
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Use of the PIXEL method to investigate gas adsorption in metal–organic frameworks†
Andrew G. P.
Maloney
ab,
Peter A.
Wood
b and
Simon
Parsons
*a
aEaStCHEM School of Chemistry and Centre for Science at Extreme Conditions, The University of Edinburgh, King's Buildings, West Mains Road, Edinburgh, EH9 3FJ, UK. E-mail: S.Parsons@ed.ac.uk; Fax: +44 (0)131 650 4743; Tel: +44 (0)131 650 5804
bCambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK
First published on 12th April 2016
Abstract
PIXEL has been used to perform calculations of adsorbate-adsorbent interaction energies between a range of metal–organic frameworks (MOFs) and simple guest molecules. Interactions have been calculated for adsorption between MOF-5 and Ar, H2, and N2; Zn2(BDC)2(TED) (BDC = 1,4-benzenedicarboxylic acid, TED = triethylenediamine) and H2; and HKUST-1 and CO2. The locations of the adsorption sites and the calculated energies, which show differences in the Coulombic or dispersion characteristic of the interaction, compare favourably to experimental data and literature energy values calculated using density functional theory.
1. Introduction
Metal–organic frameworks (MOFs) are made up of metal-based nodes connected by organic bridging ligands to form 3-dimensional porous networks. Combinations of nodes and linkers give a variety of structures with different pore sizes, topologies and chemical functionalities.1 This modular structure means that it is theoretically possible to design a vast number of different frameworks for specific applications, an approach that is not possible for other porous materials such as zeolites.2There are over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MOF structures recorded in the Cambridge Structural Database (CSD).3 Location of adsorption sites in these structures is time-consuming and requires specialist equipment,4 making computational modelling of gas adsorption a very appealing alternative. Calculations can be performed under a range of different simulated conditions on frameworks with any desired chemical functionality, ideally enabling properties to be predicted before any synthetic work is undertaken.
000 MOF structures recorded in the Cambridge Structural Database (CSD).3 Location of adsorption sites in these structures is time-consuming and requires specialist equipment,4 making computational modelling of gas adsorption a very appealing alternative. Calculations can be performed under a range of different simulated conditions on frameworks with any desired chemical functionality, ideally enabling properties to be predicted before any synthetic work is undertaken.
Adsorbate–framework interactions in MOFs can be modelled using quantum mechanical or force-field methods. The latter are computationally very efficient, but while they are flexible and have been widely used with great success, the transferability of force fields to different systems can be problematic. Calculations are very sensitive to the quality of the parameterisation and the most accurate results are obtained with tailor-made force fields developed for specific sets of conditions, but this is difficult work requiring considerable effort.
Density functional theory (DFT) has the advantage that it does not require system-specific parameterisation, but it is more computationally demanding than force-field methods. It also has the well-known disadvantage that it fails to capture dispersion, an important term for framework–guest interaction energies. Although van der Waals functionals have been developed, treatment of dispersion is most commonly accomplished by an extra semi-empirical term. Perturbation methods, such as MP2, can account for dispersion energies, but they are also computationally expensive.
The aim of the present paper is to explore and assess the applicability of the PIXEL method5–8 for evaluating the energies of adsorbate-framework interactions. The PIXEL method is a semi-empirical technique for evaluating intermolecular interactions based on integrations over calculated electron densities of molecules. It permits energetic analysis to be carried-out quickly with an accuracy comparable to quantum mechanical methods.9,10 Calculation of the electron densities is the only computationally expensive step, the actual interaction energy evaluations taking a matter of a few minutes on a desktop PC. The method requires definition of certain atomic parameters, but these are mostly physically measurable quantities such as ionisation potentials, and one of the most appealing features of the method is the transferability of parameters across many different chemical systems. The parameterisation has recently been extended to include transition metals.11 Energies are partitioned into individual Coulombic, polarisation, dispersion and repulsion contributions which enables the character of interactions to be inferred from the dominant terms, providing chemical insight. The PIXEL method is thus a potentially valuable addition to established techniques applied to metal-containing systems.
2. Computational details
2.1 System selection
The PIXEL method is currently formulated for use with systems containing discrete molecules, and the calculations were based on fragments of frameworks chosen to represent key chemical functionality. Three different MOFs, MOF-5 (Zn4O(BDC)3), Zn2(BDC)2(TED) and HKUST-1 (Cu3(BTC)2), were chosen because they display varied chemical functionalities, experimental and theoretical data for adsorption for a range of gases are available, and the framework chemistry can be represented with relatively small models.2.2 Fragment definition in MOF-5
