
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDimorphism of 1,4-dibromo-2,5-bis(bromomethyl)benzene: crystallographic and physico-chemical investigations†
Christian
Näther
*a,
Inke
Jess
a,
Piotr
Kuś
b and
Peter G.
Jones
*c
aInstitute of Inorganic Chemistry, Christian-Albrechts-University of Kiel, Max-Eyth-Str. 2, 24118 Kiel, Germany
bDepartment of Chemistry, University of Silesia, 9 Szkolna St., 40-006 Katowice, Poland
cInstitute of Inorganic and Analytical Chemistry, Technical University of Braunschweig, P.O. Box 3329, D-38023 Braunschweig, Germany. E-mail: p.jones@tu-bs.de; Fax: +49 531 5387; Tel: +49 531 5382
First published on 4th April 2016
Abstract
Two polymorphic modifications of 1,4-dibromo-2,5-bis(bromomethyl)benzene have been discovered and structurally characterized; their thermodynamic relationships and transformation behaviour have been investigated. Form I crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , whereas form II crystallizes monoclinic in space group P21/c, both with imposed inversion symmetry of the molecule. Their crystal structures involve layers, in which the molecules are linked by intermolecular Br⋯Br interactions to form similar systems of linked rings. Initial studies involved batches consisting of pure form I or a mixture of I with traces of II (as obtained by chance from the synthesis), but solvent-mediated conversion experiments in various solvents clearly prove that form II is the thermodynamically stable form at room temperature. Thermomicroscopic and heating-rate-dependent DSC measurements show that the melting point of form I is slightly higher than that of form II and that the higher melting polymorph exhibits the lower heat of fusion. Therefore, form I becomes thermodynamically stable at higher temperatures and both forms are related by enantiotropism. This is also in agreement with the density rule, because the low-temperature form II exhibits the higher density. Isothermal annealing of both modifications at different temperatures reveals a thermodynamic transition temperature of about 135 °C, which is in excellent agreement with that of 137.5 °C calculated from the melting temperatures and the heat of fusion of both forms. The high-temperature form can easily be prepared pure by solidification of the melt, which is in agreement with Ostwald's step rule, because form I crystallizes at a temperature where it is thermodynamically metastable. A qualitative energy/temperature diagram is presented.
, whereas form II crystallizes monoclinic in space group P21/c, both with imposed inversion symmetry of the molecule. Their crystal structures involve layers, in which the molecules are linked by intermolecular Br⋯Br interactions to form similar systems of linked rings. Initial studies involved batches consisting of pure form I or a mixture of I with traces of II (as obtained by chance from the synthesis), but solvent-mediated conversion experiments in various solvents clearly prove that form II is the thermodynamically stable form at room temperature. Thermomicroscopic and heating-rate-dependent DSC measurements show that the melting point of form I is slightly higher than that of form II and that the higher melting polymorph exhibits the lower heat of fusion. Therefore, form I becomes thermodynamically stable at higher temperatures and both forms are related by enantiotropism. This is also in agreement with the density rule, because the low-temperature form II exhibits the higher density. Isothermal annealing of both modifications at different temperatures reveals a thermodynamic transition temperature of about 135 °C, which is in excellent agreement with that of 137.5 °C calculated from the melting temperatures and the heat of fusion of both forms. The high-temperature form can easily be prepared pure by solidification of the melt, which is in agreement with Ostwald's step rule, because form I crystallizes at a temperature where it is thermodynamically metastable. A qualitative energy/temperature diagram is presented.
