
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Short contacts of the sulphur atoms of a 1,2,3,5-dithiadiazolyl dimer with triphenylstibine: first co-crystal with an aromatic compound†
René T.
Boeré
Department of Chemistry and Biochemistry, and the Canadian Centre for Research in Advanced Fluorine Technologies, University of Lethbridge, Lethbridge, Alberta T1K 3M4, Canada. E-mail: boere@uleth.ca; Fax: +403 329 2057; Tel: +403 329 2045
First published on 15th March 2016
Abstract
The structure of dimeric 2,7-bis[4-(trifluoromethyl)phenyl]-4λ4,5λ4,9λ4,10λ4-tetrathieto[1,2-a:3,4-a′]bis [1,2,3,5]dithiadiazole (C8H4F3N2S2)2 and its adduct with triphenylstibine, (C8H4F3N2S2)2·C18H15Sb, both have triclinic (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) symmetry. They crystallize in layers containing centrosymmetric clusters consisting of four dithiadiazolyl dimers in the parent compound and two such dimers paired with two triphenylstibine units in the aromatic co-crystal. In the co-crystal, the Ph3Sb molecules associate with an equivalent moiety from a neighbouring cluster in a geometry that is very reminiscent of other Ph3Sb-containing structures. Thus, the adduct combines structural elements from those of its component parts. Key interactions between molecules in the pure dithiadiazolyl (S to S) and the co-crystal (S to C) are significantly shorter than the sums of atom van der Waals radii.
) symmetry. They crystallize in layers containing centrosymmetric clusters consisting of four dithiadiazolyl dimers in the parent compound and two such dimers paired with two triphenylstibine units in the aromatic co-crystal. In the co-crystal, the Ph3Sb molecules associate with an equivalent moiety from a neighbouring cluster in a geometry that is very reminiscent of other Ph3Sb-containing structures. Thus, the adduct combines structural elements from those of its component parts. Key interactions between molecules in the pure dithiadiazolyl (S to S) and the co-crystal (S to C) are significantly shorter than the sums of atom van der Waals radii.
Introduction
There is an extensive chemistry of 1,2,3,5-dithiadiazolyl (DTDA) radicals because of interest in metallic conductivity and magnetism.1 Such properties depend on intermolecular contacts and thus the crystal engineering of DTDA radicals has received intensive investigation.2 DTDA radicals normally dimerize in the solid state unless there are both steric factors to prevent dimerization and secondary bonding interactions to stabilize the monomers. There are at least five recognizable dimer configurations; of these the cis-oid co-facial is by far the most common. Extensive experimental and theoretical considerations have concluded that the inter-dimer bonds are exclusively between the CN2S2 heterocycles and are dominated by S⋯S interactions,3 a strong interaction that has been effectively described as ‘pancake bonding’ which constitutes a (diffuse) quantum-chemical bond but also involves a dispersive component and contributions from diradical character.4–6 Crystal structures have been reported for more than 70 different neutral DTDA dimers and monomers in the Cambridge Structure Database (CSD, version 5.37, with updates to November 2015).7 In only a handful of cases are the structures heterogeneous. A mixed oxidation state species crystallizes as the trimer [5-PhCN2S2]3I3 (CSD refcode: HEGVOE).8 Similarly, a channel structure of HCN2S2 crystallizes with ∼0.18 iodine atoms in a partial charge-transfer species (refcode LEJFAH).9 A co-crystal of PhCN2S2 and S3N3 involves an indeterminate degree of charge transfer (refcode SIHZAK).10 The structure of 4-(3-fluoro-4-trifluoromethylphenyl)-1,2,3,5-dithiadiazolyl (refcode: UMAROP) is typical of a (distorted) cis-oid dimer, but is significant in the context of this work in that the lattice readily opens up to form channels when co-sublimed with N2, Ar, CO2 or SO2 (refcodes: UMARUV, UMASAC, UMASEG and UMASIK) to form host-guest gas-clathrates.11 This is the only other case to our knowledge where co-crystallization with neutral molecules has previously been demonstrated in DTDA chemistry, although identification of electron density for the incorporated gas molecules relied on the delocalized solvent tools of the PLATON ‘SQUEEZE’ routine.12In two recent reports, Haynes et al. and Rawson et al. reported on the preparation of fascinating mixed-radical dimers by combining slightly electron rich with electron poorer DTDAs.13,14 The successful co-crystallizations include [PhCN2S2][5-C6F5–CN2S2] (refcode: QUNQUM)13 and [PhCN2S2][NC5F5–CN2S2] (refcode: YIMNIT),14 which emphasizes the importance of perfluorination for reducing electron richness in DTDA heterocycles via purely inductive effects. Complex charge balances exist in mixed fluorinated/hydrocarbon DTDA dimers, which have been intensively investigated by experimental and computational charge density determinations.3 The co-crystallization could be achieved either from solution or by sublimation in a tube furnace. They also reported many failed attempts by mixing other DTDAs, and attempts to combine about 10 different aromatic ring compounds, incorporating a variety of functional groups, with DTDAs; no co-crystals with aromatics were obtained. It is not clear from the published report as to whether the aromatics were thought to be able to co-dimerize with the DTDA or whether some other form of association was expected. Several recent reports indicate a directive or ‘shepherding’ role for aromatic co-crystallizers with organic radicals.15–17 “End-on” interactions from the sulphur atoms of DTDAs with aromatic carbon atoms belonging to the same DTDA species have been known since at least 1991. Thus, in the lattice of [1,4-CN2S2–C6H4]2 (refcode: VINJIL),18 there is an interaction between two sulphur atoms of a DTDA dimer and the ipso and ortho carbon atoms of the di-substituted benzene ring of a neighbouring molecule. It has a shortest C⋯S contact that is 0.22 Å < ∑rvdW. Of much more recent origin are other structures showing similar interactions, as in [3-Cl-4-CH3–C6H3–CN2S2]2 (refcode: EZIQUY, shortest C⋯S contact 0.33 Å < ∑rvdW),19 in [4-F–C6H3–CN2S2]2 (refcode: QEFGIT, shortest C⋯S contact 0.20 Å < ∑rvdW),20 in [3-CH3–C6H3–CN2S2]2 (refcode: LELPUP, shortest C⋯S contact 0.29 Å < ∑rvdW),21 and in [4-CH3–C6H3–CN2S2]2 (refcode: LELPOJ, shortest C⋯S contact 0.24 Å < ∑rvdW).21
The synthesis of the fluorinated DTDA radical 5-(4-CF3C6H4)–CN2S2, 1 (Chart 1), was reported by Boeré et al.22 and the crystal structure was briefly mentioned in the context of metal coordination chemistry of DTDA radicals.23 We now report a detailed analysis of the lattice structure of 1 and the discovery that it can form a unique 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 co-crystal with triphenylstibine, [5-(4-CF3C6H4)–CN2S2]2·Ph3Sb, 2, in which a typical cis-oid co-facial radical dimer moiety – in itself of quite similar structure to that found in pure 1 – undergoes specific supramolecular contacts to a phenyl ring of the stibine. This structure is the first reported co-crystal of a DTDA dimer with an aromatic compound.