The applications of MOF-5 (ref. 12) to gas storage have been extensively studied, with both experimental and theoretical data available for H2,13 CH4,14 CO2,15 N2,16 and Ar.16 The framework consists of Zn4O13 clusters connected by benzene dicarboxylate linkers, forming a structure containing pores with diameters of up to 15 Å. Single crystal X-ray diffraction studies between 30 and 293 K by Rowsell et al.17 located eight symmetry independent adsorption sites for argon. Three of these sites are located towards the centre of the pores, the result of secondary adsorption. The remaining five sites (shown for argon in Fig. 1) are associated with the framework. Site I sits directly above the central oxygen atom of the Zn4O(CO2)6 unit, site II lies above the zinc atom in the ZnO3 unit, site III lies above the ZnO2 unit of the metal cluster, site IV lies above the phenyl ring, and site V lies to the edge of the phenyl ring. Distances from the adsorption sites for argon to selected framework atoms are shown in Table 1. The fragment of MOF-5 used for calculations (Fig. 1) was defined by capping the benzene dicarboxylate linkers with hydrogen atoms.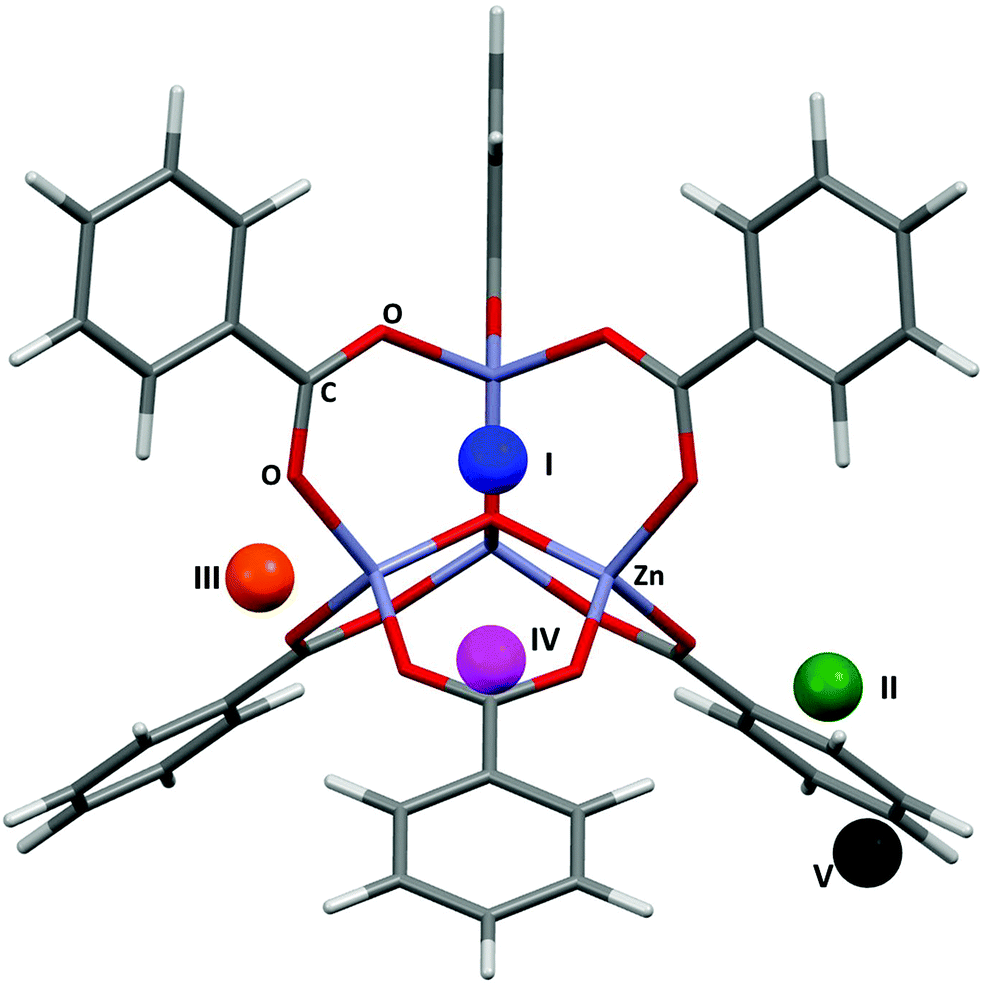 | ||
| Fig. 1 The five adsorption sites associated with the framework structure of MOF-5. The locations of these sites were obtained from single crystal diffraction data (CSD Refcode LAWFUL).17 | ||
| Site | Framework atom | Expt. dist. (Å) MOF-5/Ar | PIXEL dist. (Å) MOF-5/Ar | Expt. dist. (Å) MOF-5/N2 | PIXEL dist. (Å) MOF-5/N2 | Expt. dist. (Å) MOF-5/H2 | PIXEL dist. (Å) MOF-5/H2 |
|---|---|---|---|---|---|---|---|
| I | C (COO) | 3.572 | 3.697 | 3.655 | 3.404 | 3.373 | 3.150 |
| II | O (COO) | 3.492 | 3.527 | 3.518 | 3.379 | 3.316 | 2.812 |
| III | O (COO) | 3.792 | 3.442 | 3.967 | 3.442 | 3.409 | 2.860 |
| IV | C (Ar) | 3.637 | 3.672 | 3.770 | 3.664 | 3.476 | 3.410 |
| V | H (Ar) | 3.288 | 3.199 | 3.610 | 3.329 | — | — |
2.3 Fragment definition in Zn2(BDC)2(TED)
Zn2(BDC)2(TED) (BDC = 1,4-benzenedicarboxylic acid, TED = triethylenediamine)18 consists of Zn2(DBC)4 paddlewheels connected by triethylenediamine molecules. Several experimental and theoretical studies of adsorption in these channels exist for H2,19,20 CH4,21 N2,22 CO,22 and CO2,22 as well as the separation of liquid mixtures such as MeOH/H2O.23 The structure consists of intersecting channels with two differently-sized pore windows. The small channel has a cross section of 4.8 × 3.2 Å and the larger pore a maximum diameter of 7.5 Å.18 The structural model used for PIXEL calculations was generated by capping the BDC linkers of the paddlewheel motif with hydrogen atoms (Fig. 2).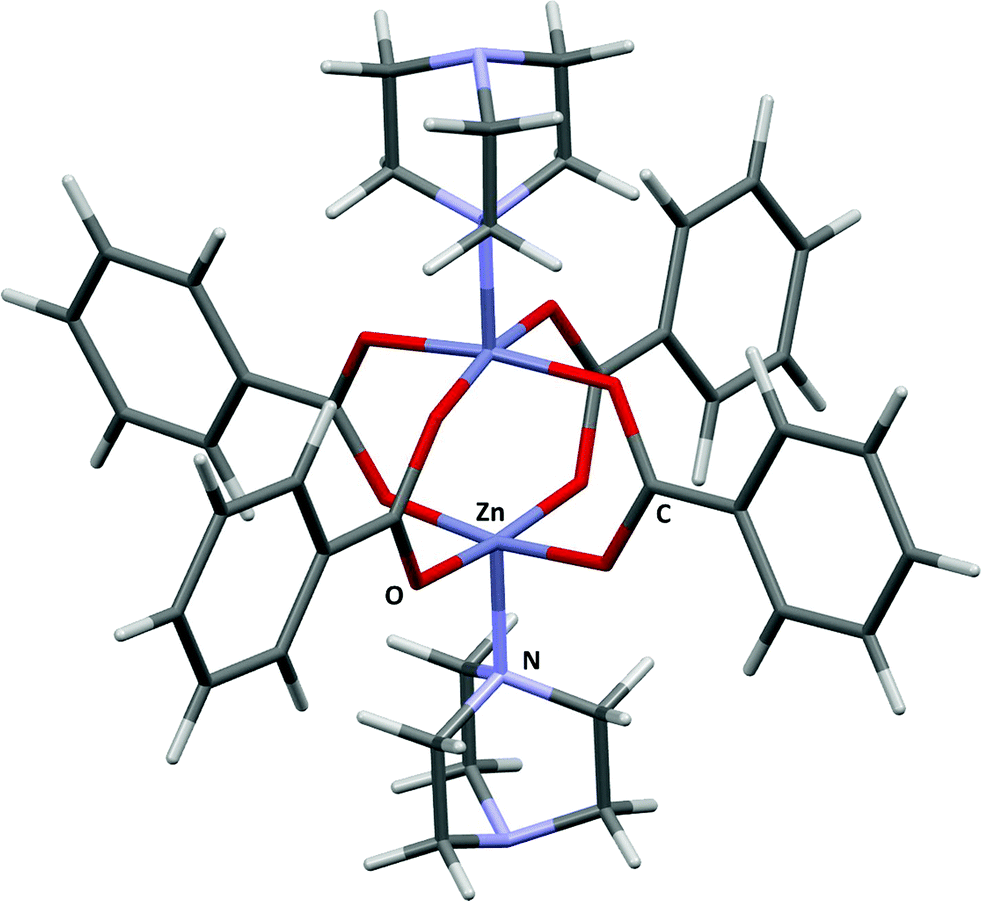 | ||
| Fig. 2 The fragment model of Zn2(BDC)2(TED). Adapted from CSD Refcode HEGKAP. | ||
2.4 Fragment definition in HKUST-1
HKUST-1 (ref. 24) is also known as Cu(BTC), where BTC is benzene-1,3,5-tricarboxylate. The large pore volumes in HKUST-1 have made it the subject of a number of experimental and theoretical studies of gas adsorption, in particular the adsorption of H2,25 N2,26 CO,26 CO2,26,27 and NO.28,29 The structure consists of Cu2(BTC)4 paddlewheel building blocks connected into a 3-dimensional porous structure leading to a large and complex structure with three distinct interconnected pores having diameters of around 5, 11 and 13.5 Å. In this study binding in the first of these pores will be compared with data obtained by Grajciar and co-workers27 using a model obtained by capping three of the copper paddlewheel motifs and three of the carboxylate groups with hydrogen (Fig. 3).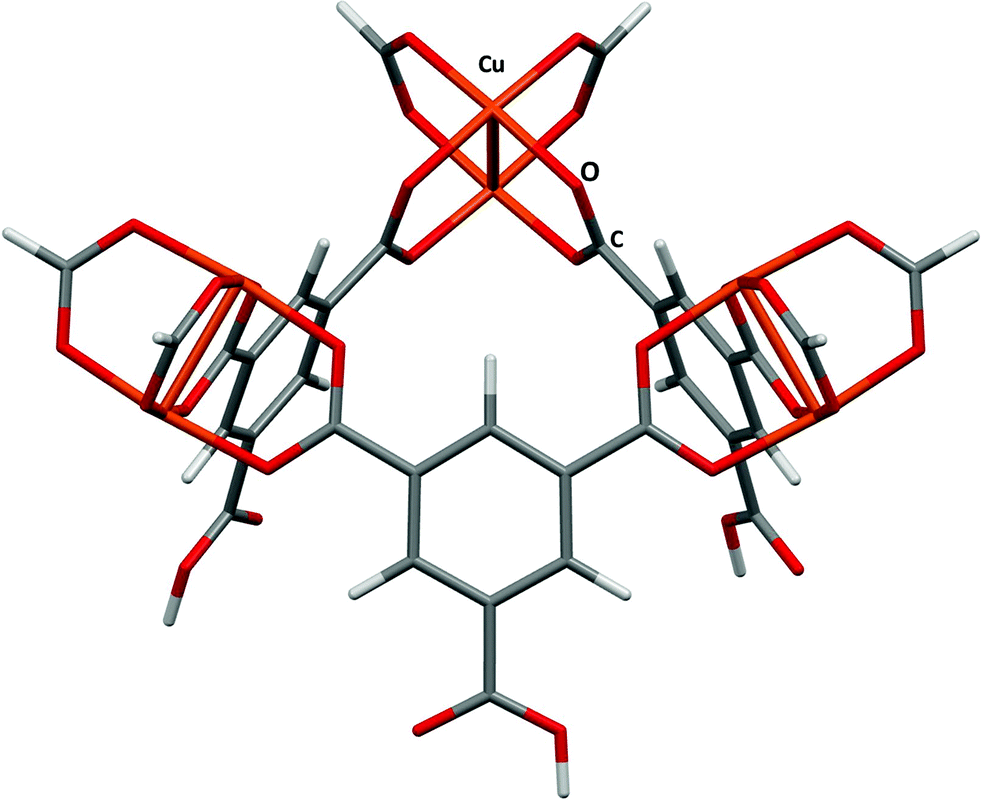 | ||
| Fig. 3 The small fragment chosen to model the small pore in HKUST-1. Adapted from CSD Refcode DOTSOV. | ||
2.5 PIXEL calculations
Crystal structures of the target MOFs were found and obtained from the Cambridge Structural Database using ConQuest30 and modified into the fragments defined above using Materials Studio31 and Mercury.32 Hydrogen atom distances were normalised to typical neutron values.33 Structural coordinates for the fragments used are given in the ESI.† Bond lengths for H2 and N2 adsorbate molecules were obtained from ref. 34, while those for CO2 were taken from ref. 35.Calculations were set up such that the MOF fragments were positioned in the centre of a 20 × 20 × 20 Å grid. The adsorbate molecule was placed at the grid origin and then moved in steps systematically along the x, y, and z axes of the grid. Typically the step size was 1 Å, but finer steps were employed in some cases (see below). The distances between the atoms in the adsorbate molecule to those in the fragment were calculated. Provided that none of these distances lay within the sum of the relevant covalent radii a PIXEL calculation was performed. Except in the case of Ar, the orientation of the adsorbate molecule was varied at each grid point. Thirteen orientations were sampled at each search point, with the molecules aligned along the following vectors: [1,0,0]; [0,1,0]; [0,0,1]; [1,1,0]; [1,0,1]; [0,1,1]; [−1,1,0]; [−1,0,1]; [0,−1,1]; [1,1,1]; [−1,1,1]; [1,−1,1] and [−1,−1,1]. The symmetry of the MOF-5 and HKUST-1 fragments allowed a reduced search to be restricted to the asymmetric unit of point group ![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m for MOF-5 (0 ≤ x ≤ 1; y ≤ min(x, 1 − x); z ≤ y) and that of a 3-fold axis along the [1,1,1] direction (0 ≤ x ≤ 1; y ≤ x; z ≤ x) for the HKUST-1 fragment.
