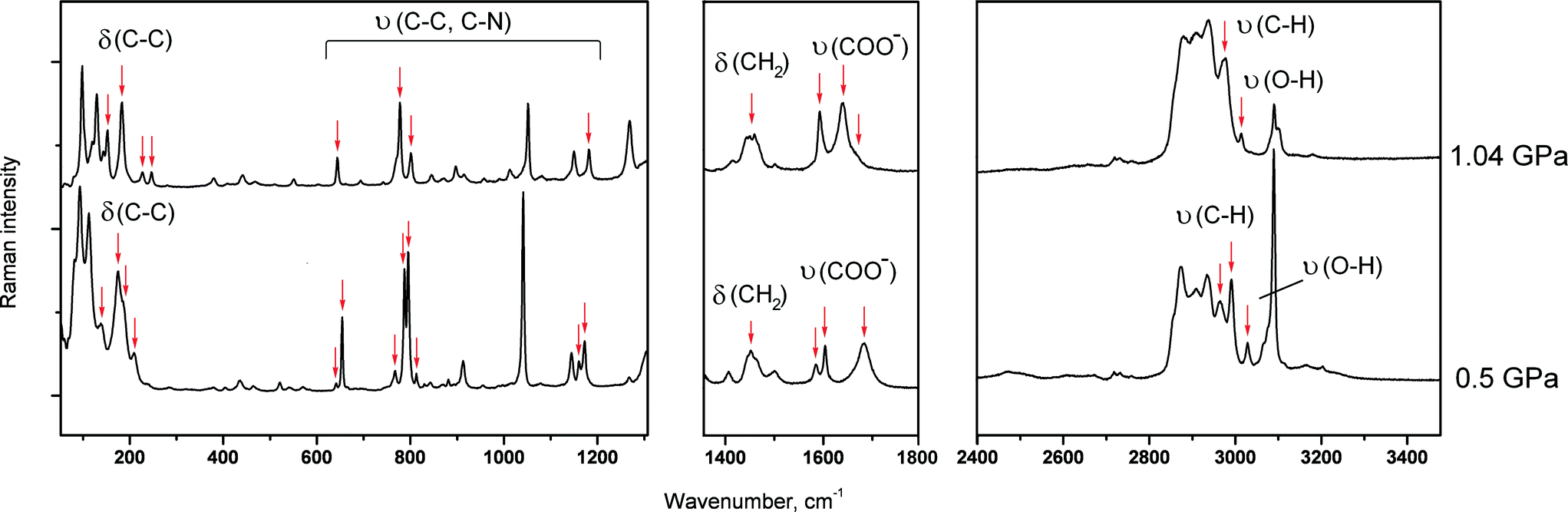 | ||
| Fig. 6 Raman spectra of the two forms of a glycine–phthalic acid co-crystal (GPh) at different pressures; before and after the phase transition. Major changes are shown by red arrows. υ – Stretching vibrations, δ – deformation vibrations. | ||
Conclusions
To date, all three known glycine–carboxylic acid co-crystals have been investigated across a wide range of temperatures (293–100 K) and pressures (0–6 GPa). All three structures undergo phase transitions at relatively low pressures (below 1 GPa), but only the glycine–glutaric acid co-crystal shows a phase transition on cooling.33 The data obtained for the glycine–glutaric acid and glycine–DL-tartaric acid co-crystals suggest that if a crystal structure includes both conformationally flexible (carboxylic acid) and rigid (glycine) components, linked in a 3D-network of hydrogen bonds, then the pressure-induced phase transition is related to conformational changes of the flexible species. If the flexible fragment is immobilised by intra- and intermolecular hydrogen bonds in the crystal structure (as in the dimer of tartaric acid), then its conformation can be preserved in the course of the phase transition. Additionally, slight changes in the position of the entire species may also be involved in such transformations. Overall, the corresponding rearrangement of the crystal structure is relatively small, and the crystals can withstand the applied mechanical stresses. In cases where the structure is formed by two rigid species (e.g. in glycine–phthalic acid co-crystals), with a robust 3D network of hydrogen bonds, changes in the crystal structure may be significantly larger and the crystal is destroyed. Similar effects have been reported for bis-glycinium oxalate.58–60Acknowledgements
This work was supported by the Russian Foundation for Basic Research (RFBR) (Grants No. 14-03-31866 mol_a and 16-33-60089 mol_a_dk), and RAS (Project 44.3.4).References
- E. V. Boldyreva, Z. Kristallogr., 2014, 229, 236 CAS.