Introduction
Polymorphism, which is defined as the ability of a compound to exist in more than one crystalline modification, continues to be an important topic in solid state chemistry.1–3 Studying the crystal structures of polymorphic modifications can provide information on intermolecular interactions between molecules and on the influence of the crystal environment on their conformation.4–6 Because the chemical composition of the different forms is identical, differences in their stability and their physical properties can be attributed to the different packing patterns, thus allowing investigations of structure–property relationships.7 Investigations of the thermodynamic stability of the various forms and their transformations are of great importance, and to this end selective preparations of specific forms must generally be discovered.8–12 In some cases these may be difficult tasks, and much effort, involving a variety of physico-chemical investigations, may be needed to establish all the thermodynamic relationships and to construct the energy-temperature diagram.13–16 The energy difference between polymorphic modifications is usually small, but may be large enough to allow for different conformations and/or to redistribute weak intermolecular interactions, thus generating a different packing.1,4 This might well be the case for solid state structures of polybrominated aromatic systems, in which we are interested particularly with respect to their intermolecular bromine-bromine interactions.17 This interest led us recently to investigate the solid-state structure of 1,4-dibromo-2,5-bis(bromomethyl)benzene, for which intermolecular bromine–bromine interactions were to be expected (Scheme 1).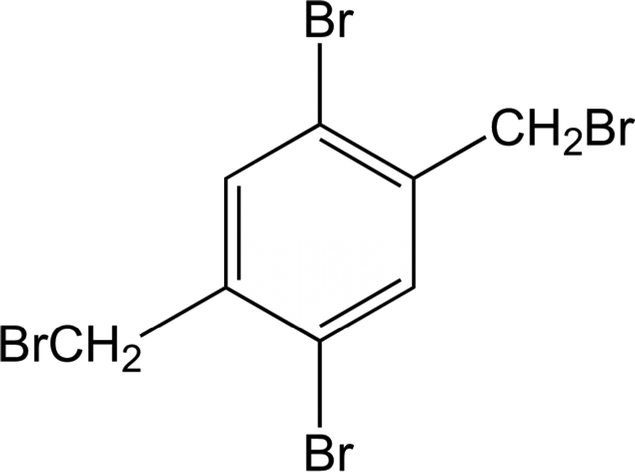 | ||
| Scheme 1 Structural formula of the title compound. | ||
Optical inspection of the sample revealed the presence of two different crystal types: both possessed a rather platy habit (sometimes elongated to form laths), but some (form I) were completely transparent and mechanically robust, whereas others (form II) were markedly crazed or opaque with a high tendency to bending or curling (Fig. 1). We subjected both forms first to single-crystal structure determination, confirming the presence of two polymorphs, and then to a variety of other physical investigative methods.
 | ||
| Fig. 1 Crystals of form I (left) and form II (right). | ||
Experimental
Preparation of 1,4-dibromo-2,5-bis(bromomethyl)benzene
The title compound was obtained from commercially available 2,5-dibromo-p-xylene (Aldrich) by radical bromination with N-bromosuccinimide in carbon tetrachloride (yield 50–80%). NMR data agree with those reported in the literature.18 Single crystals used in crystallographic studies were obtained by slow evaporation of chloroform solutions of the title compound.X-ray crystallography
Details of intensity measurements and refinements are given in Table 1. Crystals were mounted in inert oil on glass fibres. Intensity data were recorded with an Oxford Diffraction Xcalibur E diffractometer using monochromated Mo Kα radiation;19 multi-scan absorption corrections were performed. The structures were solved with direct methods using SHELXS-97, and structure refinement was performed with full-matrix least-squares on F2 using SHELXL-97.20 Hydrogen atoms were included using a riding model. Molecular graphics were prepared with XP.21| Compound | Polymorph I | Polymorph II |
| Chemical formula | C8H6Br4 | C8H6Br4 |
| M r | 421.77 | 421.77 |
| Crystal system, space group | Triclinic, P |
Monoclinic, P21/c |
| Temperature (K) | 100 | 100 |
| a (Å) | 4.3542(3) | 6.6114(5) |
| b (Å) | 7.3675(6) | 8.7776(6) |
| c (Å) | 8.7140(6) | 9.1807(6) |
| α (°) | 72.481(7) | 90 |
| β (°) | 83.722(6) | 104.041(7) |
| γ (°) | 84.917(6) | 90 |
| V (Å3) | 264.52 | 516.86 |
| d calc (g cm−3) | 2.648 | 2.710 |
| Z | 1 | 2 |
| Radiation, wavelength | Mo Kα, λ = 0.71073 Å | Mo Kα, λ = 0.71073 Å |
| F(000) | 194 | 388 |
| μ (mm−1) | 15.2 | 15.5 |
| Crystal size (mm) | 0.35 × 0.10 × 0.03 | 0.10 × 0.08 × 0.04 |
| Transmissions | 0.14–1.00 | 0.48–1.00 |
| 2θmax (°) | 61.6 | 58.8 |
| No. of measured and independent reflections | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 988, 1543 988, 1543 |
10821, 1328 |
| Completeness | 97% to 2θ 60° | 99% to 2θ 56° |
| R int | 0.040 | 0.067 |
| wR(F2) all refl., R1 [F > 4σ(F)], S(F2) | 0.051, 0.022, 1.07 | 0.042, 0.025, 0.87 |
| No. of parameters | 55 | 55 |
| Δρmax,min (e Å−3) | 0.56, −0.66 | 0.77, −0.68 |
The crystals of form II were non-merohedrally twinned, but the twin components were not related by simple 180° rotations about direct or reciprocal axes; it seems that this crystal form has a high tendency to form satellite crystals. Despite the availability of powerful routines (such as the “HKLF 5” method as implemented in SHELXL) for handling twinned data, the best results were obtained by simply choosing the reflections from the major component and omitting all overlapped reflections.