3m for MOF-5 (0 ≤ x ≤ 1; y ≤ min(x, 1 − x); z ≤ y) and that of a 3-fold axis along the [1,1,1] direction (0 ≤ x ≤ 1; y ≤ x; z ≤ x) for the HKUST-1 fragment.
Electron densities were obtained using Gaussian09 (ref. 36) at the B3LYP/6-31G** level of theory. PIXEL calculations were carried out at condensation level 5 (a pixel size of 0.2 × 0.2 × 0.2 Å) and the atomic parameters for metal and non-metal atoms were those reported in previous work.11
Upon completion, the energies at each gridpoint were ranked from highest to lowest, and the 200 strongest interactions were visualised around the fragment using Mercury. This allowed the independent adsorption sites to be located visually, with any points arising from favourable interactions in locations between these independent adsorption sites being omitted. For the MOF-5/Ar, MOF-5/N2 and MOF-5/H2 systems, once located, the positions of the independent sites were optimised by performing a 0.2 Å grid search around a reduced space (searching in the region x/y/z ± 1 Å, where x/y/z are the coordinates of the independent adsorption site). All RMS fits were calculated using Mercury, and are based on the centres of mass of the adsorbate molecules.
3. Results and discussion
3.1 Gas adsorption between MOF-5 and Ar
Fig. 4 shows the results of the PIXEL calculations on MOF-5/Ar (red) overlaid with those obtained experimentally by Rowsell (black).17 The RMS fit of the overlay between the central Zn4O and the Ar atoms of each fragment as calculated in Mercury is 0.251 Å when a 1 × 1 × 1 Å grid was used, indicating that the locations of the sites obtained from PIXEL correspond to sites I–V (see section 2). Using a smaller grid step of 0.2 × 0.2 × 0.2 Å led to a slightly improved RMS fit of 0.195 Å. Table 1 shows the distances from selected framework atoms of the adsorption sites obtained experimentally compared to those found using PIXEL with the finer grid search.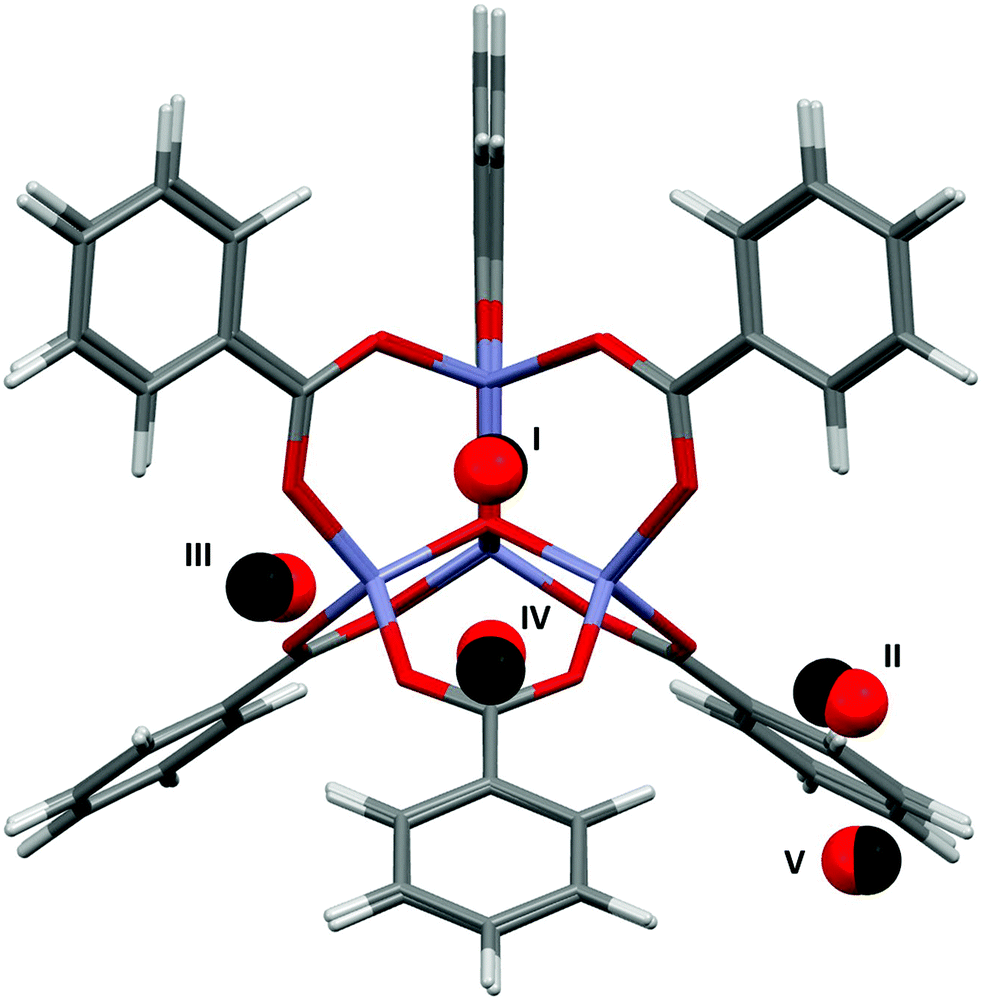 | ||
| Fig. 4 Overlay of Ar adsorption sites in MOF-5 located using PIXEL (red) with those obtained experimentally (black). Adapted from CSD Refcode LAWFUL. | ||
In a theoretical study, Monte Carlo methods were used by Dubbeldam et al.37 to probe the energies of the adsorption sites of argon in MOF-5 found in Rowsell's X-ray diffraction study.17 Energy minimizations of a single argon atom at a temperature of 30 K located preferential adsorption at sites I, II and IV. Upon higher loading, site III is filled preferentially over site II, although the binding energies for these sites are found to be almost equal. Adsorption energies are reported at these four sites, although it should be noted that Dubbeldam et al. did not identify site V, and consequently no energy data are available for this site. Since the experimental data of Rowsell et al. shows that site V is only occupied at a lower temperature than site IV, it would be expected that site V has a lower adsorption energy.
