
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChalcogen–chalcogen secondary bonding interactions in trichalcogenaferrocenophanes†
Minna M.
Karjalainen
,
Clara
Sanchez-Perez
,
J. Mikko
Rautiainen
,
Raija
Oilunkaniemi
and
Risto S.
Laitinen
*
Laboratory of Inorganic Chemistry, University of Oulu, P.O. Box 3000 FI-90014, Finland. E-mail: risto.laitinen@oulu.fi; Tel: +358 294 481611
First published on 29th April 2016
Abstract
The solid-state structures of all members in the series of trichalcogenaferrocenophanes [Fe(C5H4E)2E′] (E, E′ = S, Se, Te) (1–9) have been explored to understand the trends in secondary bonding interactions (SBIs) between chalcogen elements sulfur, selenium, and tellurium. To complete the series, the crystal structures of the four hitherto unknown complexes [Fe(C5H4S)2Te] (3), [Fe(C5H4Se)2S] (4), [Fe(C5H4Se)2Te] (6), and [Fe(C5H4Te)2S] (7) have been determined in this contribution. The packings of all complexes 1–9 were considered by DFT calculations at the PBE0/pob-TZVP level of theory using periodic boundary conditions. The intermolecular close contacts were considered by QTAIM and NBO analyses. The isomorphous complexes [Fe(C5H4S)2S] (1), [Fe(C5H4S)2Se] (2), and [Fe(C5H4Se)2Se] (5a) form dimers via weak interactions between the central chalcogen atoms of the two trichalcogena chains of adjacent complexes. In the second isomorphous series consisting of [Fe(C5H4Se)2S] (4) and 5b, the complexes are linked together into continuous chains by short contacts via the terminal selenium atoms. The intermolecular chalcogen–chalcogen interactions are significantly stronger in complexes [Fe(C5H4S)2Te] (3), [Fe(C5H4Se)2Te] (6), and [Fe(C5H4Te)2E′] (E′ = S, Se, Te) (7–9), which contain tellurium. The NBO comparison of donor–acceptor interactions in the lattices of [Fe(C5H4S)2S] (1), [Fe(C5H4Se)2Se] (5a and 5b), and [Fe(C5H4Te)2Te] (9) indeed shows that the n(5pTe)2 → σ*(Te–Te) interactions in 9 are the strongest. All other interaction energies are significantly smaller even in the case of tellurium. The computed natural charges of the chalcogen atoms indicate that electrostatic effects strengthen the attractive interactions in the case of all chalcogen atoms.
Introduction
The concept of secondary bonding interactions (SBIs)1 describes interatomic interactions that are longer than covalent single bonds, but shorter than the sum of van der Waals radii of relevant atoms. They have also been known as non-covalent, soft–soft, closed-shell, semi-bonding, non-bonding, and weakly bonding interactions. In the case of halogen, chalcogen, and pnictogen atoms, they are also known as halogen, chalcogen, and pnictogen bonds, respectively. Meanwhile, SBIs also include hydrogen bonding, and the strength of the interactions between the p-block elements becomes more significant upon going down the groups in the periodic table.Secondary bonding interactions are currently considered to be due to electrostatic and dispersion effects in which the so-called σ-hole and polarizability play important roles.2 On the other hand, the interactions involving chalcogen compounds have traditionally been described as donor–acceptor interactions n2(D) → σ*(E–X) in which the lone pair of a donor atom D interacts with the antibonding σ* orbital of the heavy atom (E) and a more electronegative atom (X). This 3c–4e arrangement is of variable strength, from a very weak interaction to that of a hypervalent single bond.3a The energy difference between the σ(E–X) and σ*(E–X) orbitals diminishes upon going down the periodic table and therefore SBIs are stronger for tellurium compared to those of selenium and sulfur.4 However, even in the case of lighter chalcogen compounds, the complete description of the SBIs also requires the consideration of orbital interactions, as well as electrostatic and dispersion contributions.2,3 The different nature of interactions even in related compounds is exemplified by the observation that telluradiazoles show predominantly covalent interactions,5 but those in isotellurazole N-oxides are electrostatic,6 and the interactions in bis(alkynyl)tellurides are mainly due to dispersion.4 It has, however, been concluded several times that there is no real difference between charge transfer and electrostatic attraction combined with polarization effects.2c,7 Furthermore, it has also been pointed out that while several procedures to decompose secondary bonding interaction energies into contributions by different components, such as “electrostatics, exchange, Pauli exclusion, polarization, charge transfer, dispersion, induction, orbital interactions, electronic correlation, delocalization, deformation, etc.”,2c have been proposed, they cannot be considered independent contributions, and the results are likely not to be even qualitatively meaningful.
When considering interactions in chalcogen compounds, the strongest SBIs are found when the donor atom is oxygen or nitrogen, but chalcogen–chalcogen interactions are also known.3 Typical examples containing chalcogen–chalcogen SBIs are hexagonal allotropes of selenium8 and tellurium,9 in which the trigonal chains show close contacts expanding the formal coordination sphere of the chalcogen atoms to an octahedron.