- B. A. Zakharov, B. A. Kolesov and E. V. Boldyreva, Phys. Chem. Chem. Phys., 2011, 13, 13106 RSC.
- B. A. Zakharov and E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 271 CAS.
- B. A. Zakharov and E. V. Boldyreva, J. Mol. Struct., 2014, 1078, 151 CrossRef CAS.
- A. K. Mishra, C. Murli, N. Garg, R. Chitra and S. M. Sharma, J. Phys. Chem. B, 2010, 114, 17084 CrossRef CAS PubMed.
- V. S. Minkov, E. V. Boldyreva, T. N. Drebushchak and C. H. Görbitz, CrystEngComm, 2012, 14, 5943 RSC.
- I. Majerz, Z. Malarski and L. Sobczyk, Chem. Phys. Lett., 1997, 274, 361 CrossRef CAS.
- T. Steiner, I. Majerz and C. C. Wilson, Angew. Chem., Int. Ed., 2001, 40, 2651 CrossRef CAS.
- C. C. Wilson, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 435 CAS.
- J. A. Cowan, J. A. Howard, G. J. McIntyre, S. M. Lo and I. D. Williams, Acta Crystallogr., Sect. B: Struct. Sci., 2003, 59, 794 Search PubMed.
- P. Macchi, N. Casati, W. G. Marshall and A. Sironi, CrystEngComm, 2010, 12, 2596 RSC.
- D. M. S. Martins, D. S. Middlemiss, C. R. Pulham, C. C. Wilson, M. T. Weller, P. F. Henry, N. Shankland, K. Shankland, W. G. Marshall, R. M. Ibberson, K. Knight, S. Moggach, M. Brunelli and C. A. Morrison, J. Am. Chem. Soc., 2009, 131, 3884 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- N. A. Tumanov, E. V. Boldyreva and N. E. Shikina, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2010, 66, o279 CAS.
- M. Subha Nandhini, R. V. Krishnakumar and S. Natarajan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 115 CAS.
- R. Chitra, V. Thiruvenkatam, R. R. Choudhury, M. V. Hosur and T. N. Guru Row, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, o274 CAS.
- E. A. Losev, B. A. Zakharov, T. N. Drebushchak and E. V. Boldyreva, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2011, 67, o297 CAS.
- K. Rajagopal, R. V. Krishnakumar, A. Mostad and S. Natarajan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, o751 CAS.
- L. Zong-Sheng and C. Jian-She, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o2857 Search PubMed.
- A. Thirunavukkarasu, A. Silambarasan, R. M. Kumar, P. R. Umarani and G. Chakkaravarthi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, o397 CAS.
- E. A. Losev, B. A. Zakharov, T. N. Drebushchak and E. V. Boldyreva, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2011, 67, o297 CAS.
- B. Riscob, M. Shakir, J. K. Sundar, S. Natarajan, M. A. Wahab and G. Bhagavannarayana, Spectrochim. Acta, Part A, 2011, 78, 543 CrossRef CAS PubMed.
- T. Balakrishnan, K. Ramamurthi and S. Thamotharan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o57 CAS.
- T. Mohandas, C. R. D. Inbaseelan, S. Saravanan and P. Sakthivel, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o236 CAS.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharmaceutics, 2007, 4, 323 CrossRef CAS PubMed.
- V. Stilinović and B. Kaitner, Cryst. Growth Des., 2012, 12, 5763 Search PubMed.
- G. Ramon, K. Davies and L. R. Nassimbeni, CrystEngComm, 2014, 16, 5802 RSC.
- A. Lemmerer, S. Govindraju, M. Johnston, X. Motloung and K. L. Savig, CrystEngComm, 2015, 17, 3591 RSC.
- A. J. Cruz-Cabeza, CrystEngComm, 2012, 14, 6362 RSC.
- N. Casati, P. Macchi and A. Sironi, Chem. Commun., 2009, 2679 RSC.