:1 co-crystal with triphenylstibine, [5-(4-CF3C6H4)–CN2S2]2·Ph3Sb, 2, in which a typical cis-oid co-facial radical dimer moiety – in itself of quite similar structure to that found in pure 1 – undergoes specific supramolecular contacts to a phenyl ring of the stibine. This structure is the first reported co-crystal of a DTDA dimer with an aromatic compound.
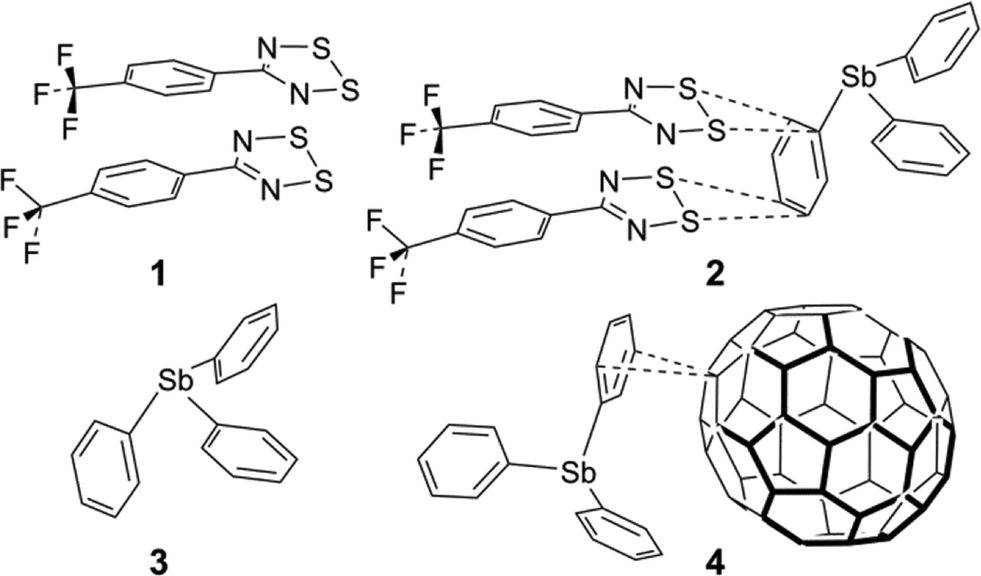 | ||
| Chart 1 | ||
Results and discussion
Sample preparation
The synthesis of 1 employed triphenylstibine, 3, as a convenient reducing agent for 1,2,3,5-dithiadiazolium chlorides and 3 is itself oxidized to Ph3SbCl2 (Scheme 1). Because 1 does not precipitate well even from concentrated CH3CN solutions, the evaporated crude reaction mixture was directly sublimed in a gradient sublimer. The neutral radical 1 is more volatile than Ph3SbCl2 and is also easy to recognize from its colour. In the sublimation, 1 presented as dark purple needles which were used for the structure determination. Since gradient sublimation in vacuum often leads to multiple crystal habits, the presence of dark purple blocks amongst the needles was not of immediate concern. When the structure of the blocks was solved using the iterative method of SHELXT24 it was shown to be a 1:1 co-crystal of 1 with 3. Evidently, some unreacted 3 was able to sublime and the mixed vapours crystallize to afford 2 in a precise ratio determined by specific intermolecular interactions. Whereas crude, powdered DTDA samples are very reactive and can inflame in air, the sublimed crystals of both 1 and 2 are sufficiently stable to handle in air for brief periods (for example, crystal selection and mounting was done on the open bench).
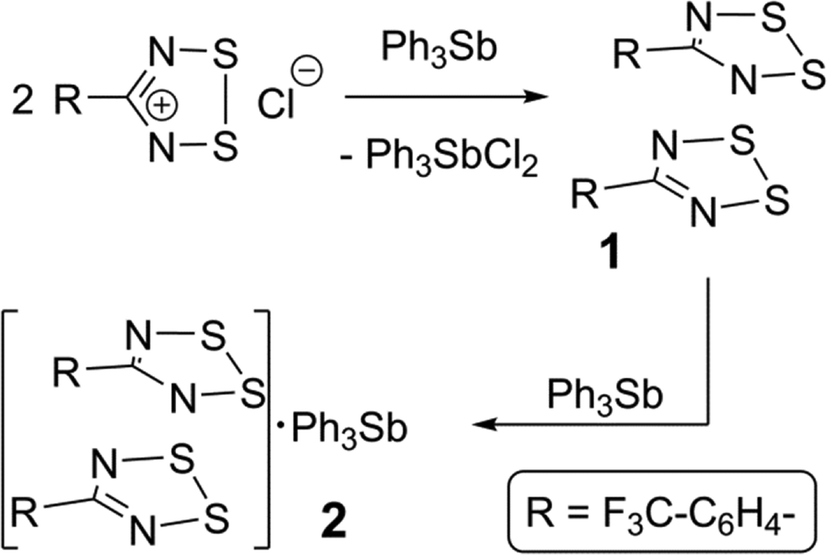 | ||
| Scheme 1 | ||
Structural commentary and supramolecular features
The geometry of the DTDA dimer in 2 consists of the common cis-oid co-facial arrangement of planar CN2S2 rings (Fig. 1 and Table 1).2 Visually, it is indistinguishable from any one of the four independent dimers found in the crystal structure of 1 (for plots of 1, see Fig. S1 in the ESI;† for an overlay structure diagram of dimers from 1 and 2, see Fig. S2). For clarity and ease of discussion, the atom numbering scheme of the single DTDA dimer in Fig. 1 will be used throughout. The average inter-dimer S⋯S distance in 2 is 3.068(1) Å, some 0.53 Å less than the sums of their v.d. Waals' radii (∑rvdW).4 The least-squares planes through the two heterocyles that constitute the dimer in 2 are inclined at 7.87(13)°; in addition the aryl rings twist out of the planes they are attached to and there is an overall miss-alignment of the upper and lower dimer constituents. Consideration of 3D models indicates that all these effects act to minimize unfavourable steric congestion of the para-CF3 groups on adjacent rings. Such a distortion is also evident in all four dimers in the asymmetric unit of 1, which crystallizes in the same space group, P, but with Z = 16 rather than two (see the Experimental section for details). In 1, each dimer has a slightly different manifestation of steric distortions to accommodate the bulky CF3 groups; the average tilt angles for the four dimer pairs is 6.1(8)° from which the value in 2 cannot be differentiated at the 99% confidence level. If for the miss-alignment of the dimer components we take the torsion angle C5–C1–C11–C15, the range for 1 is 2.7(1)–5.6(1)°, within which the value of 3.0(8)° for 2 fits comfortably. The average inter-dimer S⋯S distance for the four dimers in 1 is 3.07(5) Å, or 0.53(5) Å less than ∑rvdW.