Trichalcogenaferrocenophanes are a useful class of compounds for studying the trend in the SBI strengths of the group 16 elements (see Chart 1). While [Fe(C5H4E)2E′] (E, E′ = S, Se, Te) complexes have been known for a long time, crystal structures have been reported for only [Fe(C5H4S)2S] (1),10 [Fe(C5H4S)2Se] (2),11 [Fe(C5H4Se)2Se] (the monoclinic polymorph 5a and the orthorhombic polymorph 5b),12 [Fe(C5H4Te)2Se] (8),13 and [Fe(C5H4Te)2Te] (9).14 Structures of some related complexes in which the cyclopentadienyl rings of ferrocene have been modified are also known.15 Trithiaosmocenophane16 and triselenaruthenocenophane17 also show similar E⋯E close contacts to trichalcogenaferrocenophanes.10,12 In this contribution, we complement the structural and spectroscopic characterization of the whole series of [Fe(C5H4E)2E′] (E, E′ = S, Se, Te) containing both homo- and heteronuclear chalcogen chains by reporting the preparation and crystal structures of 3, 4, 6, and 7. All structural data for complexes 1–9 are compared and discussed in terms of trends in the chalcogen–chalcogen secondary bonding interactions. The experimental information is complemented by solid-state DFT calculations, which provide insight into the nature of SBIs in these systems.
 | ||
| Chart 1 [Fe(C5H4E)2E′] (E, E′ = S, Se, Te) (1–9). | ||
Experimental section
Syntheses
[Fe(C5H4S)2Se] (2), [Fe(C5H4S)2Te] (3), [Fe(C5H4Se)2S] (4), [Fe(C5H4Se)2Se] (5), [Fe(C5H4Se)2Te] (6), [Fe(C5H4Te)2S](7), [Fe(C5H4Te)2Se] (8), and [Fe(C5H4Te)2Te] (9) were prepared according to the literature procedures.11,13,14,18,19 The 77Se and 125Te NMR spectra of 2–9 were recorded from the crude products in THF, as appropriate. After purification using column chromatography involving silica gel as a stationary phase and a mixture of hexane and dichloromethane as an eluant, red X-ray quality crystals of 3, 6, and 7 were obtained from the hexane/CH2Cl2 mixture. Those of 4 were obtained in a similar fashion with the exception that hexane was used as an eluant in chromatographic separation, and recrystallization was carried out from the hexane solution upon evaporation of the solvent.Crystal structures
Diffraction data for [Fe(C5H4S)2Te] (3), [Fe(C5H4Se)2S] (4), [Fe(C5H4Se)2Te] (6) and [Fe(C5H4Te)2S] (7) were collected using a Nonius Kappa CCD diffractometer at 120 K (3 and 4) and at room temperature (6 and 7) using graphite monochromated MoKα radiation (λ = 0.71073 Å). Crystal data and details of the structure determination are given in Table S1 in the ESI.† Structures were solved by direct methods using SHELXS-2013 and refined using SHELXL-2013.20 After the full-matrix least-squares refinement of non-hydrogen atoms with anisotropic thermal parameters, the hydrogen atoms were placed in calculated positions in the cyclopentadienyl rings (C–H = 0.95 Å). In the final refinement, the hydrogen atoms were riding with the carbon atom they were bonded to. The isotropic thermal parameters of the hydrogen atoms were fixed at 1.2 times that of the corresponding carbon atom. The scattering factors for the neutral atoms were those incorporated with the programs.Computational details
Solid-state DFT calculations of the trichalcogenaferrocenophanes were carried out using the CRYSTAL14 software package21 using the PBE0 functional22 and localized atomic basis sets composed of Gaussian-type functions. Triple-zeta valence basis sets pob-TZVP, which were designed for solid-state calculations and include polarization functions, were used for sulfur, carbon, iron, and hydrogen.23 The selenium basis set was derived from the pob-TZVP basis set by increasing the exponent of the most diffuse s function to half of the second most diffuse s function and determining the exponent of the most diffuse p function from the other p functions using the even-tempered method.24 We used the same modified basis set for tellurium as that previously employed.25The optimization was started from the experimental X-ray structures. Both the lattice parameters and atomic positions were optimized in the calculations. Monkhorst–Pack-type grids of k-points in the reciprocal space were generated using a shrinking factor (SHRINK) of 8. For the evaluation of Coulomb and exchange integrals (TOLINTEG), tolerance factors of 7, 7, 7, 7, and 14 were used. Default optimization thresholds and integration grid for the density functional part were employed in all calculations. The effect of dispersion forces on energy was modeled using Grimme's empirical D2 model.26 The D2 model has a tendency to overemphasize weak interactions26b and therefore the SBIs in the optimized structures are shorter than the corresponding contacts in the experimental structures. Despite this overbinding, the calculated structures are considered to give meaningful relative strengths of the different chalcogen–chalcogen bonding interactions, which have been analyzed using topological analysis of the electron density.27
In previous studies, an empirical relationship between the hydrogen bond interaction energy and potential energy density at the bond critical point has been suggested by Espinosa et al. as Eint = VBCP/2.28 The use of this relationship has been extended by others to other weak closed-shell interactions29 and the relationship is adopted here as an additional descriptor for qualitatively comparing the relative strengths of chalcogen–chalcogen contacts and hydrogen bonds. It should be noted that the reliability of the results from the relationship has been questioned30 and they should be considered qualitative at most. Topological analysis was performed with the TOPOND module31 implemented in the CRYSTAL14 program. For TOPOND calculations, basis sets were modified by removing the f functions from iron and tellurium.