Table 2 shows the PIXEL adsorption energies compared to those obtained by Dubbeldam et al. The trend in the order of site energies observed is similar in both cases, and all PIXEL values lie within 3.3 kJ mol−1 of the corresponding values from Dubbeldam. The interactions are dominated by the dispersion term, and the ordering of the different adsorption sites correlates with the number of short contacts that can be made between the adsorbing argon atoms and the atoms of the framework. Since an argon atom at site I interacts with the central oxygen as well as three zinc atoms and three CO2 moieties, this site has the highest dispersion interaction. Argon atoms at sites II and III interact with one zinc atom and three and two oxygen atoms respectively, explaining the decreasing dispersion interactions for these sites. Since sites IV and V interact only with the aromatic ring of the organic linker, they have the lowest interaction energy, as is observed from both PIXEL and the classical studies.
| Adsorbate | Site | E literature | E PIXEL | E Coul | E disp | E rep | E pol | |
|---|---|---|---|---|---|---|---|---|
| Ar | I | −11.52 | −11.4 | −2.7 | −15.6 | 7.7 | −0.7 | |
| II | −8.42 | −9.8 | −3.4 | −13.5 | 8.6 | −1.4 | ||
| III | −8.44 | −8.2 | −3.0 | −12.9 | 8.8 | −1.0 | ||
| IV | −6.99 | −3.7 | −1.7 | −8.2 | 6.8 | −0.6 | ||
| V | — | −3.2 | −0.5 | −4.3 | 1.9 | −0.2 | ||
| N2 | I | −12.13 | −17.0 | −18.7 | −33.2 | 40.5 | −5.6 | |
| II | −8.70 | −10.7 | −6.7 | −16.9 | 15.3 | −2.4 | ||
| III | −10.65 | −10.1 | −4.4 | −14.0 | 9.9 | −1.6 | ||
| IV | −7.57 | −5.2 | −1.5 | −7.6 | 4.3 | −0.5 | ||
| V | — | −3.8 | −1.6 | −4.4 | 2.6 | −0.4 | ||
| H2 | I | −12.8 | −15.4 | −13.1 | −9.7 | −22.6 | 24.0 | −4.7 |
| II | −8.3 | −11.1 | −8.1 | −7.8 | −15.3 | 19.3 | −4.3 | |
| III | −5.4 | −10.4 | −7.5 | −6.4 | −11.5 | 12.7 | −2.3 | |
| IV | −8.9 | −10.2 | −3.1 | −1.6 | −5.3 | 4.5 | −0.6 | |
3.2 Gas adsorption between MOF-5 and N2
Fig. 5 shows the locations of the adsorption sites obtained from PIXEL, and Table 1 shows the average distances between the N2 centres of mass and particular framework atoms. The studies cited above in the context of Ar adsorption also report adsorption between MOF-5 and N2.17,37 Experimental measurements show that at 30 K sites I, II and V are occupied. It was found that site IV becomes occupied preferentially to site V at 50 K, and at 90 K site II is occupied instead of site III. The RMS fit between the centres of mass of the independent adsorption sites found using the PIXEL method is 0.549 Å, which is reduced to 0.295 Å upon a search over a finer grid spacing (0.2 Å step). PIXEL calculated adsorption energies corresponding to the geometries in Table 1 are shown in Table 2 along with those obtained from Monte Carlo studies. Also shown are the component energy terms from each PIXEL calculation.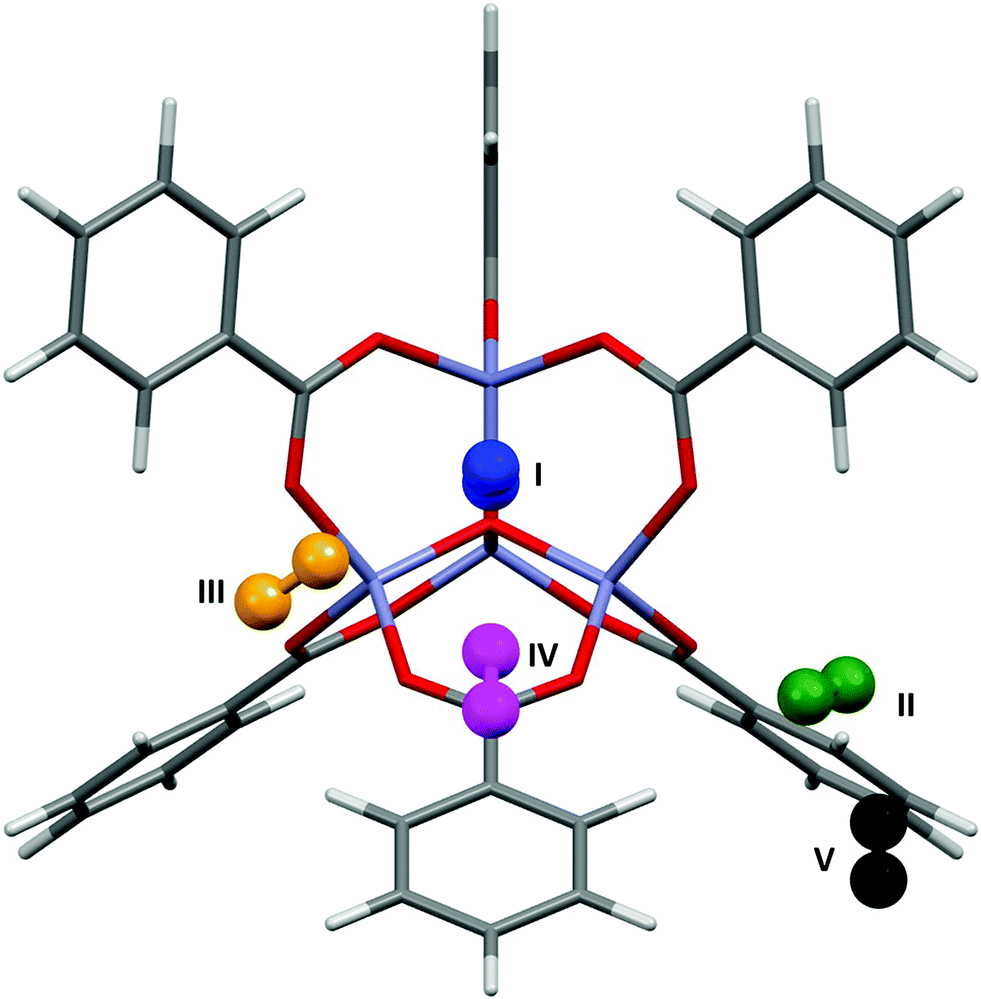 | ||
| Fig. 5 N2 adsorption sites in MOF-5 located using PIXEL. | ||
As Table 2 shows, while site I is significantly higher in energy than the other sites, and sites IV and V are the lowest in energy, the magnitudes of the PIXEL calculated energies for sites II and III are swapped with regards to the energies obtained from Monte Carlo methods. However, the results of the PIXEL calculations agree with qualitative analysis from Rowsell based on variable temperature studies,17 where site II was found to have a higher energy of adsorption than site III. Since no energetic data were available for site V it is impossible to compare values, however PIXEL calculates this site as being of a similar energy to site IV, which is consistent with the experimental observation that both sites IV and V are occupied only at low temperatures (50 K and 30 K respectively).17 Once again, the interactions are dominated by the dispersion term. In the case of site I, the Coulombic interaction is around three times larger than those of the other sites reflecting three relatively short (3.188 Å) distances from N to three Zn sites. While the charge on a nitrogen atom in N2 is formally zero, the proximity of the lone pair electron density to the three cations results in a favourable electrostatic interaction.