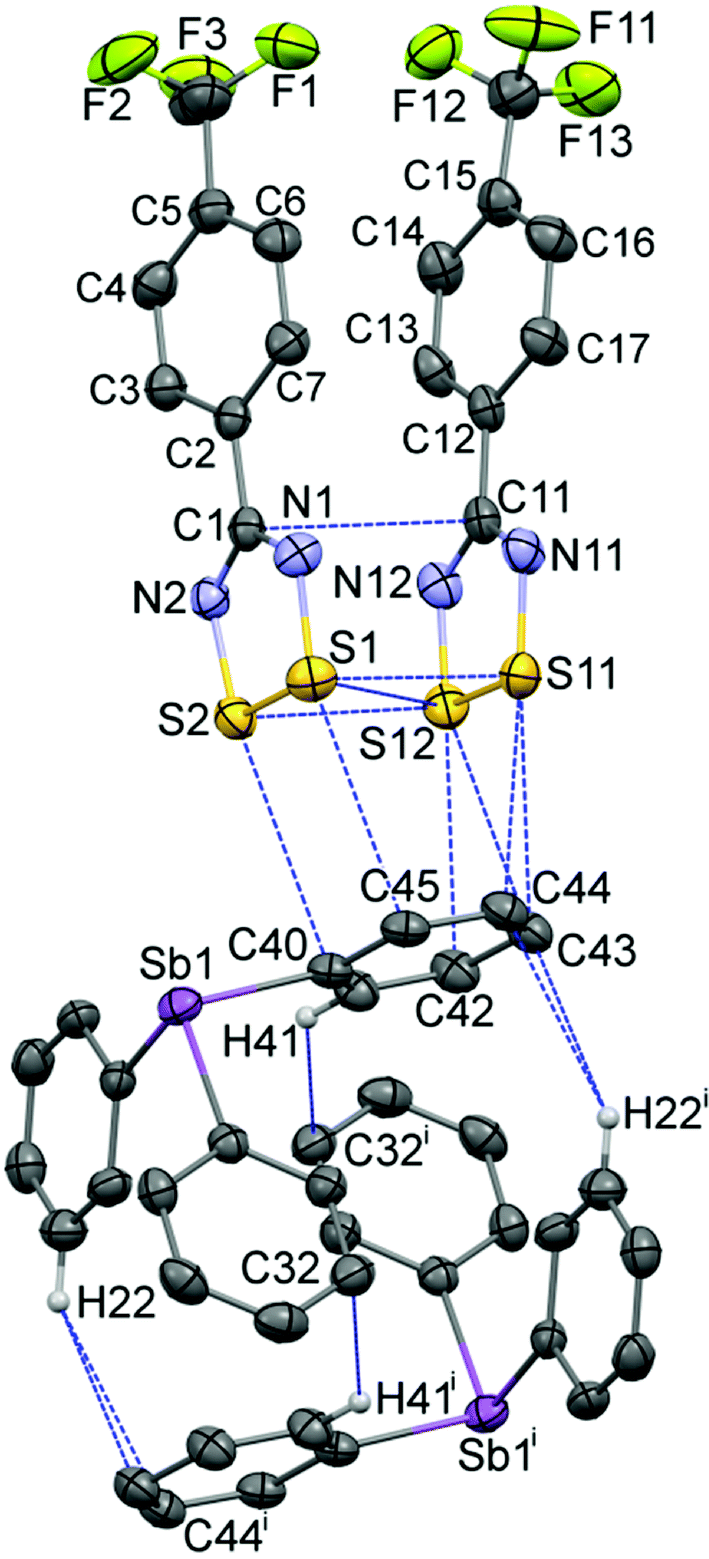 | ||
| Fig. 1 Displacement ellipsoids (30% probability level) of the 263 K structure of 2, depicting the asymmetric unit augmented by the symmetry-equivalent second component of the pseudo-cuboidal Ph3Sb entity, showing the atom numbering scheme used to discuss both 1 and 2. Intermolecular contacts up to (∑rvdW + 0.1) Å are shown by dotted lines [symmetry code: (i) 2 − x, −y, 1 − z]. The CF3 groups are rotationally disordered (see ESI†). | ||
| Parametera | 1-ib | 1-iic | 1-iiid | 1-ive | 2 |
|---|---|---|---|---|---|
| a The atom numbering scheme is that of 2, see Fig. 1. b Dimer i: S1S2; S3S4. c Dimer ii: S5, S6; S7S8. d Dimer iii: S9, S10; S11S12. e Dimer iv: S13, S14; S15S16. f CSD refcode: ZZZEHA01; 2 mol per eq. pos.25 g CSD refcode: ZZZEHA02; 2 mol per eq. pos.26 | |||||
| S1–S2 | 2.0886(18) | 2.0879(18) | 2.0881(19) | 2.0784(19) | 2.0865(9) |
| S1–N1 | 1.622(4) | 1.636(4) | 1.626(4) | 1.628(4) | 1.629(2) |
| S2–N2 | 1.626(4) | 1.624(4) | 1.625(4) | 1.622(4) | 1.629(2) |
| N1–C1 | 1.338(6) | 1.336(6) | 1.328(6) | 1.346(6) | 1.336(3) |
| N2–C1 | 1.342(6) | 1.339(6) | 1.330(6) | 1.337(6) | 1.330(3) |
| C1–C2 | 1.476(6) | 1.469(6) | 1.485(7) | 1.474(6) | 1.479(3) |
| S11–S12 | 2.0840(19) | 2.0798(18) | 2.0753(18) | 2.0841(18) | 2.0972(10) |
| S11–N11 | 1.633(4) | 1.639(4) | 1.628(4) | 1.639(4) | 1.626(2) |
| S12–N12 | 1.634(4) | 1.621(4) | 1.636(4) | 1.621(4) | 1.629(2) |
| N11–C11 | 1.337(6) | 1.339(6) | 1.339(6) | 1.338(6) | 1.339(3) |
| N12–C11 | 1.333(6) | 1.339(6) | 1.339(6) | 1.336(6) | 1.334(3) |
| C11–C12 | 1.478(6) | 1.483(6) | 1.480(6) | 1.485(6) | 1.485(4) |
| N1–S1–S2 | 94.27(16) | 94.67(16) | 94.01(16) | 95.21(16) | 94.40(8) |
| N2–S2–S1 | 94.60(15) | 94.16(15) | 94.63(16) | 94.59(16) | 94.42(8) |
| C1–N1–S1 | 115.0(3) | 114.0(3) | 114.6(3) | 113.1(3) | 114.27(18) |
| C1–N2–S2 | 114.4(3) | 114.9(3) | 114.0(3) | 114.2(3) | 114.39(17) |
| N2–C1–N1 | 121.7(4) | 122.1(4) | 122.8(4) | 122.8(4) | 122.5(2) |
| N2–C1–C2 | 117.1(4) | 119.3(4) | 118.2(4) | 118.9(4) | 118.7(2) |
| N1–C1–C2 | 121.2(4) | 118.5(4) | 119.0(4) | 118.2(4) | 118.7(2) |
| N11–S11–S12 | 94.19(16) | 94.96(15) | 94.25(16) | 94.80(15) | 94.56(8) |
| N12–S12–S11 | 94.91(15) | 94.50(15) | 95.21(15) | 94.28(16) | 94.12(8) |
| C11–N11–S11 | 114.4(3) | 113.3(3) | 114.8(3) | 113.6(3) | 114.21(19) |
| C11–N12–S12e | 113.7(3) | 114.6(3) | 113.5(3) | 115.0(3) | 114.57(19) |
| N12–C11–N11 | 122.8(4) | 122.6(4) | 122.3(4) | 122.3(4) | 122.5(2) |
| N12–C11–C12 | 118.9(4) | 117.9(4) | 120.7(4) | 117.2(4) | 119.1(2) |
| N11–C11–C12 | 118.3(4) | 119.5(4) | 117.0(4) | 120.4(4) | 118.4(2) |
| 3a-if | 3a-iif | 3b-ig | 3b-iig | 2 | |
|---|---|---|---|---|---|
| Sb1–C20 | 2.143(6) | 2.155(6) | 2.146(5) | 2.154(7) | 2.146(5) |
| Sb1–C30 | 2.150(10) | 2.170(10) | 2.143(7) | 2.148(7) | 2.143(7) |
| Sb1–C40 | 2.151(9) | 2.161(9) | 2.139(8) | 2.139(7) | 2.139(8) |
| C20–Sb1–C30 | 98.0(3) | 95.2(3) | 96.5(3) | 96.1(3) | 97.46(9) |
| C20–Sb1–C40 | 95.7(3) | 95.5(3) | 96.5(2) | 97.4(3) | 96.88(9) |
| C30–Sb1–C40 | 96.0(3) | 97.5(3) | 96.0(3) | 95.5(3) | 95.76(9) |
| ∑∠(C–Sb–C) | 289.7(4) | 288.2(4) | 289.0(3) | 289.0(4) | 290.1(11) |
Within the heterocycles, the average S–S bond length of 2.0919(9) Å in 2 (Table 1) can be compared to a mean of 2.085(3) Å for four such bonds in 1; the average S–N bond length of 1.628(1) Å with a mean of 1.629(7) Å in 1; the average N1–C1 bond length of 1.335(3) Å with a mean of 1.338(3) Å in 1 and the average C1–C2 bond length of 1.482(3) Å with a mean of 1.477(5) Å in 1. Each parameter in 2 is therefore comfortably within the statistical ranges observed for the independent values found in the structure of 1 except the S–S bond length which is statistically longer in 2; however, the difference is just 0.3%, so is unlikely to be chemically significant.