The donor–acceptor nature of the chalcogen–chalcogen interactions between trichalcogenaferrocenophanes has been studied using natural bond orbital (NBO) analysis32 on snapshots of the optimized crystal structures. Each of the snapshots included four molecules to represent the closest chalcogen–chalcogen interactions in the crystal structures (see the ESI†) and donor–acceptor interaction energies were estimated by second-order perturbation theory analysis between filled donor NBOs and vacant acceptor NBOs. The NBO analyses were carried out using NBO 5.9 software33 on Kohn–Sham orbitals from a PBE0/pob-TZVP single point Gaussian 09 calculation.34
Results and discussion
Crystal structures
Each lattice of 1–9 consists of similar discrete molecules, as shown in Fig. 1 together with the numbering scheme of atoms, which has been used in the case of complexes 3, 4, 6, and 7 determined in this contribution. Selected bond lengths of each member in the series [Fe(C5H4E)2E′] (E, E′ = S, Se, Te) are presented in Table S2 in the ESI.† It can be seen that the lengths of all bonds are close to those of single bonds (see covalent radii in ref. 35). The PBE0/pob-TZVP-optimized solid-state structures are also presented in Table S2† and show very good agreement with experimentally determined values.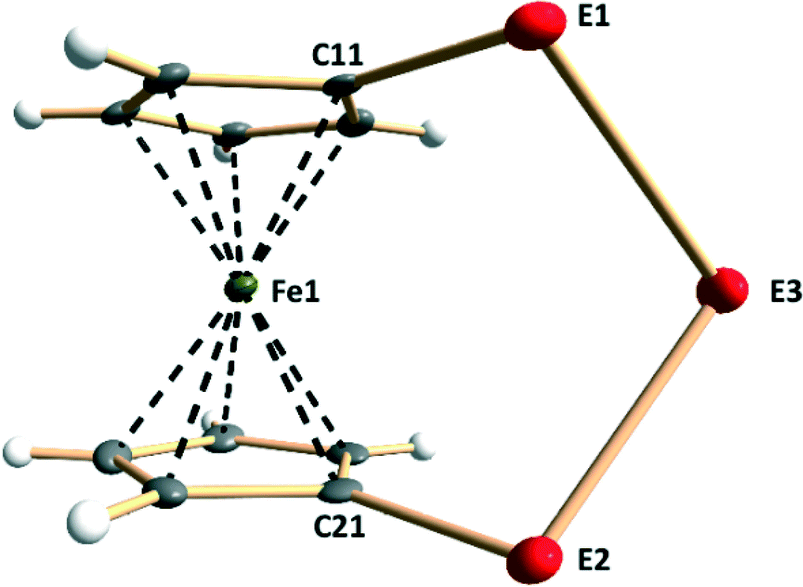 | ||
| Fig. 1 Molecular structure of [Fe(C5H4E)2E′] (E, E′ = S, Se, Te) (1–9) indicating the labeling of the atoms. The crystal structures of 3, 4, 6, and 7 have been determined in this contribution. | ||
The variation in the packing of the [Fe(C5H4E)2E′] (E, E′ = S, Se, Te) (1–9) complexes is shown in Fig. 2. [Fe(C5H4S)2S] (1),10 [Fe(C5H4S)2Se] (2),11 and [Fe(C5H4Se)2Se] (5a)12c are isomorphic, crystallizing in the monoclinic space group P21/c [see Fig. 2(a)].
 | ||
| Fig. 2 Packing of [Fe(C5H4E)2E′] (E, E′ = S, Se, Te) (1–9). (a) [Fe(C5H4S)2S] (1),10 [Fe(C5H4S)2Se] (2),11 and [Fe(C5H4Se)2Se] (5a),12c (b) [Fe(C5H4Se)2S] (4) and [Fe(C5H4Se)2Se] (5b),12a (c) [Fe(C5H4S)2Te] (3) and [Fe(C5H4Se)2Te] (6), (d) [Fe(C5H4Te)2S] (7), (e) [Fe(C5H4Te)2Se] (8),9 and (f) [Fe(C5H4Te)2Te] (9).10 The packings of 1, 2, 5a, 5b, 8, and 9 have been redrawn from crystallographic information in the appropriate references. | ||
They form loosely linked dimers with close contacts between the central chalcogen atoms. In 1, the S⋯S distance is 3.7056(11) Å (ref. 10) and in 2 and 5a, the corresponding Se⋯Se close contacts are 3.6394(8) (ref. 11) and 3.6348(11) Å,12c respectively [see Fig. 2(a)]. [Fe(C5H4Se)2S] (4) and [Fe(C5H4Se)2Se] (5b)12a are also mutually isomorphic (orthorhombic space group Pca21). These complexes form quasi-planar chains linked by terminal selenium atoms with the respective distances of 3.8705(12) Å and 3.9572(13) Å (ref. 12a) [see Fig. 2(b)]. Complexes 3 and 6–9 [see Fig. 2(c–f)] containing tellurium show supramolecular frameworks with more numerous and shorter intermolecular chalcogen–chalcogen contacts than complexes where the trichalcogena chain consists only of sulfur and selenium atoms. [Fe(C5H4S)2Te] (3) and [Fe(C5H4Se)2Te] (6) are mutually isomorphic (different monoclinic lattices from those of 1, 2, and 5a, though crystallizing also in the space group P21/c). The S⋯S, S⋯Te, and Te⋯Te close contacts in 3 are 3.5749(17), 3.4693(14) and 3.5119(13), and 3.8517(7) Å, respectively. The analogous Se⋯Se, Se⋯Te, and Te⋯Te distances in 6 are 3.5989(16) Å, 3.6239(15) Å and 3.6908(16) Å, and 3.8795(13) Å, respectively. The S⋯Te distance in 7 is 3.446(3) Å, the shortest Se⋯Te distance in 8 is 3.7044(14) Å,13 and the shortest Te⋯Te contacts in 7–9 are 3.5559(13), 3.7241(13)–3.9168(18),13 and 3.4552(17)–3.8691(17) Å,14 respectively. All these distances are well below the sums of van der Waals' radii of the elements in question.35
All complexes 1–9 are also linked together with H⋯E (E = chalcogen) bonds and also show E⋯π and H⋯π electron interactions involving the cyclopentadienyl rings. The shortest close contacts are exemplified in Fig. S1 (see the ESI†) for [Fe(C5H4S)2S] (1), [Fe(C5H4Se)2Se] (5a and 5b), and [Fe(C5H4Te)2Te] (9). The strength of these interactions does not seem to vary within the series. However, as the strength and the number of chalcogen–chalcogen interactions increase, the number of H⋯E, E⋯π and H⋯π interactions diminishes. Only the tellurium-containing complexes 3, 6, 8, and 9 exhibit π-stacking of the cyclopentadienyl rings.