3.3 Gas adsorption between MOF-5 and H2
Fig. 6 shows the most favourable orientations of the H2 molecules at sites I–IV as calculated by PIXEL, and Table 1 shows the distances of the centres of mass of these sites to selected framework atoms. The initial RMS fit of the PIXEL sites from the 1.0 Å grid search against the experimental positions was 0.359 Å, which was improved slightly to 0.328 Å with a finer 0.2 Å grid. The distances for sites II and III are consistent with those calculated for single H2 molecule adsorption at the MP2 level by Sillar, where H⋯O distances of 2.988 Å and 2.969 Å were obtained for sites II and III, respectively. Sites II and III in the computation studies are found significantly closer to the framework than those observed experimentally. However, if sites II and III as found by PIXEL are occupied simultaneously, a short H⋯H distance of 2.149 Å exists, which is smaller than the sum of the van der Waals radii (2.2 Å), indicating that it is the interactions between the adsorbate molecules at these sites affecting their locations. This interaction was not considered in either the PIXEL work reported here or in Sillar's study.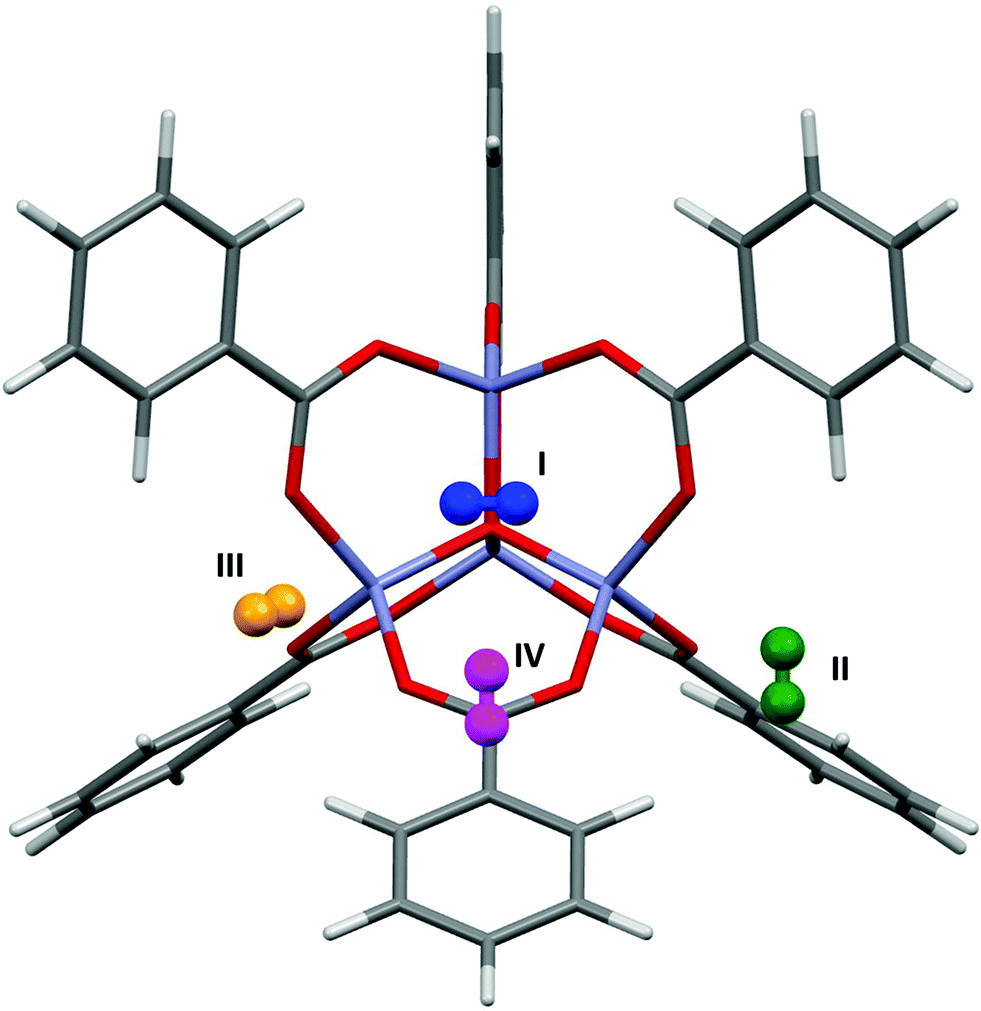 | ||
| Fig. 6 The most favourable orientations of the H2 molecules found using PIXEL at sites I–IV in MOF-5. | ||
Neutron powder diffraction studies at 3.5 K by Yildirim and Hartman38 of hydrogen adsorption in MOF-5 revealed occupancy of sites I–IV. Periodic DFT (LDA) calculations39 were used to calculate the binding energies. Two different orientations of the gas molecules were simulated, one where the H2 molecule lay parallel to the 3-fold symmetry axis near the adsorption site (Epar), and the other where it lay perpendicular to this axis (Eperp), the most favourable interactions occurring for the perpendicular orientation of the H2 molecules. Table 2 shows the energies of the interactions at sites I–IV as calculated by PIXEL, along with the breakdown of individual terms, compared to the DFT energies.
The PIXEL energies are mostly around 4 kJ mol−1 lower than the DFT energies, and the trend mirrors that for the DFT Eperp energies, which are the more favourable in each case. However, since the coordinates used for the DFT calculations were not published, it is difficult to compare more exactly the effect of the orientation of the H2 molecule using the two methods. The interaction energy for site IV calculated with PIXEL is significantly lower than the DFT value, although it is similar to that obtained by Dubbeldam (−3.1 versus −3.78 kJ mol−1),37 suggesting this discrepancy could arise from cooperative stabilisation of site IV by loadings in the other sites, an effect not accounted for by PIXEL.
It is noticeable from the data in Table 1 that distances tend to be underestimated in PIXEL calculations. The approximations of the PIXEL method are presumably responsible for much of this disparity, though it is also possible that the differences are to some extent ascribable to the effects of thermal libration, which are present in the experimental values but not accounted for in the PIXEL calculations which are performed at formally 0 K conditions.
3.4 Gas adsorption between Zn2(BDC)2(TED) and H2
A study by Kong et al.20 investigated hydrogen adsorption in Zn2(BDC)2(TED) (where BDC is benzenedicarboxylate and TED is triethylenediamine). Using both experimental and theoretical techniques they found that in this system there are no specific adsorption sites, but rather two channels, denoted A and B, that run through the structure within which the binding energies are almost equal. Channel A runs parallel to the Zn(TED) axis and channel B runs perpendicular to this (Fig. 7, black and blue lines respectively).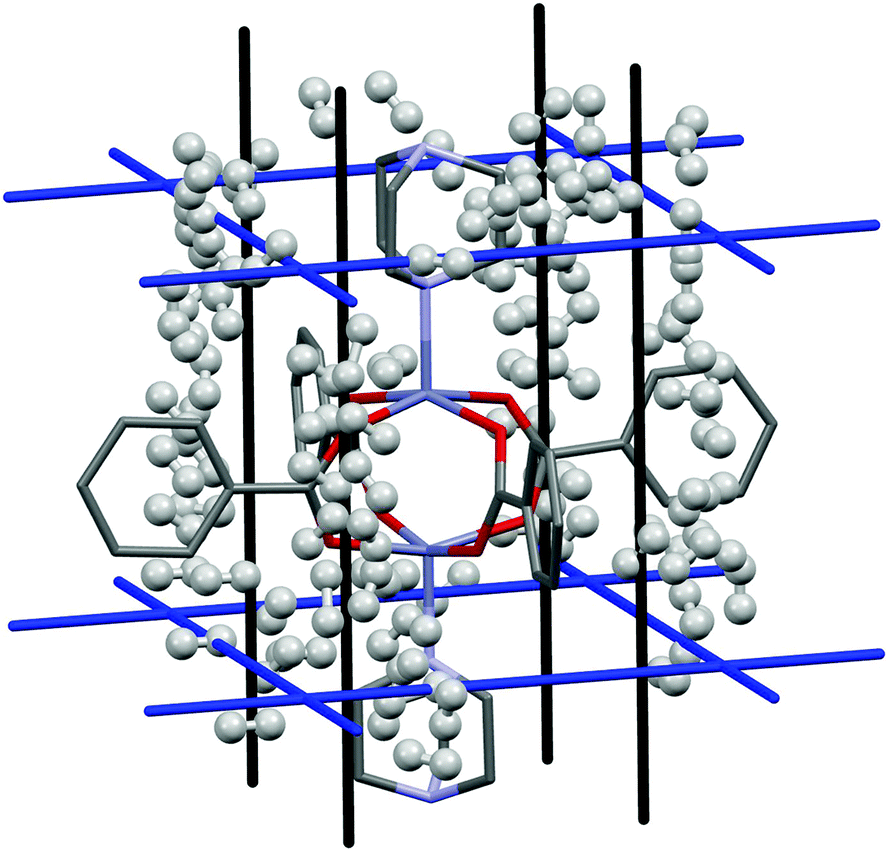 | ||
| Fig. 7 H2 adsorption in Zn2(BDC)2(TED) for the first 100 binding sites. Hydrogen atoms are omitted from the framework structure for clarity. | ||
In a DFT study using a recently developed non-empirical van der Waals functional,40,41 it was found that throughout both channels the hydrogen binding energy was around −10 kJ mol−1, with a variation of only around 1 kJ mol−1 depending on channel and H2 orientation. This was found to be consistent with experimental data, where a single Langmuir isotherm was used to fit the uptake data, and from which binding energies of around −7 kJ mol−1 were obtained.
Fig. 7 shows the results of the PIXEL calculation for the 100 highest energy H2 interactions, and Fig. 8 shows the frequencies of these energies.