Triphenylstibine, 3, is a long-known compound; structures have been reported in triclinic (refcode: ZZZEHA01)25 and monoclinic (refcode: ZZZEHA02) polymorphs,26 both of which have two independent molecules per asymmetric unit. The Ph3Sb geometry is remarkably uniform amongst all of these structures (Table 1). Thus the mean Sb–C distance in 2 of 2.153(6) Å is well within the s.u. of the mean values for the five independent molecules in the comparison set at 2.150(8) Å, whilst the mean C–Sb–C pyramidal angles in 2 at 96.7(7)° is also within s.u. of 96.3(8)° in the comparison set. The close-to-90° angles at antimony, which is a feature of heavy Group 15 element chemistry, are possibly of importance for stabilizing the pseudo-cuboidal dimerization of Ph3Sb also depicted in Fig. 1. This geometry is almost indistinguishable from that in the monoclinic form of 3 (see Fig. S7 in the ESI†). The shortest contacts are “T-interactions” from a ring C atom to a CH of the other component, with lengths in 2 and 3 of 2.915 and 2.862 Å. This association of two strongly pyramidal triphenyl components is reminiscent of the supramolecular organization of Ph4P+ cations which has been dubbed the “sextuple phenyl embrace” with an estimated attraction energy of 60–85 kJ mol−1.27
The supramolecular architecture of 1, beyond its cis-oid dimerization,4 is dominated by a ‘pin-wheel’ arrangement of four such DTDA dimers into a square pattern, with short inter-molecular contacts between dimers, from the ‘end’ of one set to the ‘side’ of the next, continuing around the square. To start the discussion, consider the simplified diagram in Fig. 2. There are two such sets of centrosymmetric pin-wheels, (A → D) and (A′ → D′), each composed of four different monomers that are symmetry duplicated. Thus, in Fig. 2a, dimers A and C are the same two molecules but reversed in this top-down view, as are B and D; the second pinwheel is similarly composed of A′/C′ and B′/D′. This type of pin-wheel motif has been observed in several DTDA crystal structures;2 it is most common for structures that adopt the tetragonal space group I41/a. Examples include [2,6-F2–C6H3–CN2S2]2 (refcode: VUXZEU02);28 [2,5-F2–C6H3–CN2S2]2 (refcodes: NIHBAH and NIHBAH01);29,30 and [1,3-(S2N2C)2–C6H4]2 (refcode: SOBSOR).31 There is one report of pin-wheels in space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2m, [1,3-CN2S2-5-tBu-C6H3]2 (refcode: POYXAC).32 The lattice of 1appears as if it should be tetragonal (i.e. thereby rendering the two pin-wheels equivalent) but it is undoubtedly the distortions induced by the bulky CF3 groups that frustrate full adoption of such symmetry. Indeed, there are precedents for this too: in [1,3,5-(S2N2C)3–C6H3]2, pin wheels exist in space group P21/c although its lattice is metrically close to tetragonal (refcode: KUFDUK),33 whilst in [3,5-Cl2–C6H3–CN2S2]2 (refcode: DIXNEF) in space group P, the lattice contains a mixture of tetrameric pin-wheels of dimers and isolated doublets of dimers.34
2m, [1,3-CN2S2-5-tBu-C6H3]2 (refcode: POYXAC).32 The lattice of 1appears as if it should be tetragonal (i.e. thereby rendering the two pin-wheels equivalent) but it is undoubtedly the distortions induced by the bulky CF3 groups that frustrate full adoption of such symmetry. Indeed, there are precedents for this too: in [1,3,5-(S2N2C)3–C6H3]2, pin wheels exist in space group P21/c although its lattice is metrically close to tetragonal (refcode: KUFDUK),33 whilst in [3,5-Cl2–C6H3–CN2S2]2 (refcode: DIXNEF) in space group P, the lattice contains a mixture of tetrameric pin-wheels of dimers and isolated doublets of dimers.34
 | ||
| Fig. 2 Simplified “cartoons” depicting the arrangements of clusters within one double-layer, which occur in the crystal lattices of 1 (a) and 2 (b); for detailed diagrams of these layers see the ESI† (Fig. S3 and S5). | ||
The most remarkable supramolecular feature of 2 is the series of ‘end-on’ short contacts between the four sulphur atoms of the DTDA dimer and the aryl ring atoms C42–C45, which range from 3.168(3)–3.463(3) Å [0.33 to 0.04 Å < ∑rvdW] as shown in Fig. 1. All of these carbon atoms are part of one phenyl ring belonging to a Ph3Sb and the mutual orientation of the components in 2 precludes interaction with the antimony donor electron pair. There are additional aryl ring “T-interactions” between the DTDA aryl H atoms and ring carbon atoms of the stibine, which results in an alternating pattern of (DTDA)2 → Ph3Sb → (DTDA)2 → Ph3Sb which, although somewhat rectangular, strongly resembles the pin-wheel arrays in 1 (Fig. 2b). This cluster is also centrosymmetric, so that dimer G is the inverse of E, and H the inverse of F. In both structures, the assemblies occur within well-defined layers. Thus, one way to describe the supramolecular architecture of 2 is that Ph3Sb molecules, each also part of their own pseudo-cuboidal dimers, replace every second DTDA dimer specifically at the site of the “end-on” bonding (Fig. 2b).