Secondary bonding chalcogen–chalcogen interactions in [Fe(C5H4E)2E′] (1–9)
The intermolecular E⋯E′ (E, E′ = S, Se, Te) bond orders for the experimental structures have been estimated using Pauling's relationship,36 and the relative bond orders for the optimized structures were calculated from the bond critical point electron densities determined using the QTAIM theory (see Table 1).27 The corresponding bond critical points are shown in Fig. 3 (see the ESI† for a more detailed description of the calculations of the QTAIM bond orders). The estimated interaction energies for the different chalcogen–chalcogen contacts are also presented in Table 1.| Complex | BCPb | Contact | X-ray (Å) | R | Pauling BOd | DFT (Å) | Anglee | ρ BCP (e Å−3) | QTAIM BOg | V(bcp) (hartree bohr−3) | E INT (kJ mol−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
a The entries of different complexes in the table have been sorted in the order of increasing QTAIM bond orders.
b BCP = bond critical point.
c Ratio of the interatomic distance and the sum of van der Waals' radii.35
d Pauling bond orders N have been calculated from intermolecular close contacts using the relationship  ,36 in which Ro is the sum of covalent radii35 of the two atoms in question and Re is the interatomic distance in the experimental X-ray structure.
e Optimized ∠E–E⋯E angle.
f Electron density at bond critical point.
g Relative QTAIM bond orders of intermolecular close contacts have been determined from bond critical point electron densities ρBCP calculated for the optimized structures using ρBCP of the intramolecular chalcogen–chalcogen bonds in [Fe(C5H4E)2E′] (1–9) as references for bonds with bond order of 1 (see the ESI).
h
Ref. 10.
i
Ref. 11.
j
Ref. 12.
k
Ref. 13.
l
Ref. 14. ,36 in which Ro is the sum of covalent radii35 of the two atoms in question and Re is the interatomic distance in the experimental X-ray structure.
e Optimized ∠E–E⋯E angle.
f Electron density at bond critical point.
g Relative QTAIM bond orders of intermolecular close contacts have been determined from bond critical point electron densities ρBCP calculated for the optimized structures using ρBCP of the intramolecular chalcogen–chalcogen bonds in [Fe(C5H4E)2E′] (1–9) as references for bonds with bond order of 1 (see the ESI).
h
Ref. 10.
i
Ref. 11.
j
Ref. 12.
k
Ref. 13.
l
Ref. 14.
|
|||||||||||
| [Fe(C5H4S)2S] (1) | A | S⋯S–S | 3.7056(11)h | 1.00 | 0.10 | 3.562 | 161.5 | 0.006 | 0.04 | −0.00282 | −3.7 |
| [Fe(C5H4S)2Se] (2) | A | Se⋯Se–S | 3.6394(8)i | 0.91 | 0.16 | 3.375 | 159.0 | 0.014 | 0.13 | −0.00782 | −10.3 |
| [Fe(C5H4Se)2Se] (5a) | A | Se⋯Se–Se | 3.6348(11) | 0.91 | 0.16 | 3.402 | 155.0 | 0.013 | 0.12 | −0.00746 | −9.8 |
| [Fe(C5H4Se)2S] (4) | B | Se⋯Se–S | 3.8705(17) | 0.97 | 0.12 | 3.607 | 159.0 | 0.010 | 0.09 | −0.00507 | −6.7 |
| [Fe(C5H4Se)2Se] (5b) | B | Se⋯Se–Se | 3.9572(13)j | 0.99 | 0.10 | 3.606 | 156.6 | 0.010 | 0.09 | −0.00509 | −6.7 |
| [Fe(C5H4S)2Te] (3) | C | S⋯S–Te | 3.5751(15) | 0.97 | 0.12 | 3.359 | 150.9 | 0.010 | 0.07 | −0.00634 | −8.3 |
| D | Te⋯S–C | 3.5121(11) | 0.87 | 0.21 | 3.431 | 164.6 | 0.012 | 0.12 | −0.00645 | −8.5 | |
| F | Te⋯Te–S | 3.8517(7) | 0.88 | 0.21 | 3.792 | 177.2 | 0.021 | 0.16 | −0.00454 | −6.0 | |
| E | S⋯Te–S | 3.4692(13) | 0.86 | 0.23 | 3.287 | 177.4 | 0.018 | 0.19 | −0.00957 | −12.6 | |
| [Fe(C5H4Se)2Te] (6) | C | Se⋯Se–Te | 3.5997(16) | 0.90 | 0.17 | 3.336 | 146.5 | 0.016 | 0.14 | −0.00945 | −12.4 |
| D | Te⋯Se–C | 3.6240(14) | 0.86 | 0.22 | 3.423 | 163.5 | 0.015 | 0.18 | −0.00825 | −10.8 | |