- N. Casati, NATO Science for Peace and Security Series B: Physics and Biophysics, 2010, p. 161 Search PubMed.
- P. Macchi, N. Casati, W. G. Marshall and A. Sironi, CrystEngComm, 2010, 12, 2596 RSC.
- B. A. Zakharov, E. A. Losev, B. A. Kolesov, V. A. Drebushchak and E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 287 CAS.
- B. A. Zakharov, E. A. Losev and E. V. Boldyreva, CrystEngComm, 2013, 15, 1693 RSC.
- H. Bhatt, A. K. Mishra, C. Murli, A. K. Verma, N. Garg, M. N. Deo and S. M. Sharma, Phys. Chem. Chem. Phys., 2016, 18, 8065 RSC.
- H. Bhatt, C. Murli, A. K. Mishra, A. K. Verma, N. Garg, M. N. Deo, R. Chitra and S. M. Sharma, J. Phys. Chem. B, 2016, 120, 851 CrossRef CAS PubMed.
- Stoe and Cie, X-AREA and X-RED32, Stoe and Cie, Darmstadt, Germany, 2006 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- R. J. Angel, Win_Strain, 2011, http://www.rossangel.com Search PubMed.
- R. Boehler, Rev. Sci. Instrum., 2006, 77, 115103 CrossRef.
- R. A. Forman, G. J. Piermarini, J. D. Barnett and S. Block, Science, 1972, 176, 284 CAS.
- G. J. Piermarini, S. Block, J. D. Barnett and R. A. Forman, J. Appl. Phys., 1975, 46, 2774 CrossRef CAS.
- Oxford Diffraction, CrysAlis Pro, Oxford Diffraction Ltd, Abington, England, 2010 Search PubMed.
- R. J. Angel, J. Appl. Crystallogr., 2004, 37, 486 CrossRef CAS.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, J. Appl. Crystallogr., 2015, 48, 306 CrossRef CAS.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895 CrossRef CAS PubMed.
- T. N. Drebushchak, V. A. Drebushchak and E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., 2011, 67, 163 CAS.
- Y. V. Seryotkin, T. N. Drebushchak and E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 77 CAS.
- R. D. L. Johnstone, M. Leva, A. R. Lennie, H. McNab, E. Pidcock, J. E. Warren and S. Parsons, CrystEngComm, 2010, 12, 2520 RSC.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997, p. 12 Search PubMed.
- T. N. Drebushchak, H. Sowa, Y. V. Seryotkin, E. V. Boldyreva and H. Ahsbahs, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o4052 CAS.
- B. A. Zakharov, B. A. Kolesov and E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 275 CAS.
- E. V. Boldyreva, Phase Transitions, 2009, 82, 303 CrossRef CAS.
- E. V. Boldyreva, H. Sowa, Yu. V. Seryotkin, T. N. Drebushchak, H. Ahsbahs, V. V. Chernyshev and V. P. Dmitriev, Chem. Phys. Lett., 2006, 429, 474 CrossRef CAS.
- B. A. Zakharov, N. A. Tumanov and E. V. Boldyreva, CrystEngComm, 2015, 17, 2074 RSC.
- H. Bhatt, C. Murli, N. Garg, M. N. Deo, R. Chitra, R. R. Choudhury and S. M. Sharma, Chem. Phys. Lett., 2012, 532, 57 CrossRef CAS.
- A. K. Mishra, C. Murli, N. Garg, R. Chitra and S. M. Sharma, J. Phys. Chem. B, 2010, 114, 17084 CrossRef CAS PubMed.
- H. Bhatt, C. Murli, N. Garg, A. K. Mishra, M. N. Deo and S. M. Sharma, AIP Conf. Proc., 2014, 1591, 230 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Includes the structural data for all the phases at multiple temperatures and pressures, the data on H-bonds, figures presenting crystal structures, the pKa values of carboxylic acids and ΔpKa of some glycine salts and co-crystals, and the data on cell parameters, volumes, and strain ellipsoids at multiple temperatures and pressures. CCDC 1435507, 1437082–1437089, 1437185–1437193, 1451443–1451454, and 1450716–1450717. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce00561f |
| This journal is © The Royal Society of Chemistry 2016 |