In Fig. 3, one of the two essentially equivalent pin-wheels in the structure of 1 is shown in molecular detail. For a more extended view of the lattice, please see the ESI† (Fig. S3), where several sets of the two symmetry-independent pin-wheels are depicted from a top view and a side view. The latter emphasizes the “double-layer” structure consisting of slices of the lattice that are parallel to the (1 1 0) Miller planes and are about 8.3 Å thick. Metric data for the intermolecular contacts both between the monomers and between the dimers that are shown in Fig. 3 are available in Table S1† (for a different perspective, see Fig. S4 in ESI†). Noteworthy is the relative shortness of all these contacts, i.e. all the blue lines in Fig. 3 are from contacts shorter than (∑rvdW − 0.2 Å). By contrast, the pin-wheels in the slices above and below the one that is drawn in Fig. S3† are partly offset and the shortest contacts from one slice to the next are S3⋯S5′ at 3.711(2) and S10⋯S14′ at 3.765(2) Å, much weaker interactions that are longer than ∑rvdW.
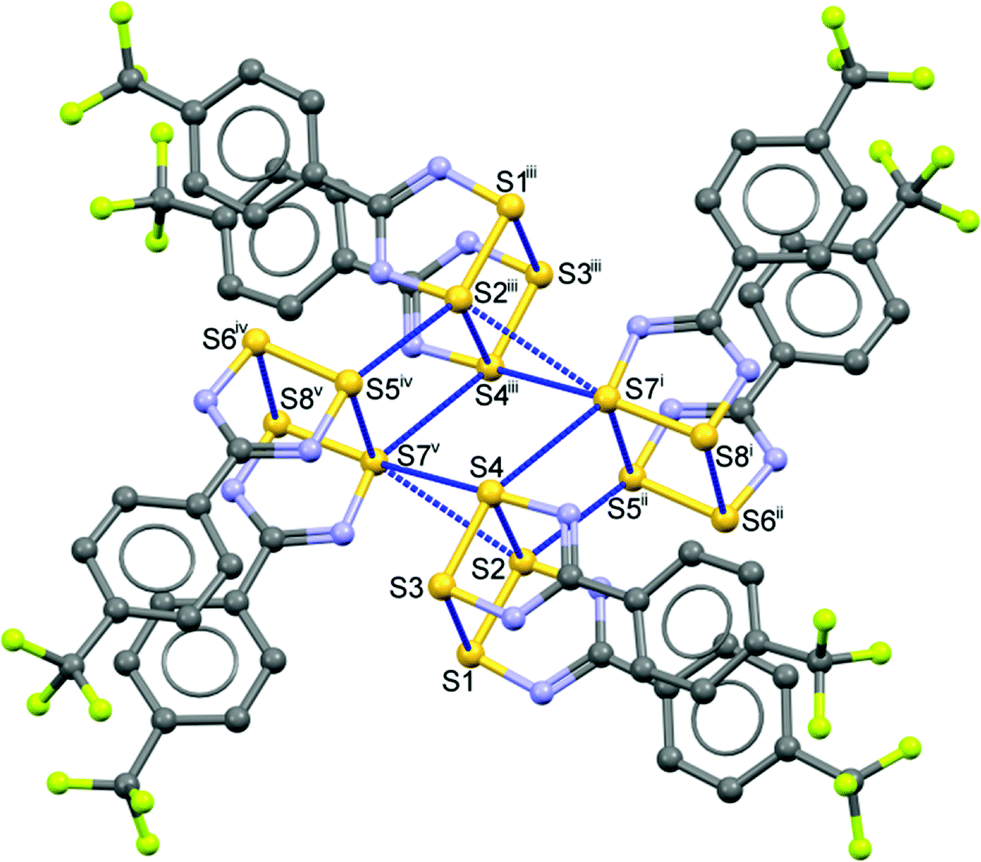 | ||
| Fig. 3 One of two symmetry-independent, centrosymmetric, pin-wheel clusters in the asymmetric unit of 1 showing intermolecular contacts shorter than (∑rvdW − 0.2 Å). H atoms have been removed to enhance visibility [symmetry codes: (i) x, −1 + y, z; (ii) 1 + x, −1 + y, z, (iii) 1 + x, −1 + y, z, (iv) −x, 1 − y, 1 − z, (v) 1 − x, 1 − y, 1 − z]. The CF3 groups belonging to molecules iv and v are rotationally disordered (for details, see the ESI†). | ||
Similarly, Fig. 4 presents a more detailed view of the intermolecular contacts that support the supramolecular architecture of the crystal lattice of 2. A more extended view of the lattice and a side-view is provided in the ESI† (Fig. S5) Metric data for the intermolecular contacts shown by the blue dotted-lines in Fig. 4 are reported in Table S2.† Noteworthy here is that the shortest sulphur–carbon interaction of 3.168(3) Å is as short when expressed as (distance < ∑rvdW) to the sulphur–sulphur inter-molecular contacts in 1 (see Tables S1 and S2 in the ESI†), i.e. they appear to be of comparable strength.