| F | Te⋯Te–Se | 3.8790(13) | 0.88 | 0.20 | 3.743 | 179.3 | 0.012 | 0.18 | −0.00503 | −6.6 | |
| E | Se⋯Te–Se | 3.6915(16) | 0.88 | 0.20 | 3.408 | 175.1 | 0.018 | 0.21 | −0.00898 | −11.8 | |
| [Fe(C5H4Te)2S (7) | G | Te⋯Te | 4.0070(13) | 0.91 | 0.16 | 3.764 | −/73.2 | 0.015 | 0.22 | −0.00630 | −8.3 |
| H | S⋯Te–S | 3.446(3) | 0.85 | 0.23 | 3.137 | 175.6 | 0.023 | 0.24 | −0.01363 | −17.9 | |
| I | Te⋯Te–S | 3.5559(13) | 0.81 | 0.32 | 3.267 | 161.9 | 0.028 | 0.41 | −0.01430 | −18.8 | |
| [Fe(C5H4Te)2Se (8) | J | Se⋯Te–C | 3.9634(15)k | 0.94 | 0.14 | 3.816 | 135.6 | 0.009 | 0.11 | −0.00451 | −5.9 |
| K | Te⋯Se–Te | 4.005(3)k | 0.95 | 0.12 | 3.752 | 138.6 | 0.010 | 0.12 | −0.00441 | −5.8 | |
| L | Te⋯Se–Te | 4.117(3)k | 0.98 | 0.11 | 3.769 | 140.2 | 0.010 | 0.12 | −0.00429 | −5.6 | |
| M | Se⋯Te–C | 3.7044(14)k | 0.88 | 0.19 | 3.582 | 161.9 | 0.013 | 0.15 | −0.00626 | −8.2 | |
| N | Te⋯Te–C | 3.8756(13)k | 0.88 | 0.20 | 3.848 | 137.4 | 0.010 | 0.15 | −0.00421 | −5.5 | |
| O | Te⋯Te–C | 3.7241(13)k | 0.85 | 0.25 | 3.623 | 169.4 | 0.011 | 0.16 | −0.00527 | −6.9 | |
| P | Te⋯Te–Se | 3.9168(18)k | 0.89 | 0.19 | 3.769 | 164.0 | 0.013 | 0.19 | −0.00475 | −6.2 | |
| Q | Te⋯Te–Se | 3.727(2)k | 0.85 | 0.25 | 3.493 | 173.8 | 0.019 | 0.28 | −0.00865 | −11.4 | |
| [Fe(C5H4Te)2Te] (9) | R | Te⋯Te–Te | 3.8806(19)l | 0.88 | 0.20 | 3.923 | 147.7 | 0.008 | 0.12 | −0.00319 | −4.2 |
| S | Te⋯Te–C | 3.8691(17)l | 0.88 | 0.20 | 3.877 | 172.4 | 0.010 | 0.15 | −0.00403 | −5.3 | |
| T | Te⋯Te–C | 3.7472(17)l | 0.85 | 0.24 | 3.659 | 161.9 | 0.012 | 0.18 | −0.00538 | −7.1 | |
| U | Te⋯Te–Te | 3.7124(18)l | 0.84 | 0.25 | 3.663 | 176.2 | 0.014 | 0.20 | −0.00602 | −7.7 | |
| V | Te⋯Te–Te | 3.7673(18)l | 0.86 | 0.24 | 3.677 | 165.6 | 0.015 | 0.22 | −0.00589 | −7.7 | |
| W | Te⋯Te–Te | 3.5493(18)l | 0.81 | 0.32 | 3.487 | 177.8 | 0.016 | 0.23 | −0.00734 | −9.6 | |
| X | Te⋯Te–Te | 3.6828(18)l | 0.84 | 0.27 | 3.503 | 172.4 | 0.019 | 0.28 | −0.00836 | −11.0 | |
| Y | Te⋯Te–Te | 3.4552(17)l | 0.78 | 0.36 | 3.388 | 176.7 | 0.024 | 0.35 | −0.01087 | −14.3 | |
 | ||
| Fig. 3 The QTAIM bond critical points in the optimized structures of (a) [Fe(C5H4S)2S] (1), [Fe(C5H4S)2Se] (2), and [Fe(C5H4Se)2Se] (5a), (b) [Fe(C5H4Se)2S] (4) and [Fe(C5H4Se)2Se] (5b), (c) [Fe(C5H4S)2Te] (3) and [Fe(C5H4Se)2Te] (6), (d) [Fe(C5H4Te)2S] (7), (e) [Fe(C5H4Te)2Se] (8), and (f) [Fe(C5H4Te)2Te] (9). For the intermolecular distances and bond orders, see Table 1. | ||
Consistent with the relatively long close contacts in complexes 1, 2, 4, and 5, which are near to the sums of the corresponding van der Waals radii of the atoms in question, the electron density values at bond critical points, as well as both the Pauling and QTAIM bond orders and interaction energies, indicate that the intermolecular chalcogen–chalcogen interactions in these complexes are weak (see Table 1). The electron density values at bond critical points, the bond orders, and the interaction energies are higher in complexes 3, 6, and 7–9, which contain tellurium. This is in accordance with the conclusions of previous studies.3,4
A simple model of donation of the p lone-pair electrons to the anti-bonding σ* orbital has been used to describe the chalcogen–chalcogen SBIs qualitatively,3,4 though they can also be accounted for by the presence of a σ-hole together with electrostatic and polarization effects.2 The correlation between QTAIM bond orders and the collinearity of the E⋯E′–X moieties (E, E′ = S, Se, Te; X = S, Se, Te, C) shown by the interactions (see Table 1) are in agreement with both models of the interactions.