X-ray powder diffraction
X-ray powder diffraction (XRPD) experiments were performed with Cu Kα radiation (λ = 1.5406 Å) using a Transmission Powder Diffraction System from Stoe & Cie, equipped with a position-sensitive detector (Mythen K1) from Stoe & Cie. Temperature-dependent XRPD measurements were performed with the same instrument, equipped additionally with a high-temperature furnace and connected to an Imaging Plate detector.Because the crystal structures were determined at low temperatures but the experimental XRPD patterns were recorded at room temperature, serious differences were observed between the reflection positions of the calculated and the measured XRPD patterns. Therefore, the crystal structures were redetermined at room temperature and these data were used to calculate the powder pattern. This led to a reasonable agreement between the experimental and the calculated patterns, which is an important prerequisite because several strong reflections used for the identification of both forms are accidentally overlapped.
Differential scanning calorimetry
The DSC experiments were performed using a DSC 1 Star System with STARe Excellence Software from Mettler-Toledo AG. All measurements were performed in aluminium crucibles under a continuous flow of nitrogen, using heating rates between 0.1 and 100 °C min−1. The instrument was calibrated using standard reference materials.Thermomicroscopy
Thermomicroscopic measurements were performed using a hot stage FP82 from Mettler and a BX60 microscope from Olympus, using the Analysis software package from Mettler.Results and discussion
Crystal structures
Polymorph I crystallizes in the triclinic space group P; the asymmetric unit consists of half a molecule, which is extended to form a complete molecule via inversion symmetry (Fig. 2). Molecular dimensions may be regarded as normal; the main degree of freedom is the torsion angle C2–C1–C4–Br2 −88.1(2)°, describing the orientation of the bromomethyl group as essentially perpendicular to the ring system.
 | ||
| Fig. 2 The molecule of polymorph I in the crystal. Ellipsoids correspond to 50% probability levels. | ||
The packing of polymorph I (Fig. 3) involves two contacts Br1⋯Br2 (Table 2), which connect the molecules to form layers parallel to (101). The C–Br⋯Br angles, one of ca. 90° and one of ca. 180° at each bromine, identify the contacts as “type 2”,22–24 thought to represent the favourable interactions between a region of positive charge in the extension of one C–Br vector with the negative charge surrounding the other bromine cylindrically; this is now regarded as a special case of a “halogen bond”.25
 | ||
| Fig. 3 Packing diagram of polymorph I with view direction perpendicular to (101). Bromine⋯bromine contacts are drawn as thick dashed lines. Hydrogen atoms are omitted for clarity. Numbering corresponds to the asymmetric unit. | ||
| C–Br⋯Br–C system | d(Br⋯Br) | ∠(C–Br⋯Br) | ∠(Br⋯Br–C) | Operator for second Br–C unit | |
|---|---|---|---|---|---|
| a We note that there is no clear cutoff distance for such contacts; the intramolecular Br1⋯Br2 contact is 3.9698(5) Å, while the shortest contact to the next layer is Br1⋯Br2 (1 − x, 1 − y, −z) 4.0535(5) Å. | |||||
| 1 | C2–Br1⋯Br2–C4 | 3.7610(4) | 84.45(7) | 158.25(7) | x, 1 + y, z |
| 2 | C2–Br1⋯Br2–C4 | 3.5663(5) | 164.25(7) | 91.87(7) | 2 – x, 1 – y, −z |
A notable feature of the packing is the formation of Br4 squares (angles at Br1 93.02(1) and at Br2 87.98(1)°). We have suggested17 modifying the familiar graph sets for hydrogen bonds26 for use with halogen contacts, such that a ring with n atoms, m of which are halogens, would be termed R(n,m). The squares are then R(4,4) and the rings adjacent to these are R(10,4) and R(12,4).
Two closely related halogen derivatives, both with Br1 replaced by iodine and one also with Br2 replaced by chlorine, have been structurally characterized and their shortest contacts reported; both are isotypic to I.27
Polymorph II crystallizes in the monoclinic space group P21/c; again the asymmetric unit consists of half a molecule, and the complete molecule displays inversion symmetry. The torsion angle C2–C1–C4–Br2 is −81.0(4)°, so that the molecules of both polymorphs are closely similar.