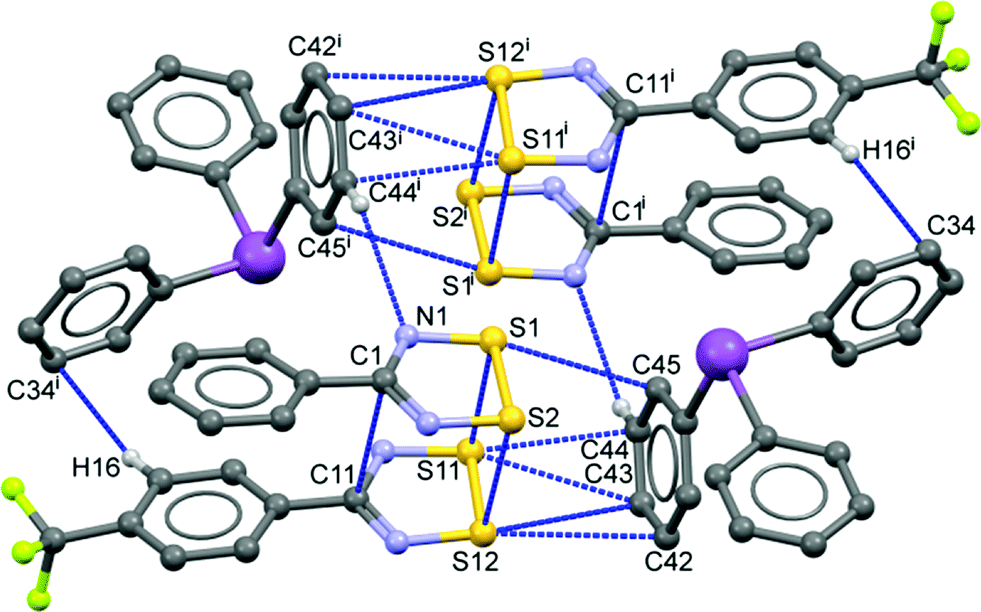 | ||
| Fig. 4 Unit cell contents of the centrosymmetric crystal structure of 2 showing intermolecular contacts shorter than ∑rvdW. H atoms except those involved in contacts and the CF3 groups on front upper and back lower DTDA have been removed to enhance visibility [symmetry code: (i) 1 − x, 1 − y, 1 − z]. | ||
For the sole other example of supramolecular interactions to Ph3Sb of the type observed in 2 we must turn (Fig. 5) to a co-crystal with fullerene, 4. This structure (refcode: YIKVET)35 displays a side-on interaction from the face of one of the three phenyl rings over a 6:5 ring junction of C60 (there are altogether six Ph3Sb associated with each C60 molecule, see the ESI,† Fig. S6). The contact distances are on the order of the ∑rvdW (3.48(1)–3.65(1) Å) and were attributed to an electrostatic interaction between a region of partial negative charge in the center of the phenyl ring and a region of partial positive charge on the C60 surface,35 although there is almost certainly a significant contribution from dispersion. To test this hypothesis, a PBEPBE/6-311+g(2df,2p) DFT calculation was undertaken (see Fig. S8 and Table S4, ESI†) on a somewhat simplified model. The average NPA charge of the (model) benzene C atoms is −0.183e and of the 6:5 junction C atoms is +0.006, so that Δq is 0.189e. When a similar calculation is performed on a model system for 2, the sulphur atoms bear an average NPA charge of +0.465e (Fig. 6 and Table 2) and the average benzene C charge is −0.182e, so that Δq is 0.647e. The net dipole moment of 6.9 Debye is oriented along the middle of the DTDA dimer and is directed to the benzene ring face.
 | ||
| Fig. 5 Interaction of a Ph3Sb phenyl ring with a 6:5 junction bond of C60 in the adduct structure (refcode: YIKVET).19 The carbon atoms in the fullerene are rendered orange for contrast. | ||
 | ||
| Fig. 6 Computed NPA atomic charges from a PBEPBE/6-311+g(2df,2p) DFT calculation and the net dipole moment of a model structure in which the pendant Ph2Sb group has been removed. Red indicates regions of negative charge and green is positive. | ||
| Atom | Charge | Atom | Charge |
|---|---|---|---|
| a Data taken from PBEPBE/6-311+g(2df,2p) DFT calculations. A full listing is provided in Table S3, ESI. | |||
| S1 | 0.463 | C8 | 1.010 |
| S2 | 0.467 | F1 | −0.317 |
| N1 | −0.716 | F2 | −0.323 |
| N2 | −0.705 | F3 | −0.347 |
| C1 | 0.497 | C40 | −0.203 |
| C2 | −0.114 | C41 | −0.173 |
| C3 | −0.135 | C42 | −0.170 |
| C4 | −0.158 | C43 | −0.179 |
| C5 | −0.107 | C44 | −0.182 |
| C6 | −0.160 | C45 | −0.187 |
| C7 | −0.130 | ||
Experimental
General
Unless otherwise indicated, all procedures were performed under an atmosphere of purified N2 using a glovebox, Schlenkware, and vacuum-line techniques. Solvents used were reagent-grade or better. Acetonitrile (HPLC grade) was double-distilled from P2O5 and CaH2 and diethyl ether was distilled from sodium/benzophenone. SCl2 was distilled under protection from moisture (5 mL crude containing 1 mL PCl3), stored on ice, and used within a few hours. Infrared spectra were obtained as Nujol mulls between CsI plates and were recorded on a Bomem MB102 Fourier transform spectrometer. Melting points (capillaries) were determined on an Electrothermal melting point apparatus and are uncorrected. Combustion analysis was performed by M-H-W Laboratories, Phoenix, AZ. Gradient sublimation was undertaken using a home-build 3-zone tube furnace under dynamic vacuum for initial purification followed by slow sublimation in a sealed, evacuated Pyrex tube (15 mm i.d. × 600 mm). The zone temperatures were adjusted based on visual inspection of the progress of sublimation. The silylated amidine 4-F3CC6H4C(![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) NTMS)N(TMS)2 was prepared by the literature method.36
NTMS)N(TMS)2 was prepared by the literature method.36
Preparation of 1
In a typical preparation, 5.0 g (13.5 mmol) of 4-F3CC6H4C(NTMS)N(TMS)2 was warmed into 40 mL of CH3CN, whereupon excess, freshly distilled, SCl2 (2 mL, excess) was added through the top of a reflux condenser with vigorous agitation. After several hours refluxing, the solution was cooled to ambient and filtered under inert gas. The dried 4-F3CC6H4CN2S2+Cl− was re-suspended in a minimum quantity of warm acetonitrile, freeze–thaw degassed 3×, and then 2.5 g solid Ph3Sb (7 mmol, slight excess based on the amidine) was added from a solids addition funnel. After refluxing for 30 min, the solution was cooled to ambient after which volatiles were removed using vacuum. The dried cake was transferred (caution: glove box!) to a borosilicate glass sublimation tube (20 mm i.d. × 600 mm) and sublimed in a dynamic vacuum in a horizontal tube furnace. The crude, black, sublimed material was then placed in a narrower tube, evacuated and sealed by melting the constricted neck. Careful gradient sublimation using three heating zones resulted in some colourless crystals near the origin and well-formed but small needles amongst large blocks of purple to black crystals. Crystals were harvested in a glove box by sacrificing the glass tube.