In order to compare the relative donor–acceptor interaction strengths, we carried out NBO analyses for [Fe(C5H4S)2S] (1), [Fe(C5H4Se)2Se] (5a and 5b), and [Fe(C5H4Te)2Te] (9). The main interaction modes have been shown in Fig. 4.
 | ||
| Fig. 4 The main intermolecular n2 → σ* interactions in [Fe(C5H4E)2E] [E = S (1), Se (5a, b), Te (9)]. (a) n(p)2 → σ*(E–E), (b) n(s)2 → σ*(E–E), (c) n(p)2 → σ*(E–C), (d) n(s)2 → σ*(E–E). The donor orbital is shown in blue and the acceptor orbital in red. | ||
[Fe(C5H4S)2S] (1) shows very weak interactions of types n(3p)2 → σ*(S–S) and n(3s)2 → σ*(S–S) shown in Fig. 4(a) and (d), respectively. In both cases, the NBO interaction energies are approximately 1.5 kJ mol−1. The corresponding interaction energies between the selenium atoms in the isomorphic [Fe(C5H4Se)2Se] (5a) are around 2.9 kJ mol−1. The interaction energies in 5b are comparable, though the donor–acceptor interaction n(3p)2 → σ*(Se–C) is somewhat stronger being 7.9 kJ mol−1. It is only with [Fe(C5H4Te)2Te] (9) that the interaction energies are significantly higher. The largest interaction energies in 9 are shown in Fig. 5 together with the observed and computed intermolecular distances. It can be seen that they correlate very well with the closest intermolecular contacts observed in the crystal structures and also with the relative QTAIM electron density values, bond orders, and interaction energies (see Table 1).
 | ||
| Fig. 5 Main NBO n(5pTe)2 → σ*(Te–Te) interaction energies and the corresponding short intermolecular contacts in [Fe(C5H4Te)2Te] (9). The experimental values from the crystal structure are given in upright font and the corresponding parameters from the DFT calculations in italics. | ||
All significant interaction energies in 9, which are shown in Fig. 5, are found for n(5pTe)2 → σ*(Te–Te) interactions involving the donation from the terminal tellurium in the Te3 chain [see Fig. 4(a)]. All other interactions shown in Fig. 4 are below 16 kJ mol−1.
Electrostatic effects involving the lone-pair electrons and the σ-holes of the chalcogen–chalcogen bonds strengthen the attractive interaction between the complexes in each case. This effect may be inferred by the PBE0/pob-TZVP natural charges of the chalcogen atoms, as shown in Table 2. The charge on the central chalcogen atom is nearly zero, as expected for atoms in the middle of homonuclear chains. The terminal chalcogen atoms, however, carry a significant positive charge. It is reasonable that these atoms are attracted by lone-pair electrons. Also, this effect is most prominent in the case of tellurium.
| Complex | Bridging chalcogen atom | Terminal chalcogen atoms | ||
|---|---|---|---|---|
| Range | Average | Average | Range | |
| [Fe(C5H4S)2S] (1) | −0.041 to −0.065 | −0.049 | +0.159 to +0.176 | +0.166 |
| [Fe(C5H4Se)2Se] (5a) | −0.019 to −0.054 | −0.036 | +0.199 to +0.221 | +0.207 |
| [Fe(C5H4Se)2Se] (5b) | −0.029 to −0.059 | −0.041 | +0.196 to +0.216 | +0.205 |
| [Fe(C5H4Te)2Te] (9) | −0.043 to −0.113 | −0.084 | +0.204 to +0.355 | +0.284 |
H⋯E, E⋯π and H⋯π interactions in [Fe(C5H4E)2E] [E = S (1), Se (5a and 5b), Te (9)].
The interaction energies involving hydrogen bonds and π-stacking in [Fe(C5H4E)2E] [E = S (1), Se (5a and 5b), Te (9)] are compared in Table 3. The interaction energies of the hydrogen bonds seem to be of the same order of magnitude in all three complexes. It can be concluded that H⋯E and H⋯π hydrogen bonds play a determining role in the lattice of 1, but their significance diminishes in 5a and 5b. In the case of [Fe(C5H4Te)2Te] (9), the hydrogen bonds have a minor effect and the structure of the lattice is determined by Te⋯Te SBIs. This seems to be the case in all complexes containing tellurium.| Complex | Contact | Exptl. (Å) | Calc. (Å) | ρ (e Å−3) | V(bcp) (hartree bohr−3) | E INT (kJ mol−1) |
|---|---|---|---|---|---|---|
| a Ct denotes the centroid of the carbon atoms in the cyclopentadienyl ring. b Ref. 10. c Ref. 12. d Ref. 14. | ||||||
| [Fe(C5H4S)2S] (1) | S⋯H | 2.9897(8)–3.1018(9)b | 2.568–2.738 | 0.0057–0.0138 | −0.00258–(−0.00687) | −3.4–(−9.0) |
| S⋯Cta | 3.5803(10)b | 3.370 | ||||
| H⋯Cta | 3.2626(9)–3.4998(11)b | 2.867–3.322 | ||||
| [Fe(C5H4Se)2Se] (5a) | Se⋯H | 3.0271(6)–3.2309(6) | 2.636–2.865 | 0.0092–0.0157 | −0.00448–(−0.00794) | −5.9–(−10.4) |
| Se⋯Cta | 3.6701(6)–3.9977(9) | 3.429–3.653 | ||||
| H⋯Cta | 3.2477(6) | 2.781 | ||||
| [Fe(C5H4Se)2Se] (5b) | Se⋯H | 3.0789(6)–3.2436(7)c | 2.664–3.039 | 0.0140 | −0.00779 | −10.2 |
| Se⋯Cta | 3.8875(8)c | 3.603 | ||||
| H⋯Cta | 3.2770(8)c | 2.812 | ||||
| [Fe(C5H4Te)2Te] (9) | Te⋯H | 3.3573(12)–3.6521(13)d | 2.980–3.432 | 0.0061–0.0084 | −0.00249–(−0.00354) | −3.3–(−4.6) |
| H⋯Cta | 3.2721(12)–3.4430(13)d | 3.031–3.346 | ||||
| Ct⋯Cta | 3.7991(14)d | 3.741 | ||||
It can also be verified in Table 3 that the cyclopentadienyl rings are only involved in π-stacking in the case of 9.