The packing of polymorph II is shown in Fig. 4; it consists of layers parallel to the ab plane, again involving bromine⋯bromine contacts (Table 3). The layers of I and II clearly have some features in common; the vertical (in the paper plane) columns of molecules are connected by the R(12,4) rings, and the square R(4,4) rings (angles at Br1 94.39(1) and at Br2 85.61(1)°) connect two rows of horizontally adjacent molecules. However, differences in the relative position and orientation of molecules of the two polymorphs lead to the formation of an extra, somewhat longer, contact Br2⋯Br2, formally arising across the long diagonal of the R(10,4) ring of I and splitting this into two rings R(6,3) that share two central bromine atoms.
 | ||
| Fig. 4 Packing diagram of polymorph II with view direction perpendicular to (001). Bromine⋯bromine contacts are drawn as thick dashed lines, except for the longer Br2⋯Br2 contacts, which are shown as thin dashed lines. Hydrogen atoms are omitted for clarity. Numbering corresponds to the asymmetric unit. | ||
| C–Br⋯Br–C system | d(Br⋯Br) | ∠(C–Br⋯Br) | ∠(Br⋯Br–C) | Operator for second Br–C unit | |
|---|---|---|---|---|---|
| a The intramolecular Br1⋯Br2 contact is 3.8215(6) Å; there are no further Br⋯Br contacts <4.47 Å. b To next layer. c Equal by symmetry. | |||||
| 1 | C2–Br1⋯Br2–C4 | 3.5230(5) | 161.48(10) | 92.04(10) | −1 + x, y, z |
| 2 | C2–Br1⋯Br2–C4 | 3.6617(6) | 102.28(10) | 171.60(10) | 1 − x, 1 − y, 1 − z |
| 3 | C2–Br1⋯Br2–C4b | 3.9155(6) | 65.89(10) | 113.71(10) | 1 − x, −½ + y, 1½ − z |
| 4 | C4–Br2⋯Br2–C4 | 3.8202(8) | 113.92(10)c | 113.92(10)c | 1 − x, 1 − y, 1 − z |
Thermodynamic relationships and transformations between the two polymorphs
Two batches of 1,4-dibromo-2,5-bis(bromomethyl)benzene were synthesized and investigated by XRPD. Comparison of the experimental powder pattern with those calculated for both forms proved that batch 1 consisted of a mixture of form I and II with form I as the major phase, whereas batch 2 seemed to consist of form I exclusively (Fig. S1†). This is also clear from microscopic inspection of batch 1, because crystals of both forms can easily be distinguished (Fig. 1 and S2†).Investigation of batch 1 by differential scanning calorimetry (DSC) at 10 °C min−1 showed one very low endothermic peak at a peak temperature (Tp) of 149.5 °C followed by an intense endothermic peak at Tp = 160.8 °C (Fig. 5). The peak at higher temperatures might correspond to the melting point of one of these forms, whereas the small and broad endothermic event at lower temperatures might correspond to an endothermic polymorphic transition or to melting of one of these forms. If the melt is cooled down, supercooling is observed and, if consecutive heating and cooling cycles are measured, the small endothermic peak is absent in the second run and the material melts at the same temperature as observed in the first run (Fig. S3†).
 | ||
| Fig. 5 DSC curve of batch 1 (top) and batch 2 (bottom) at 10 °C min−1. Peak temperatures (Tp) are given in °C. | ||
In contrast, for batch 2, which according to XRPD consists only of pure form I, one endothermic event occurs at Tp = 158.2, which might correspond to the melting point of this form. However, a very broad signal is observed, which implies a more complicated process (Fig. 5).
To investigate the origin of the small endothermic peak observed for batch 1, a second DSC measurement was performed and stopped after this event at 155 °C. Investigation of the residue by XRPD showed that all reflections of form II had disappeared and modification I was obtained as a pure phase (Fig. S4†). This proves that form II has been transformed into I, either by a solid-to-solid polymorphic transition, which might be feasible based on the similarity of the packing motifs (see above) or via melting of II followed by solidification of the melt and crystallization of I on further heating, as observed e.g. for trimethylthiourea.13 Based on these experiments, it can be concluded that the strong endothermic peak observed in the DSC measurement at Tp = 160.8 °C of batch 1 must correspond to the melting of form I. It is therefore surprising that this value is higher than Tp = 158.2 °C as determined from batch 2, which consists of form I exclusively.
In an attempt to resolve this problem, heating-rate-dependent measurements on batch 2 were performed, but very broad peaks were always observed and there was no indication that additional transformations are involved that might be resolved at different heating rates (Fig. S5 and Table S1†). Moreover, if consecutive heating and cooling cycles were measured for this batch at 10 °C min−1, very broad signals and lower melting points were always observed (Fig. S6†). To rule out any further transformation before melting, a further measurement was performed and stopped at 120 °C, but the experimental XRPD pattern was identical to that of the pristine material (Fig. S7†). This is consistent with temperature-dependent XRPD measurements, which showed no changes up to the melting point (Fig. S8†).