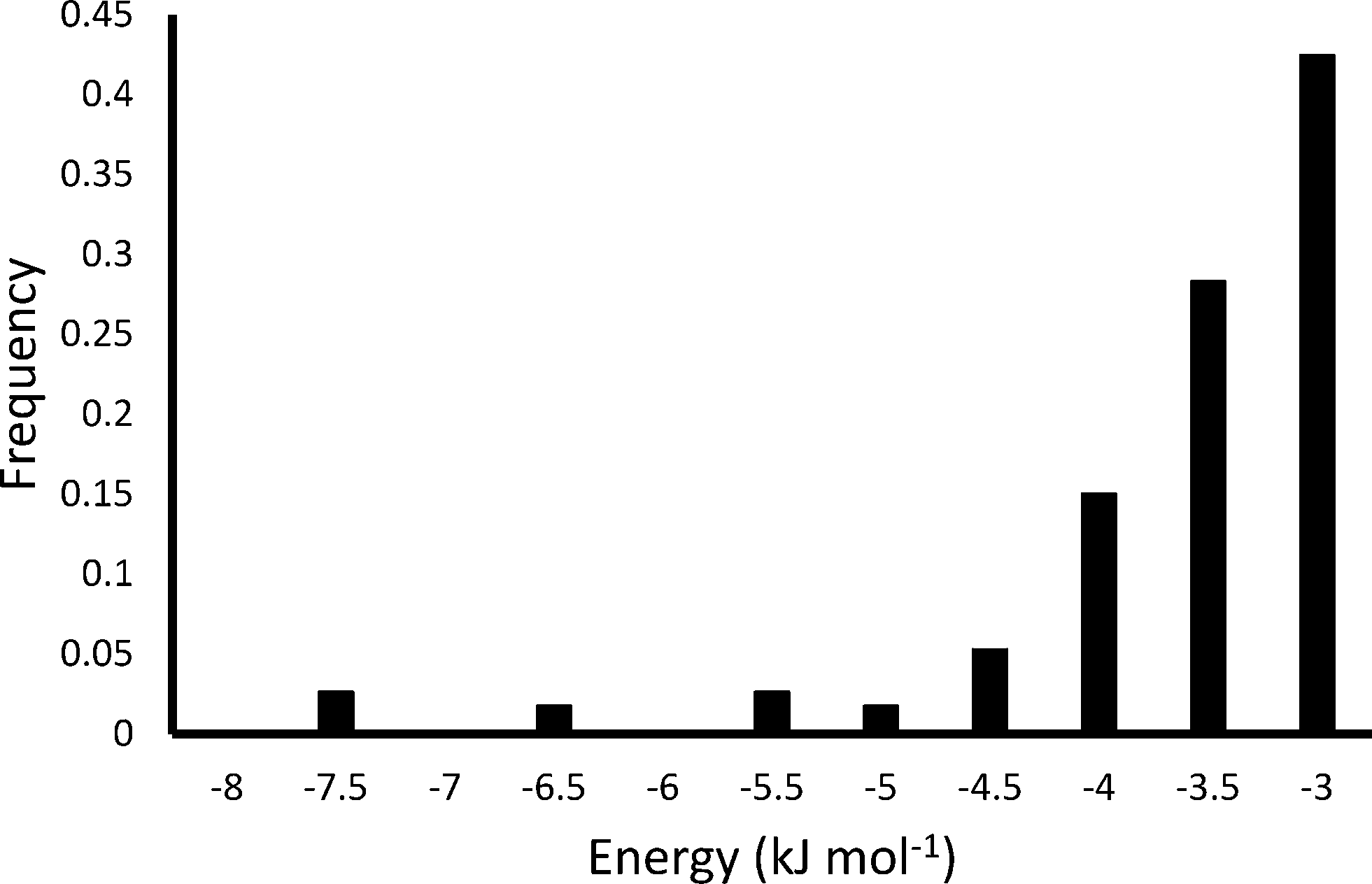 | ||
| Fig. 8 Energy frequency for the first 100 binding sites of H2 in Zn2(BDC)2(TED) as calculated by PIXEL. | ||
The PIXEL calculated adsorption energies of the H2 binding sites shown in Fig. 7 and 8 all lie in the range of −7.6 kJ mol−1 to −3.1 kJ mol−1, with an average interaction of −3.9 kJ mol−1. Additionally, the orientation of the H2 molecule seems to have very little impact on the energy calculated at each point. This indicates that in this system the H2 molecules simply condense in the channels of the framework, and as such it does not present a strong candidate for a hydrogen storage material under the criteria of Bhatia and Myers.42
3.5 Gas adsorption between HKUST-1 and CO2
Fig. 9 shows the location of the three independent sites for CO2 in HKUST-1 found in the PIXEL analysis. Table 3 compares the interaction energies with the theoretical coupled-cluster DFT results of Grajciar et al.43 and data obtained from microcalorimetry experiments.27 The latter study concluded that at low CO2 coverage (up to a pressure of 15 bar of CO2 at 300 K) there are three distinct adsorption sites situated around the window of the small cage: one at the open copper site (denoted CU); one at the window of the small cage (denoted CW); and one in the centre of the cage (denoted CC).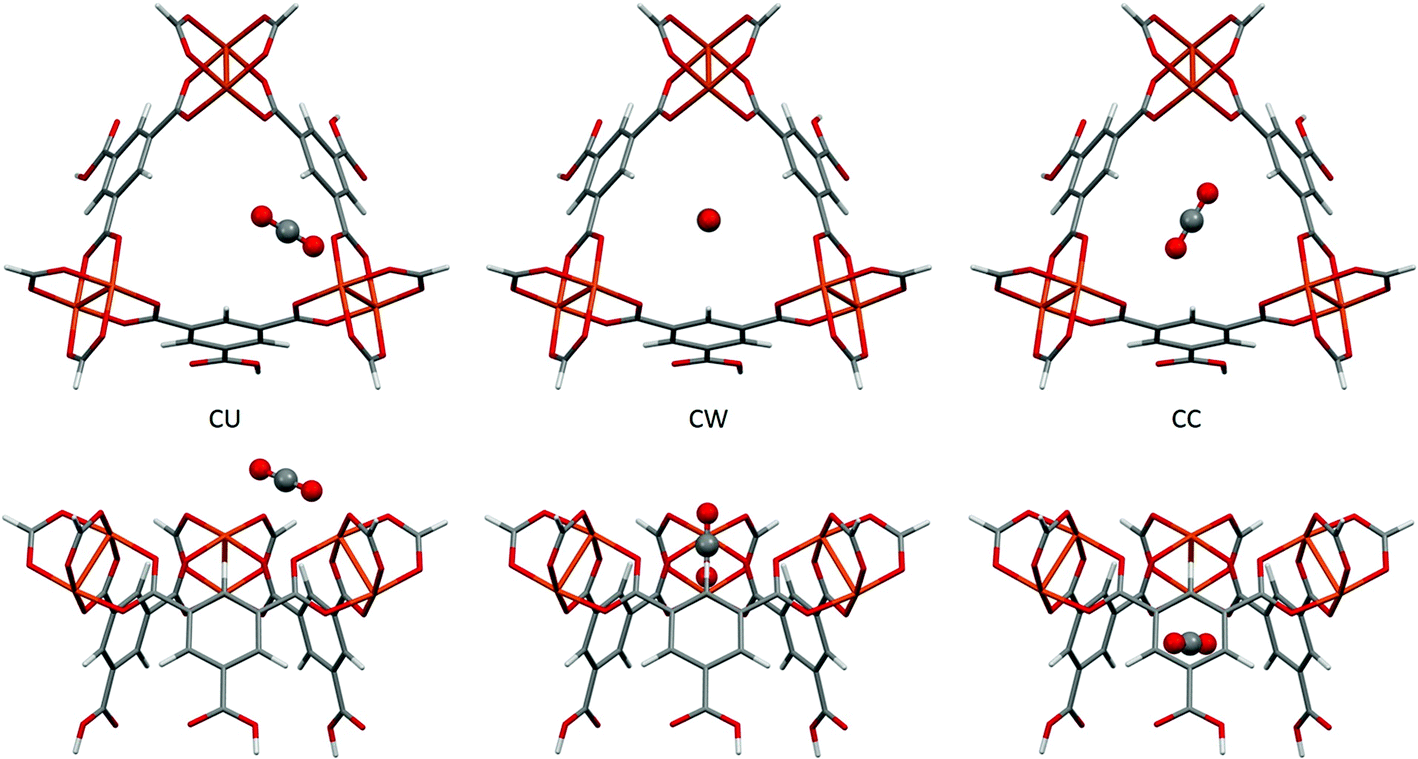 | ||
| Fig. 9 The three independent CO2 adsorption sites found in HKUST-1 viewed perpendicular (top) and parallel (bottom) to the small cage opening. | ||
| Site | E DFT/CC | E PIXEL | E Coul | E disp | E rep | E pol |
|---|---|---|---|---|---|---|
| CU | −28.2 | −42.7 | −48.1 | −29.5 | 61.0 | −26.1 |
| CW | −23.1 | −23.2 | −6.9 | −19.9 | 5.1 | −1.5 |
| CC | −23.2 | −20.3 | −9.0 | −24.1 | 15.6 | −2.9 |
The coupled-cluster DFT calculations find the CU site to be located close to the copper paddlewheel with a Cu–O distance of 2.39 Å and a Cu–O–C angle of 123.0°. PIXEL calculations find this site with a Cu–O distance of 2.34 Å and a Cu–O–C angle of 148.9°. It should be noted that the PIXEL results were obtained in a grid search without further optimisation and some differences in the orientation are to be expected. The CO2 molecule is oriented along the edge of the small window, interacting with the neighbouring paddlewheel as well as the aromatic hydrogen of the organic linker. This explains the dominance of the Coulombic term for this interaction, as shown in Table 3. The total energy of this site obtained from PIXEL calculations is −42.7 kJ mol−1, some 14.5 kJ mol−1 higher than the DFT energy of −28.2 kJ mol−1, but as Fig. 9 shows, the CU site is rather exposed and the energy is likely to be quite sensitive to the fragment chosen to represent the framework.