Conclusions
Trichalcogenaferrocenophanes [Fe(C5H4E)2E′] (E, E′ = S, Se, Te) (1–9) are a useful series for studying the trends in secondary bonding interactions (SBIs) between chalcogen elements sulfur, selenium, and tellurium. In this contribution, we have compared the structures and packings of all [Fe(C5H4E)2E′] complexes (1–9). We have prepared and determined the crystal structures of all hitherto missing members of the series (3, 4, 6, and 7). The comparison of the experimental crystal lattices with those optimized at the PBE0/pob-TZVP level of theory utilizing periodic boundary conditions has enabled inferences on the geometries and relative strengths of the intermolecular interactions that have been explored by the use of QTAIM and NBO analyses.Complexes 1, 2, 4, 5a and 5b show only weak intermolecular interactions. The isomorphous 1, 2, and 5a form dimers where the most significant interaction is between the central chalcogen atoms of the two trichalcogena chains of adjacent complexes. In the second isomorphous series consisting of 4 and 5b, the complexes are linked together into continuous chains by short contacts via the terminal selenium atoms.
The intermolecular interactions are expectedly stronger in complexes 3, 6, and 7–9, which contain tellurium. The NBO comparison of donor–acceptor interactions in the lattices of [Fe(C5H4S)2S] (1), [Fe(C5H4Se)2Se] (5a and 5b), and [Fe(C5H4Te)2Te] (9) show that the n(5pTe)2 → σ*(Te–Te) interactions in 9 are the strongest. All other interaction energies are significantly smaller even in the case of tellurium. The computed natural charges of the chalcogen atoms indicate that electrostatic effects strengthen the attractive interactions in the case of all chalcogen atoms.
Acknowledgements
Financial support from the Academy of Finland (The Inorganic Materials Chemistry Graduate Program), The Emil Aaltonen Foundation (M. M. K.), and The Fortum Foundation (C. S.-P.) is gratefully acknowledged. We are also grateful to Finnish CSC-IT Center for Science Ltd for their generous provision of computational resources.References
- N. W. Alcock, Adv. Inorg. Chem. Radiochem., 1972, 15, 1 CrossRef CAS.
- (a) J. S. Murray, P. Lane and P. Politzer, J. Mol. Model., 2009, 15, 723–729 CrossRef CAS PubMed; (b) M. E. Brezgunova, J. Lieffrig, E. Aubert, S. Dahaoui, P. Fertey, S. Lebègue, J. G. ÁngyÁn, M. Fourmigué and E. Espinosa, Cryst. Growth Des., 2013, 13, 3283–3289 CrossRef CAS; (c) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC; (d) G. E. Garrett, G. L. Gibson, R. N. Straus, D. S. Seferos and M. S. Taylor, J. Am. Chem. Soc., 2015, 137, 4126–4133 CrossRef CAS PubMed; (e) P. Politzer, J. S. Murray and T. Clark, Top. Curr. Chem., 2015, 358, 19–42 CrossRef CAS PubMed; (f) M. D. Esrafili and H. Akhgarpour, Mol. Phys., 2016 DOI:10.1080/00268976.2016.1158421; (g) M. H. Kolár and P. Hobza, Chem. Rev., 2016, 116, 5155–5187 CrossRef PubMed; (h) P. B. Lutz and C. A. Bayse, J. Inorg. Biochem., 2016, 157, 94–106 CrossRef CAS PubMed; (i) H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS PubMed.
- For recent reviews, see (a) A. F. Cozzolino, P. J. W. Elder and I. Vargas-Baca, Coord. Chem. Rev., 2011, 255, 1426 CrossRef CAS; (b) W.-W. du Mont and C. G. Hrib, in Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, ed. F. A. Devillanova and W.-W. du Mont, RSC Publishing, U. K., 2nd edn, 2013, ch. 12.1, vol. 2, pp. 273–316 Search PubMed; (c) T. Chivers and R. S. Laitinen, Chem. Soc. Rev., 2015, 44, 1725 RSC.
- C. Bleiholder, D. B. Werz, H. Köppel and R. Gleiter, J. Am. Chem. Soc., 2006, 128, 2666 CrossRef CAS PubMed.
- (a) A. F. Cozzolino, I. Vargas-Baca, S. Mansour and A. H. Mahmooudkhani, J. Am. Chem. Soc., 2005, 127, 3184–3190 CrossRef CAS PubMed; (b) A. F. Cozzolino, P. J. W. Elder, L. M. Lee and I. Vargas-Baca, Can. J. Chem., 2013, 91, 338–347 CrossRef CAS, and references cited therein.
- J. Kübel, P. J. W. Elder, H. A. Jenkins and I. Vargas-Baca, Dalton Trans., 2010, 39, 11126–11128 RSC.
- (a) A. J. Stone and S. L. Price, J. Phys. Chem., 1988, 92, 3325–3335 CrossRef CAS; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS; (c) W. A. Sokalski and S. M. Roszak, THEOCHEM, 1991, 234, 387–400 CrossRef; (d) J. Chen and T. J. Martínez, Chem. Phys. Lett., 2007, 438, 315–320 CrossRef CAS.