However, when the solidified melt of batch 1 was investigated by XRPD, it was clear that form I has been obtained exclusively (Fig. S9†). Moreover, if consecutive heating and cooling cycles were measured for this batch at 10 °C min−1, very narrow melting peaks were always observed, and always at higher melting points compared to those measured for batch 2 (Fig. S3†); there is also no influence of the actual heating rate (Fig. S10†). Consequently, it must be assumed that batch 2 is contaminated either by an amorphous material or by a very small amount of a crystalline material undetectable by XRPD. This is reasonable, because in some other batches we detected some monobrominated 2,5-dibromo-p-xylene (4-bromomethyl-2,5-dibromotoluene), which has a lower melting point, as a by-product using TLC and NMR spectroscopy. Only a very small amount of contamination, difficult to detect by other methods, would be needed to reduce the melting point by a few degrees.
The melting point and melting enthalpy of pure form I, obtained from melting of batch 1, was determined from several DSC runs at various heating rates, and at 5 °C min−1 average values for the peak temperature Tp of 159.1 °C, for the onset temperature To of 156.7 °C and for the heat of fusion of ΔHfus = 27.0 kJ mol−1 were obtained (Table S2†).
Because we had no access to pure form II, we tried to prepare this modification by kinetic control. Therefore, the compound was dissolved in dichloromethane, which because of its low boiling point can be vaporized very rapidly. We obtained a batch that consisted of a mixture of both forms, and in contrast to all other batches contained form II as the major phase, but when the experiment was repeated, form I was obtained exclusively (Fig. S11†).
To determine the relative thermodynamic stability at room temperature, a saturated solution with an excess of form I (obtained by melting the material) and of form II (prepared by fast crystallization from dichloromethane) was stirred for one week in ethanol. Investigation of the residues thus obtained by XRPD showed that form I had been completely transformed into form II, which proves that form II represents the thermodynamically stable modification at room temperature (Fig. 6). The same result is obtained if such an experiment is performed in other solvents (Fig. S12†). We conclude that form I was accidentally obtained from the synthesis by kinetic control.
 | ||
| Fig. 6 Experimental XRPD pattern of a mixture of form I and II (B) and of the residue after stirring this mixture in ethanol for one week (C) together with the calculated pattern for form I (A) and form II (D). | ||
For the following thermomicroscopic investigations, crystals of both forms were selected by hand, their identity was checked by single crystal X-ray diffraction and fragments of these crystals were investigated. On heating form II at 1 °C min−1 (Fig. 7, top set of 3 × 3 images) the crystal became brighter between about 146 and 162 °C, whereas no changes were observed for the crystal of form I. The change in appearance may be attributed to the transformation from form II to form I. On further heating, both crystals melted at the same temperature (Fig. 7, top set, last two images), which is to be expected if the crystal of form II transforms into form I before melting. From this experiment it is difficult to decide if the transformation is a solid-to-solid transition that proceeds via nucleation and growth of a new phase, or if the transformation takes place by melting of form II, crystallization of form I and melting of this form at higher temperatures. The latter would be in accordance with the observation that the crystals become brighter during the transition and, because both melting points are very similar, it is not necessary for the whole crystal of II to melt before crystallization of I takes place; the transformation from II to I can take place gradually throughout the entire crystal without changing its habit.
 | ||
| Fig. 7 Microscopic images, obtained by thermomicroscopy at heating rates of 1 °C min−1 (top set of 3 × 3 images) and 10 °C min−1 (bottom set of 3 × 3 images), of selected crystals of form I (top right and bottom left) and form II (top left and bottom right). The temperature is given in °C N.B. The apparently higher melting temperature observed at 10 °C min−1 can be attributed to the higher heating rate. The differences in the melting temperatures between the thermomicroscopic and DSC measurements originate from an imprecise calibration of the hot stage, which is not directly relevant for the outcome of this experiment; it should be remembered that the DSC temperatures are the reliable values. | ||
However, at the faster heating rate of 10 °C min−1, in one experiment (Fig. 7, bottom set of 3 × 3 images) crystals of form II did not become noticeably clearer, but instead were observed to melt before the melting of form I, which clearly shows that the melting point of form II is lower than that of form I. This is expected, because form I exhibits the higher melting point and therefore should be thermodynamically stable at higher temperatures. At lower temperatures such as room temperature, however, the solvent-mediated conversion experiments have proved that form II is thermodynamically stable, and thus both forms should be related by enantiotropism, with form II as the lower melting polymorph. In most such cases the low-temperature phase transforms into the high-temperature phase before melting, so that one cannot measure the melting point of the former, but in a few cases, e.g. at higher heating rates, this transformation can be suppressed, which was obviously the case for the thermomicroscopic measurement at the higher heating rate.
Interestingly, a similar observation was made in our temperature-dependent XRPD measurements (Fig. S13 and S14†). If a mixture of both modifications is investigated, the transformation of form II into I occurs at about 140 °C, but if pure form II is measured, the sample melts without any previous transformation. It seems that the presence of crystals of form I in the mixture induces transformation to this modification.