X-ray crystallography
A thin, dark purple-black, needle corresponding to 1 was selected, coated in Paratone™ oil, mounted on the end of a thin glass capillary and cooled on the goniometer head to 173(2) K with the Bruker low-temperature accessory. A large red-purple block corresponding to 2 was likewise selected and mounted, but the best dataset could be obtained at 263(2) K. A full hemisphere of data was collected for each on a Bruker APEX-II diffractometer using Mo Kα radiation (λ = 0.71073 Å) controlled by APEX2 software.37 A multi-scan absorption correction (SADABS)37 was applied to the data, scaled and corrected for polarization (SAINT-Plus),37 where after the structure was solved by direct methods (SHELXS or SHELXT)24,38 and refinement was conducted with full-matrix least-squares on F2 using SHELXL-2014.39 H atoms attached to carbon were observed in a fine-focused Fourier map and were treated as riding on their attached aromatic carbon atoms with C–H = 0.95 Å and Uiso = 1.2Ueq(C) for the purpose of model refinement. The structure of 1 has disorder of the CF3 groups in one of four independent DTDA dimers. An adequate two part disorder model was developed; restraints were required to ensure reasonable geometries. For a detailed description and graphics, see the ESI† (Fig. S9). The structure of 2 displays a similar disorder applying to both CF3 groups for which a model akin to that used for 1 was developed. Details are in the ESI† (Fig. S10). In the refinement of both structures, the displacement ellipsoids were globally restrained using the newly developed RIGU code in SHELX-2014.39,40 This was necessary to prevent oblate or NPD fluorine displacement ellipsoids and was also valuable for the increased thermal motion in the structure of 2 determined at 263 K. Crystal and experimental parameters are compiled in Table 3, and selected interatomic distances are available in Table 1. More detailed crystal structure reports are available in Tables S5 and S6, ESI.† Structures were visualized and the lattice geometrical properties were analyzed with the use of Mercury v3.7.41 Structure depositions: 1, CCDC 1452129 and 2 CCDC 1452130, contain the supplementary crystallographic data for this paper.| Parameter | 1 | 2 |
|---|---|---|
| a Full-matrix least-squares on F2. | ||
| Formula | C8H4F3N2S2 | C34H23F6N4S4Sb |
| FW (amu) | 249.25 | 851.55 |
| Temperature (K) | 173(2) | 263(2) K |
| Radiation, λ (Å) | Mo, 0.71073 | Mo, 0.71073 |
| Crystal system | Triclinic | Triclinic |
| Space group |
P |
P |
| a (Å) | 9.4916(9) | 11.4543(10) |
| b (Å) | 18.1887(17) | 11.7399(10) |
| c (Å) | 22.275(2) | 13.9480(12) |
| α (°) | 91.5790(10) | 73.3640(10) |
| β (°) | 97.3290(10) | 73.2470(10) |
| γ (°) | 102.7550(10) | 82.5790(10) |
| Volume (Å3) | 3713.8(6) | 1718.6(3) |
| Z | 16 | 2 |
| D calc (g cm−3) | 1.783 | 1.646 |
| μ (mm−1) | 0.583 | 1.110 |
| F(000) | 2000 | 848 |
| Crystal size (mm3) | 0.18 × 0.10 × 0.04 | 0.460 × 0.420 × 0.280 |
| θ range (°) | 1.847 to 26.220° | 1.813 to 28.578° |
| Index ranges | −11 ≤ h ≤ 11 | −15 ≤ h ≤ 15 |
| −22 ≤ k ≤ 22 | −15 ≤ k ≤ 15 | |
| −27 ≤ l ≤ 27 | −18 ≤ l ≤ 18 | |
| Total rfl. | 39140 |
19773 |
| Indep. rfl. | 14828 |
8041 |
| R (int) | 0.0812 | 0.0195 |
| Compl. θ 25.5° | 99.6% | 99.6% |
| Abs. corr. | Semi-empirical from equivalents | Semi-empirical from equivalents |
| Max. and min. transmission | 0.900 | 0.900 |
| 0.811 | 0.717 | |
| Data/restraints/parametersa | 14828/1034/1137 |
8041/594/516 |
| GOF, F2 | 0.973 | 1.054 |
| Final R indices [I > 2σ] | R 1 = 0.0540, wR2 = 0.0930 | R 1 = 0.0313, wR2 = 0.0789 |
| R indices (all data) | R 1 = 0.1396, wR2 = 0.1185 | R 1 = 0.0404, wR2 = 0.0864 |
| Larg. pk (e Å3) | 0.470 | 0.664 |
| Larg. hole (e Å3) | −0.456 | −0.590 |
Computation
For the DFT calculations, a simplified model with benzene representing the phenyl group of 2 was employed, using the crystal structure geometry to define the shape. The pendant Ph2Sb group was removed and replaced by an H atom at standard C–H distances using the program GaussView 5.0. A density functional theory (DFT) calculation was undertaken at this static geometry at the PBEPBE/6-311+G(2df,2p) level of theory in Gaussian W03 on a personal computer under Windows 7.42 The lack of availability of good parameters for antimony in high-level basis sets was the main reason for excluding it from these calculations. The Normal Population Analysis atomic charges and the calculated dipole moment were visualized in GaussView (Fig. 6). In an analogous fashion, a model of C60 surrounded by a hexagonal array of six benzene rings in the location of Ph3Sb phenyl rings was computed as a model for 4 (see ESI,† Fig. S8 and Table S4).Conclusions
Co-sublimation of rather volatile 4-CF3-substituted DTDA 1 with triphenylstibine 3 results in a well-defined 1:1 adduct 2 that is linked by supramolecular contacts between the electropositive heterocycle sulphur atoms and the negative charge associated with the phenyl ring π-system. The structure determined for 2 shows remarkable similarity to that of the parent DTDA dimer; in place of the ‘pin-wheel’ arrangement of four such dimers in the lattice of 1, the adduct consists of two DTDA dimers and two Ph3Sb units, resulting in a slightly rectangular arrangement in place of the symmetrical square. The aromatic interactions do not disrupt the ‘pancake bonding’ within DTDA dimers, but involve the sulphur terminus of the rings in a longitudinal interaction of a type that dominates DTDA crystal engineering.2 A preliminary investigation of NPA charges shows a significantly larger electrostatic component to the interaction in 2 compared to the C60 adduct 4, consistent with shorter intermolecular contact distances in 2 compared to 4.
Ph3Sb may be a very suitable complexing agent for many thiazyl radicals;35 the resulting supramolecular architectures may be capable of further optimization to achieve desirable solid-state properties. Further progress in DTDA-aromatic supramolecular chemistry may be anticipated by concentrating on very electron rich aromatics – mesitylene or durene as benzene derivatives – but also PAHs such as triphenylene43 or perylene. By employing radical aromatics such as phenalenyl, it may indeed be possible to engineer mixed DTDA/aromatic pancake dimers.44,45
Acknowledgements
The Natural Sciences and Engineering Research Council of Canada (NSERC) is gratefully acknowledged for generous support through the Discovery Grants program. The diffractometer was purchased with the help of NSERC and the University of Lethbridge. Many students have worked on dithiadiazolyl chemistry in my laboratory over the years. Their names appear in the cited references. The anonymous referees are thanked for several helpful suggestions.References
- R. T. Boeré and T. L. Roemmele, Chalcogen–Nitrogen Radicals, in Comprehensive Inorganic Chemistry II, ed. Jan Reedijk and Kenneth Poeppelmeier, Elsevier, Oxford, 2013, vol. 1, pp. 375–411 Search PubMed.
- D. A. Haynes, CrystEngComm, 2011, 13, 4793–4805 RSC.
- S. Domagala, K. Kosc, S. W. Robinson, D. A. Haynes and K. Woźniak, Cryst. Growth Des., 2014, 14, 4834–4848 CAS.
- K. E. Preuss, Polyhedron, 2014, 79, 1–15 CrossRef CAS.