- R. Keller, W. B. Holzapfel and H. Schulz, Phys. Rev. B: Solid State, 1977, 16, 4404 CrossRef CAS.
- C. Adenis, V. Langer and O. Lindqvist, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 941 CrossRef.
- B. R. Davis and I. Bernal, J. Cryst. Mol. Struct., 1972, 2, 143 CrossRef CAS.
- A. G. Osborne, R. E. Hollands, J. A. K. Howard and R. F. Bryan, J. Organomet. Chem., 1981, 205, 395 CrossRef CAS.
- (a) P. G. Jones, C. Thöne and C. O. Kienitz, Z. Kristallogr. - New Cryst. Struct., 1997, 212, 118 CAS; (b) R. F. Bryan and S. N. Lockhart, Eur. Cryst. Meeting, 1983, vol. 8, p. 200 Search PubMed; (c) J. Kahr, C. Moser, S. Spirk, F. Belaj and R. Pietschnig, Phosphorus, Sulfur Silicon Relat. Elem., 2014, 189, 1467 CrossRef CAS.
- M. M. Karjalainen, R. Oilunkaniemi and R. S. Laitinen, Inorg. Chim. Acta, 2012, 390, 79 CrossRef CAS.
- M. Herberhold, P. Leitner and U. Thewalt, Z. Naturforsch., B: J. Chem. Sci., 1990, 45, 1503 CAS.
- (a) I. R. Butler, W. R. Cullen, F. G. Herring, N. R. Jagannathan, F. W. B. Einstein and R. Jones, Inorg. Chem., 1986, 25, 4534 CrossRef CAS; (b) P. F. Brandt, D. L. Compton and T. B. Rauchfuss, Organometallics, 1998, 17, 2702 CrossRef CAS; (c) S. Zürcher, J. Petrig, V. Gramlich, M. Wörle, C. Mensing, D. von Arx and A. Togni, Organometallics, 1999, 18, 3679 CrossRef; (d) D. L. Compton and T. B. Rauchfuss, Organometallics, 1994, 13, 4367 CrossRef CAS; (e) G. Thaler, B. Klotz, K. Wurst and F. Sladky, J. Organomet. Chem., 2001, 637, 745 CrossRef; (f) S. Fukuzawa and D. Wachi, Heteroat. Chem., 2006, 17, 118 CrossRef CAS.
- E. W. Abel, N. J. Long, A. G. Osborne, M. B. Hursthouse and M. A. Mazid, J. Organomet. Chem., 1992, 430, 117 CrossRef CAS.
- A. J. Blake, R. O. Gould and A. G. Osborne, J. Organomet. Chem., 1986, 308, 297 CrossRef CAS.
- R. Broussier, A. Abdulla and B. Gautheron, J. Organomet. Chem., 1987, 332, 165 CrossRef CAS.
- M. Herberhold and P. Leitner, J. Organomet. Chem., 1991, 411, 233 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- (a) R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-Wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre, P. D'Arco, Y. Noël, M. Causà, M. Rérat and B. Kirtman, Int. J. Quantum Chem., 2014, 114, 1287 CrossRef CAS; (b) R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D'Arco, M. Llunell, M. Causà and Y. Noël, CRYSTAL14 User's Manual, University of Torino, Torino, 2014 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- M. F. Peintinger, D. V. Oliveira and T. Bredow, J. Comput. Chem., 2013, 34, 451 CrossRef CAS PubMed.
- D. C. Young, Computational Chemistry, Wiley-Interscience, New York, 2001 Search PubMed.
- S. M. Närhi, J. Kutuniva, M. K. Lajunen, M. K. Lahtinen, H. M. Tuononen, A. J. Karttunen, R. Oilunkaniemi and R. S. Laitinen, Spectrochim. Acta, Part A, 2014, 117, 728 CrossRef PubMed.
- (a) S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed; (b) L. A. Burns, Á. Vázquez-Mayagoitia, B. G. Sumpter and C. D. Sherrill, J. Chem. Phys., 2011, 134, 084107 CrossRef PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- (a) Y.-S. Chen, A. I. Stash and A. A. Pinkerton, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 309–318 CAS; (b) M. S. Pavan, R. Pal, K. Nagarajan and T. N. G. Row, Cryst. Growth Des., 2014, 14, 5477–5485 CrossRef CAS; (c) M. Bai, S. P. Thomas, R. Kottokkaran, S. K. Nayak, P. C. Ramamurthy and T. N. G. Row, Cryst. Growth Des., 2014, 14, 459–466 CrossRef CAS.
- M. A. Spackman, Cryst. Growth Des., 2015, 15, 5624–5628 CAS.
- C. Gatti and S. Casassa, TOPOND14 User's Manual, CNR-ISTM Milano, Milano, 2014 Search PubMed.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS; (b) E. D. Glendening, C. R. Landis and F. Weinhold, WIREs Comput. Mol. Sci., 2012, 2, 1 CrossRef CAS; (c) F. Weinhold, J. Comput. Chem., 2012, 33, 2363 CrossRef CAS PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.9., Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2012, http://www.chem.wisc.edu/~nbo5 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- J. Emsley, The Elements, Oxford University Press, Oxford, UK, 3rd edn, 1998 Search PubMed.
- L. Pauling, The Nature of The Chemical Bond, Cornell University Press, Ithaca, NY, 3rd edn, 1960 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic information, QTAIM bond orders and NBO analysis, and NMR spectroscopic information. CCDC 1455391–1455394, CIF files of PBE0/def2-TZVPP optimized crystal structures. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce00451b |
| This journal is © The Royal Society of Chemistry 2016 |