The interaction energies at the CW and CC sites are reproduced to within 3 kJ mol−1. Their locations are defined with respect to the plane of the upper Cu2+ ions of the three paddlewheel motifs. The DFT calculations position the carbon atom of the CW site 1.19 Å below this plane, the CO2 molecular axis making an angle of 84.5°. For the CC site, the carbon atom is 3.47 Å below the plane and the angle is 23.5°. PIXEL calculations with a 1 Å grid-spacing find the distances and angles of the CW site to be 0.35 Å and 90° respectively, the distance increasing to 1.21 Å with a grid-spacing of 0.5 Å. Corresponding values for the CC site are 3.81 Å and 0°, again using the smaller grid spacing. The CW and CC interactions are dominated by the dispersion term.
3.6 Investigations of “novel” frameworks
One of the advantages to using a fragment to represent a MOF structure is that no knowledge of the space group of the framework is necessary. As such, “novel” metal–organic frameworks can be modelled simply by making modifications to the fragment, be it functionalising the organic linkers or changing the metal used in the clusters. This means that screening of the adsorption capabilities of a number of possible target structures can be carried out relatively easily.Lofti and Saboohi used dispersion-corrected DFT (DFT-D) calculations to explore the effects on H2-binding of functionalising the benzene-dicarboxylate linkers in MOF-5 with different halogens.44 They found that, while no improvements are observed upon replacing all four aromatic hydrogen atoms with fluorine, adsorption energies increase by an order of magnitude upon functionalization with chlorine and bromine, with adsorption energies ranging from 3.8 kJ mol−1 for fluorine functionalisation up to 38 kJ mol−1 for bromine.
PIXEL calculations were performed on three modified versions of the model used to investigate the adsorption of H2 with MOF-5 as described in section 3.3, with four aromatic hydrogen atoms on each linker being replaced by fluorine, chlorine and bromine. The same sites as were located in MOF-5 were found in each case, and Table 4 shows the relative energies of these sites for the functionalised structures. Fig. 10 shows the variation in the different energy terms from the PIXEL calculations.
| Site | MOF-5 | F-MOF-5 | Cl-MOF-5 | Br-MOF-5 |
|---|---|---|---|---|
| I | −11.9 | −11.8 | −13.2 | −13.7 |
| II | −7.1 | −8.1 | −7.0 | −8.6 |
| III | −6.4 | −5.9 | −5.5 | −3.9 |
| IV | −2.2 | −3.1 | −4.5 | −5.0 |
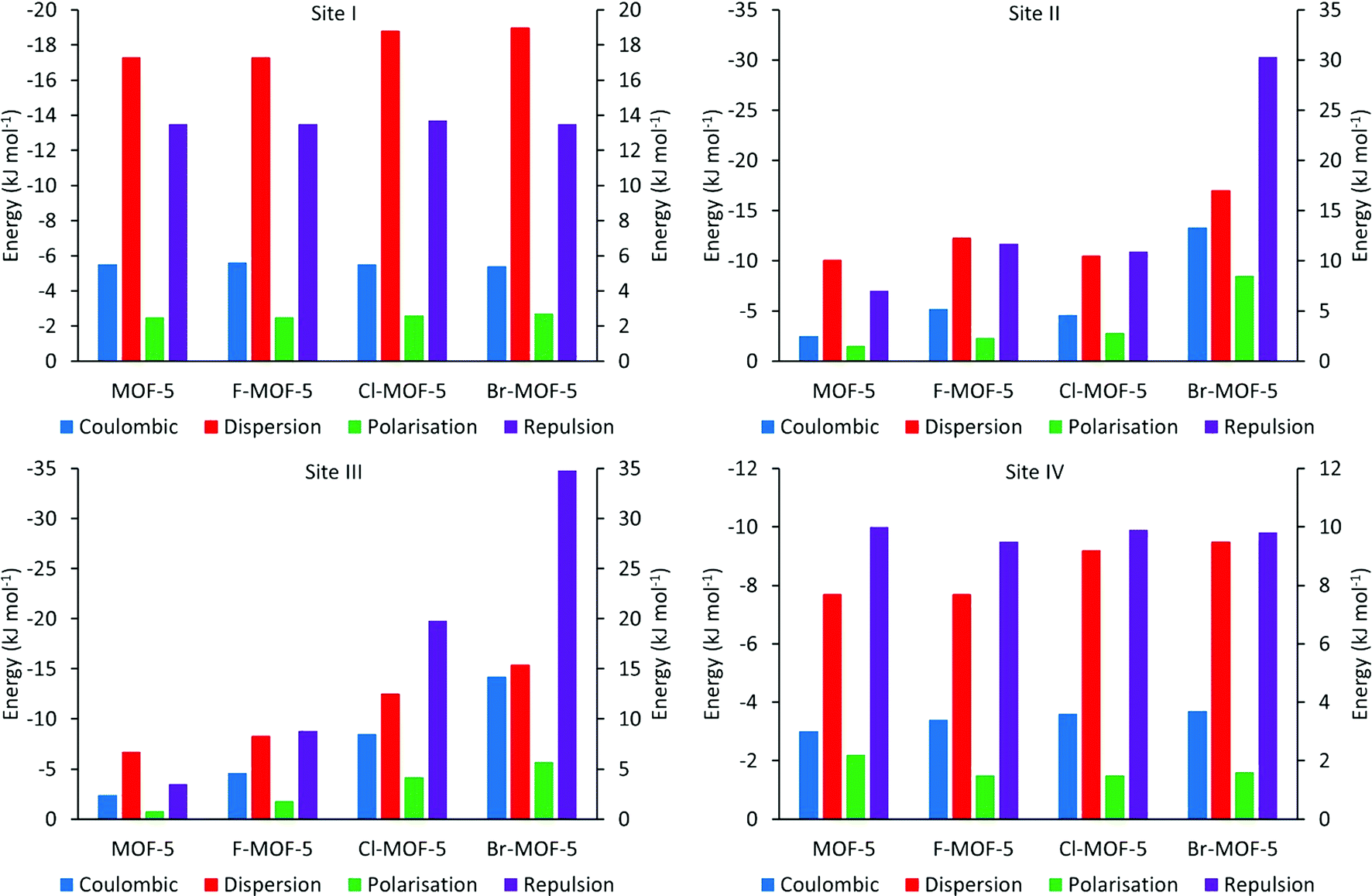 | ||
| Fig. 10 PIXEL component energies for the four H2 adsorption sites in MOF-5 and its halogen functionalised analogues. Repulsion bars correspond to the right hand axis, others to the left axis. | ||
While there is little difference observed upon functionalization of the linker with fluorine, adsorption energies generally increase over sites I, II and IV for chlorine and bromine, while the binding becomes weaker at site III upon halogenation (Table 4). Fig. 10 shows the reasons for these differences. The terms for site I remain almost constant upon functionalization, except for the dispersion term which increases slightly. Since site I is the furthest removed from the linkers, interacting predominantly with the zinc oxide cluster, halogenation has little effect on this site, with the exception of the observed increase in dispersion caused by the additional electron density of the halogens. Sites II and III are located at the edge and face of the metal cluster respectively, and as such will be much more sensitive to the halogenation. This can be seen from Fig. 10, where there is a general increase in the magnitude of all the terms. While this is somewhat compensated for with site II by the increase in the Coulombic and dispersion terms, the proximity of site III to the halogen atoms means that it becomes increasingly less favourable. In site IV, the terms again remain relatively constant, except from the dispersion term which increases with the size of the halogen used for functionalization. As the interaction at this site is dominated by the dispersion term, this is to be expected as such an interaction will become more favourable with increasing electron density.