- H. Z. Beneberu, Y.-H. Tian and M. Kertesz, Phys. Chem. Chem. Phys., 2012, 14, 10713–10725 RSC.
- R. T. Oakley, Prog. Inorg. Chem., 1988, 36, 299–391 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- C. D. Bryan, A. W. Cordes, R. C. Haddon, R. G. Hicks, R. T. Oakley, T. T. M. Palstra, A. S. Perel and S. R. Scott, Chem. Mater., 1994, 6, 508–515 CrossRef CAS.
- C. D. Bryan, A. W. Cordes, R. C. Haddon, R. G. Hicks, D. K. Kennepohl, C. D. MacKinnon, R. T. Oakley, T. T. M. Palstra, A. S. Perel, S. R. Scott, L. F. Schneemeyer and J. V. Waszczak, J. Am. Chem. Soc., 1994, 116, 1205–1210 CrossRef CAS.
- A. J. Banister, M. I. Hansford, Z. V. Hauptman, A. W. Luke, S. T. Wait, W. Clegg and K. A. Jorgensen, J. Chem. Soc., Dalton Trans., 1990, 2793–2808 RSC.
- C. S. Clarke, D. A. Haynes, J. M. Rawson and A. D. Bond, Chem. Commun., 2003, 2774–2775 RSC.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- C. Allen, D. A. Haynes, C. M. Pask and J. M. Rawson, CrystEngComm, 2009, 11, 2048–2050 RSC.
- S. W. Robinson, D. A. Haynes and J. M. Rawson, CrystEngComm, 2013, 15, 10205–10211 RSC.
- H. Akpinar, J. T. Mague and P. M. Lahti, CrystEngComm, 2013, 15, 831–835 RSC.
- G. M. Espallargas, A. Recuenco, F. M. Romero, L. Brammer and S. Libri, CrystEngComm, 2012, 14, 6381–6383 RSC.
- H. Akpinar, J. T. Mague, M. A. Novak, J. R. Friedman and P. M. Lahti, CrystEngComm, 2012, 14, 1515–1526 RSC.
- A. W. Cordes, R. C. Haddon, R. T. Oakley, L. F. Schneemeyer, J. V. Waszczak, K. M. Young and N. M. Zimmerman, J. Am. Chem. Soc., 1991, 113, 582 CrossRef CAS.
- J. M. Cole, C. M. Aherne, J. A. K. Howard, A. J. Banister and P. G. Waddell, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o2514 CAS.
- J. M. Cole, C. M. Aherne, P. G. Waddell, A. J. Banister, A. S. Batsanov and J. A. K. Howard, Polyhedron, 2012, 45, 61–70 CrossRef CAS.
- Y. Beldjoudi, D. A. Haynes, J. J. Hayward, W. J. Manning, D. R. Pratt and J. M. Rawson, CrystEngComm, 2013, 15, 1107–1113 RSC.
- R. T. Boeré, K. H. Moock, S. Derrick, W. Hoogerdijk, K. Preuss, J. Yip and M. Parvez, Can. J. Chem., 1993, 71, 473–486 CrossRef.
- H. F. Lau, P. C. Y. Ang, V. W. L. Ng, S. L. Kuan, L. Y. Goh, A. S. Borisov, P. Hazendonk, T. L. Roemmele, R. T. Boeré and R. D. Webster, Inorg. Chem., 2008, 47, 632–644 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- E. A. Adams, J. W. Kolis and W. T. Pennington, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 917–919 CrossRef.
- Effendy, W. J. Grigsby, R. D. Hart, C. L. Raston, B. W. Skelton and A. H. White, Aust. J. Chem., 1997, 50, 675–682 CrossRef CAS.
- I. Dance and M. Scudder, Chem. – Eur. J., 1996, 2, 481–486 CrossRef CAS.
- C. S. Clarke, D. A. Haynes, J. N. B. Smith, A. S. Batsanov, J. A. K. Howard, S. I. Pascu and J. M. Rawson, CrystEngComm, 2010, 12, 172–185 RSC.
- A. J. Banister, A. S. Batsanov, O. G. Dawe, P. L. Herbertson, J. A. K. Howard, S. Lynn, I. May, J. N. B. Smith, J. M. Rawson, T. E. Rogers, B. K. Tanner, G. Antorrena and F. Palacio, J. Chem. Soc., Dalton Trans., 1997, 2539–2541 RSC.
- L. Beer, A. W. Cordes, D. J. T. Myles, R. T. Oakley and N. J. Taylor, CrystEngComm, 2000, 2, 109–114 RSC.
- M. P. Andrews, A. W. Cordes, D. C. Douglass, R. M. Fleming, S. H. Glarum, R. C. Haddon, P. Marsh, R. T. Oakley, T. T. M. Palstra, L. F. Schneemeyer, G. W. Trucks, R. Tycko, J. V. Waszczak, K. M. Young and N. M. Zimmerman, J. Am. Chem. Soc., 1991, 113, 3559–3568 CrossRef CAS.
- R. A. Beekman, R. T. Boeré, K. H. Moock and M. Parvez, Can. J. Chem., 1998, 76, 85–93 CrossRef CAS.
- A. W. Cordes, R. C. Haddon, R. G. Hicks, R. T. Oakley, T. T. M. Palstra, L. F. Schneemeyer and J. V. Waszczak, J. Am. Chem. Soc., 1992, 114, 5000–5004 CrossRef CAS.
- C. P. Constantinides, D. J. Eisler, A. Alberola, E. Carter, D. M. Murphy and J. M. Rawson, CrystEngComm, 2014, 16, 7298 RSC.
- M. Fedurco, M. M. Olmstead and W. R. Fawcett, Inorg. Chem., 1995, 34, 390–392 CrossRef CAS.
- R. T. Boeré, R. T. Oakley and R. W. Reed, J. Organomet. Chem., 1987, 331, 161–167 CrossRef.
- Bruker, APEX2, SAINT-Plus and SADABS, Bruker AXS Inc., Madison Wisconsin, USA, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- A. Thorn, B. Dittrich and G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2012, 68, 448–451 CrossRef CAS.
- D. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- J. C. Collings, K. P. Roscoe, R. L. Thomas, A. S. Batsanov, L. M. Stimson, J. A. K. Howard and T. B. Marder, New J. Chem., 2001, 25, 1410–1417 RSC.
- S. K. Pal, M. E. Itkis, F. S. Tham, R. W. Reed, R. T. Oakley, B. Donnadieu and R. C. Haddon, J. Am. Chem. Soc., 2007, 129, 7163–7174 CrossRef CAS PubMed.
- Pradip Bag, Mikhail E. Itkis, Dejan Stekovic, Sushanta K. Pal, Fook S. Tham and Robert C. Haddon, J. Am. Chem. Soc., 2015, 137, 10000–10008 CrossRef CAS PubMed , and references therein..
Footnote |
| † Electronic supplementary information (ESI) available: CIF for all X-ray structures have been deposited. CCDC 1452129–1452130. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce00351f |
| This journal is © The Royal Society of Chemistry 2016 |