To investigate the thermal behavior in more detail, heating-rate-dependent DSC measurements were performed for form II (Fig. S15 and Table S3†). Heating rates up to 100 °C min−1 were used, because in some cases a transition can be suppressed on fast heating, leading to more precise values for the transition enthalpies. However, for all heating rates clearly two transitions are visible, and these can only successfully be resolved at 1 and 5 °C min−1. Interestingly, the intensity of the first peak increases with increasing heating rates and becomes comparable with that of the second peak, which is not consistent with a solid-to-solid polymorphic phase transition, where the heat of transformation does not change with the heating rate. It is more likely that the polymorphic transition is more effectively suppressed with increasing heating rates and that in this case melting of form II is observed. However, in these experiments even at very low heating rates melting of form II might be observed, which is indicated from the measurement at 1 °C min−1, where directly after the first maximum a very small exothermic peak is observed, which implies crystallization of modification I formed by melting of form II during the first thermal event. This is in agreement with a measurement at 0.1 °C min−1, where several very small consecutive endothermic and exothermic peaks were observed, indicating a stepwise melting and crystallization of this compound (Fig. S16†). Finally, when DSC curves were measured for a very pure sample of form II prepared by stirring the material in ethanol, in one measurement at 3 °C min−1 two well-resolved thermal events were observed, of which the first was more intense than the second, showing that two melting points are involved (Fig. 8, top). In a second such measurement at 1 °C min−1, only one peak was observed, which corresponds to the melting of form II (Fig. 8, bottom). Obviously the purity of each form and the kinetics of the transition has a significant impact on the thermal behavior.
 | ||
| Fig. 8 DSC curve of pure form II measured at 3 (top) and 1 °C min−1 (bottom). The peak temperatures (Tp) are given in °C. | ||
All experiments discussed above prove that form II is stable at lower temperatures and that the melting point of form I is higher than that of form II. Therefore, both forms must behave enantiotropically and in this case the higher melting polymorph should exhibit the lower heat of fusion.28 For form I the heat of fusion was determined at 5 °C min−1 to be 27.0 kJ mol−1 (Table S2†) but precise values for form II are lacking, because in nearly all measurements the two thermal events cannot be resolved successfully. However, if the sum of both peaks is calculated for each measurement shown in Fig. S15,† an average value of 33.1 kJ mol−1 is obtained, which agrees nicely with that obtained from the measurement shown in Fig. 8 of 32.1 kJ mol−1. These values are higher than those for form I and thus both modifications are indeed related by enantiotropism.29
To determine the thermodynamic transition temperature, mixtures of both forms were annealed at different temperatures until one form had been converted into the other. If such an experiment is performed at 130 °C, form I was converted to II within 4 days, but if the same experiment was performed at 140 °C, all crystals of form II were transformed into form I (Fig. 9). When this experiment was repeated at 135 °C, no complete transformation was observed even after one week. Therefore, the thermodynamic transition temperature must be close to this temperature.
 | ||
| Fig. 9 Experimental XRPD pattern of a mixture of form I and II (C) and of the residue obtained after annealing this mixture at 130 °C (B) and 140 °C (D), together with the calculated pattern for form II (A) and form I (E). | ||
From all these experiments a qualitative energy/temperature diagram can be drawn that shows the thermodynamic relationships between both modifications (Fig. 10). From very low temperatures up to 135 °C, form II is thermodynamically stable, which is also in agreement with its higher density compared to that of form I.29 At about 135 °C the free energy temperature curves cross, and above this temperature form I represents the thermodynamically stable form, whereas form II is metastable. In this diagram the curve for the relative free energy of the melt is also shown, from which it is clear that form I exhibits the higher and form II the lower melting point and that in the case of enantiotropism the heat of fusion of the higher melting polymorph I must be lower.