While the improvement in binding energies observed with PIXEL are much more modest than those seen using DFT-D, the trend of increasing adsorption upon functionalization of MOF-5 with heavier halogens is consistent. Since sites II and III are seldom occupied simultaneously,37 the loss in binding energy observed at site III is compensated for by the increase in the three other sites. Consequently, functionalization by halogenation of the linker molecules in MOF-5 presents itself as a route for improvements in H2 adsorption in this system.
4. Conclusions
Understanding adsorption interactions between molecular species and metal–organic frameworks is an important component in the design of new systems for gas storage and separation. While several different methods for calculating the energies of such interactions exist, we have shown that the PIXEL method is a potentially valuable tool in such analysis, and almost any MOF can be studied, though the systems chosen in this work are those for which extensive theoretical and experimental data are available. It is important to note that the PIXEL method is currently formulated for use with systems containing discrete molecules, and the calculations need to be based on fragments of frameworks chosen to represent key chemical functionality. While the implementation of PIXEL used in this work was intended to calculate interaction energies between two independent fragments, more complex systems could in principle be studied with the PIXEL-d module. Cooperative adsorption between two or more molecules as well as secondary adsorption could therefore be captured with this alternative method, although the additional dimensions required in the grid search would increase the computational cost considerably.Notwithstanding the limitations of the method, adsorption sites calculated using the PIXEL method reproduce those observed experimentally, with RMS values between 0.195 Å and 0.328 Å. The trends of the PIXEL adsorption energies correlate reasonably well with experimental and theoretical data, except in cases where cooperativity arises between adsorption sites.
The PIXEL method is potentially applicable to all elements, and the system-specific parameterisation associated with force field calculations is not necessary. This also means that calculations use the same sets of parameters and are therefore comparable. Furthermore, the simplicity of modifying the fragment means that the adsorption characteristics of almost any conceivable metal–organic framework can be investigated whether or not the crystal structure has been determined.
The short computing times mean that screening of different MOFs can be performed rapidly. All calculations were performed over 13 processors distributed over two desktop computers. Full screening of the interactions between a simple adsorbate such as argon and MOF-5 can be accomplished in around 30 minutes. A more complicated system, such as CO2 and HKUST-1 can be completed in around 24 hours. These calculation times are considerably faster than corresponding quantum mechanical calculations, and one application of the methods we have described would be to identify energy minima which then become the subject of more extensive calculations in a more limited search space.
Acknowledgements
We thank the Cambridge Crystallographic Data Centre and the EPSRC for studentship funding, and The EaStCHEM Research Computing Facility for access to computing resources.References
- S. L. James, Chem. Soc. Rev., 2003, 32, 276 RSC.
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626 RSC.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- N. Nijem, J.-F. Veyan, L. Kong, K. Li, S. Pramanik, Y. Zhao, J. Li, D. Langreth and Y. J. Chabal, J. Am. Chem. Soc., 2010, 132, 1654 CrossRef CAS PubMed.
- A. Gavezzotti, J. Phys. Chem. B, 2002, 106, 4145 CrossRef CAS.
- A. Gavezzotti, J. Phys. Chem. B, 2003, 107, 2344 CrossRef CAS.
- A. Gavezzotti, Z. Kristallogr., 2005, 220, 499 CAS.
- A. Gavezzotti, Molecular Aggregation - Structure Analysis and Molecular Simulation of Crystals and Liquids, Oxford University Press, 2007 Search PubMed.
- L. Maschio, B. Civalleri, P. Ugliengo and A. Gavezzotti, J. Phys. Chem. A, 2011, 115, 11179 CrossRef CAS PubMed.
- W. B. Schweizer and J. D. Dunitz, J. Chem. Theory Comput., 2006, 2, 288 CrossRef CAS PubMed.
- A. G. P. Maloney, P. A. Wood and S. Parsons, CrystEngComm, 2015, 17, 9300 RSC.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276 CrossRef CAS.
- J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670 CrossRef CAS PubMed.
- T. Duren, L. Sarkisov, O. M. Yaghi and R. Q. Snurr, Langmuir, 2004, 20, 2683 CrossRef PubMed.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998 CrossRef CAS PubMed.
- M. Eddaoudi, H. L. Li and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 1391 CrossRef CAS.
- J. L. C. Rowsell, E. C. Spencer, J. Eckert, J. A. K. Howard and O. M. Yaghi, Science, 2005, 309, 1350 CrossRef CAS PubMed.
- J. Y. Lee, D. H. Olson, L. Pan, T. J. Emge and J. Li, Adv. Funct. Mater., 2007, 17, 1255 CrossRef CAS.
- J. Liu, J. Y. Lee, L. Pan, R. T. Obermyer, S. Simizu, B. Zande, J. Li, S. G. Sankar and J. K. Johnson, J. Phys. Chem. C, 2008, 112, 2911 CAS.
- L. Kong, V. R. Cooper, N. Nijem, K. Li, J. Li, Y. J. Chabal and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 081407 CrossRef.
- V. Krungleviciute, S. Pramanik, A. D. Migone and J. Li, Microporous Mesoporous Mater., 2012, 161, 134 CrossRef CAS.
- J. R. Karra and K. S. Walton, J. Phys. Chem. C, 2010, 114, 15735 CAS.
- Y. F. Chen, J. Y. Lee, R. Babarao, J. Li and J. W. Jiang, J. Phys. Chem. C, 2010, 114, 6602 CAS.
- Y. Wu, A. Kobayashi, G. J. Halder, V. K. Peterson, K. W. Chapman, N. Lock, P. D. Southon and C. J. Kepert, Angew. Chem., Int. Ed., 2008, 47, 8929 CrossRef CAS PubMed.
- Y. Liu, C. M. Brown, D. A. Neumann, V. K. Peterson and C. J. Kepert, J. Alloys Compd., 2007, 446, 385 CrossRef.
- S. Bordiga, L. Regli, F. Bonino, E. Groppo, C. Lamberti, B. Xiao, P. S. Wheatley, R. E. Morris and A. Zecchina, Phys. Chem. Chem. Phys., 2007, 9, 2676 RSC.
- L. Grajciar, A. D. Wiersum, P. L. Llewellyn, J.-S. Chang and P. Nachtigall, J. Phys. Chem. C, 2011, 115, 17925 CAS.
- B. Xiao, P. S. Wheatley, X. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203 CrossRef CAS PubMed.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef.
- Materials Studio, Release 7.0, Accelrys Software Inc., San Diego, 2013 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- F. H. Allen and I. J. Bruno, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 380 CrossRef CAS PubMed.
- H. Lipson, Endeavour, 1966, 25, 53 CrossRef.
- A. Simon and K. Peters, Acta Crystallogr., Sect. B: Struct. Sci., 1980, 36, 2750 CrossRef.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Wallingford, 2009 Search PubMed.
- D. Dubbeldam, H. Frost, K. S. Walton and R. Q. Snurr, Fluid Phase Equilib., 2007, 261, 152 CrossRef CAS.
- T. Yildirim and M. R. Hartman, Phys. Rev. Lett., 2005, 95, 215504 CrossRef CAS PubMed.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045 CrossRef CAS.
- M. Dion, H. Rydberg, E. Schroder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- T. Thonhauser, V. R. Cooper, S. Li, A. Puzder, P. Hyldgaard and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 125112 CrossRef.
- S. K. Bhatia and A. L. Myers, Langmuir, 2006, 22, 1688 CrossRef CAS PubMed.
- O. Bludsky, M. Rubes, P. Soldan and P. Nachtigall, J. Chem. Phys., 2008, 128, 114102 CrossRef PubMed.
- R. Lotfi and Y. Saboohi, Comput. Theor. Chem., 2014, 1044, 36 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ce00555a |
| This journal is © The Royal Society of Chemistry 2016 |