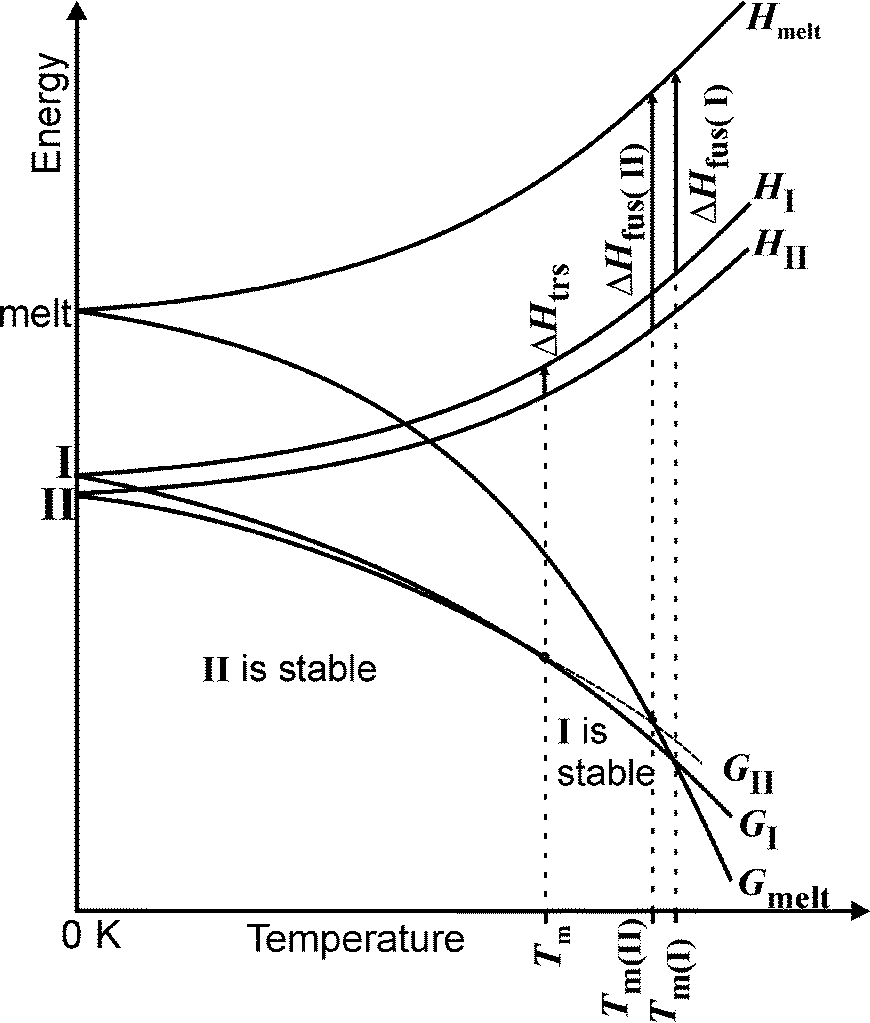 | ||
| Fig. 10 Qualitative energy temperature diagram for the enantiotropically related modifications I and II (G = free energy; H = enthalpy; Ttrs = transition temperature; Tm = melting point; ΔHfus = enthalpy of fusion). | ||
Finally, in those cases where the melting point and the heat of fusion for two modifications are known, the transition temperature can be estimated using the following equation, in which differences in the heat capacity are neglected:30

At 1 °C min−1 the melting points of form I were determined to be To = 156.9 and Tp = 158.5 °C and for the heat of fusion ΔHfus a value of 28.0 kJ mol−1 was obtained. As pointed out above, for one batch of form II melting of only this form was observed (Fig. 8). From three such DSC runs values for To of 154.7 °C, for Tp of 155.7 °C and for ΔHfus of 32.1 kJ mol−1 were obtained, with the latter value in nice agreement with that of 33.1 kJ mol−1 determined from the heating-rate-dependent measurements. Because the onset temperature cannot be determined very precisely, but data were measured for the same heating rate, the peak temperatures might preferably be used for the calculation. Using these values the thermodynamic transition temperature Ttrs of 137.5 °C is calculated, which is in excellent agreement with that determined by experiment. However, it should be borne in mind that the values for form II are not very precise and that even small differences in the experimental values will affect this temperature appreciably.
Conclusions
Investigations of the thermodynamic relationships between both forms of 1,4-dibromo-2,5-bis(bromomethyl)benzene have shown that form II (monoclinic) is thermodynamically stable at lower temperatures, where form I (triclinic) is metastable. Because the density of form II is higher than that of form I it should also be stable at 0 K.13 Above about 135 °C form I becomes thermodynamically stable and therefore both polymorphs are enantiotropically related. In several experiments, melting of form II is first observed, followed by melting of form I; however, at very low heating rates there are strong indications that form I crystallizes from the melt of form II, because at this temperature form I is thermodynamically stable and its melting point has not been reached. If mixtures are used, both thermal events are visible by DSC, but if very pure form II is investigated, melting of only this form can be observed exclusively. In the former case, crystals of form I might induce crystallization of this form after melting of form II. From our thermomicroscopic and DSC investigations it is difficult to decide if the transition of form II into I is a solid-to-solid polymorphic transition or if it proceeds via melting of the low temperature form with subsequent crystallization of the high temperature form. Obviously both events can be observed, and any particular observation will depend on the purity of each form and the kinetics of these reactions. The fact that form I is always formed on solidification of the melt is in full accordance with Ostwald's rule;31 because of supercooling this compound always crystallizes below the thermodynamic transition temperature, where it is metastable.It is to be noted that initial experiments provided contradictory results, because one batch was contaminated with a very small amount of a further compound that was difficult to detect and that led to a lower melting point. This clearly shows that for the investigations of polymorphism, and for the determination of the thermodynamic relationships between different forms, great experimental effort using different analytical techniques must be made and very pure samples are needed, especially if the melting points of two different modifications are similar.
Acknowledgements
We are grateful to Prof. Dr. W. Bensch for access to his powder diffractometer.References
- A. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170–2191 CrossRef CAS PubMed
.
- A. Y. Lee, D. Erdemir and A. S. Myerson, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 259–280 CrossRef CAS PubMed
- S. Aitipamula, P. Shan Chowa and R. Tan, CrystEngComm, 2014, 16, 3451–3465 RSC
-
J. Bernstein, Conformational Polymorphism, in Organic Solid State Chemistry, ed. G. R. Desiraju, Elsevier, Amsterdam, 1987, p. 471 Search PubMed
-
Crystal Engineering, Mat. Sci. Monogr., ed. G. R. Desiraju, Elsevier, Amsterdam, 1989 Search PubMed
- C. Näther, I. Jess, Z. Havlas, N. Nagel, M. Bolte and S. Nick, Solid State Sci., 2002, 4, 859–871 CrossRef
- J. Bernstein, J. Phys. D: Appl. Phys., 1993, 26, B66–B76 CrossRef CAS
- J. Bernstein, R. Davey and O. Henck, Angew. Chem., Int. Ed., 1999, 38, 3440–3461 CrossRef
-
R. Hilfiker, Polymorphism in the Pharmaceutical Industry, Wiley-VCH, 2006 Search PubMed
-
H. G. Brittain, Polymorphism in Pharmaceutical Solids, Marcel Dekker Inc., New York, 1999, vol. 23 Search PubMed
- J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193–201 CrossRef CAS
- C. Döring, C. Näther, I. Jess, K. Ibrom and P. G. Jones, CrystEngComm, 2015, 17, 5206–5215 RSC
- C. Näther, I. Jess, P. G. Jones, C. Taouss and N. Teschmit, Cryst. Growth Des., 2013, 13, 1676–1684 Search PubMed
- H. Schödel, C. Näther, H. Bock and F. Butenschön, Acta Crystallogr., Sect. B: Struct. Sci., 1996, 52, 842–853 CrossRef
- C. Näther, I. Jess, L. Seyferth, K. Bärwinkel and J. Senker, Cryst. Growth Des., 2015, 15, 366–373 Search PubMed
- A. Carletta, C. Meinguet, J. Wouters and A. Tilborg, Cryst. Growth Des., 2015, 15, 2461–2473 CAS
- P. G. Jones, P. Kuś and I. Dix, Z. Naturforsch., 2012, 67b, 1273–1281 Search PubMed
- M. C. Bonifacio, C. R. Robertson, J.-Y. Jung and B. T. King, J. Organomet. Chem., 2005, 70, 8522–8526 CAS
-
Agilent, CrysAlis PRO, Agilent Ltd., Yarnton, England, 2014 Search PubMed
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
-
Siemens XP, Version 5.03. Siemens Analytical X–Ray Instruments, Madison, Wisconsin, U.S.A, 1994 Search PubMed
- V. R. Pedireddi, D. S. Reddy, B. S. Goud, D. C. Craig, A. D. Rae and G. R. Desiraju, J. Chem. Soc., Perkin Trans. 2, 1994, 2353–2360 RSC
- G. R. Desiraju and R. Parthasarathy, J. Am. Chem. Soc., 1989, 111, 8725–8726 CrossRef CAS
- The first report of distinctive geometric types of chlorine⋯chlorine contacts was published much earlier, but it did not use the type 1/type 2 nomenclature: T. Sakurai, M. Sundaralingam and G. A. Jeffrey, Acta Crystallogr., 1963, 16, 354–363 CrossRef CAS
- For a general review of halogen bonding, see e.g. P. Metrangelo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef PubMed
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS
- G. Gaefke, V. Enkelmann and S. Höger, Synthesis, 2006, 2971–2973 CAS
- A. Burger and R. Ramberger, Microchim. Acta, 1979, 2, 259–271 CrossRef CAS
-
A. I. Kitaigorodski, Organic Chemical Crystallography, Consultants Bureau, New York, 1961 Search PubMed
-
R. Hilfiker, Physico-Chemical Methods in Drug Discovery and Development, ed. Z. Mandić, IAPC Publishing, Zagreb, 2012, pp. 349–383 Search PubMed
- W. Ostwald, Z. Phys. Chem., 1887, 22, 289–330 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1453756 (polymorph I), 1453757 (polymorph II), contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce00438e |
| This journal is © The Royal Society of Chemistry 2016 |