
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudies on the crystal structure and arrangement of water in sitagliptin L-tartrate hydrates†
Eszter
Tieger
*ab,
Violetta
Kiss
b,
György
Pokol
a,
Zoltán
Finta
b,
Michal
Dušek
c,
Jan
Rohlíček
c,
Eliška
Skořepová
bd and
Petr
Brázda
c
aDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Szt. Gellért tér 4, H-1111 Budapest, Hungary. E-mail: tiegereszter@gmail.com
bZentiva k.s., U kabelovny 130, 102 37, Prague, Czech Republic
cInstitute of Physics ASCR, v. v. i., Na Slovance 2, 182 21 Prague, Czech Republic
dDepartment of Solid State Chemistry, University of Chemistry and Technology, Prague, Technicka 5, 166 28 Prague, Czech Republic
First published on 9th May 2016
Abstract
The hydration/dehydration behavior of four distinct channel hydrates of sitagliptin L-tartrate (SLT) was investigated by thermoanalytical methods, dynamic vapour sorption analysis and variable humidity X-ray powder diffraction. The crystal structures were determined from single crystal and powder X-ray diffraction data. A survey of the forms revealed that SLT hydrates exhibit both stoichiometric and non-stoichiometric features demonstrating that the characterization of channel hydrates can be challenging as their behavior is not inevitably unambiguous. Upon dehydration, the parent hydrates retain their structures, and the lattices do not collapse; isostructural dehydrates are formed. The solved crystal structures of the packing polymorphs SLT phase 1 and phase 2 provide an effective basis to rationalize the observed hydration/dehydration pathways. The structures are dominated by infinite sheets formed by hydrogen tartrate anions, linked by hydrogen bonds. These layers separate the parallel, infinite chains of water molecules. The water molecules stabilize the structures by providing additional hydrogen bonds between the cation and the anion. This interaction substantiates the high affinity of water molecules to the API framework and explains the stoichiometric characteristics observed by solid state analytical methods. On the other hand, their non-stoichiometric character is evidenced by the non-destructive dehydration processes.
1. Introduction
Polymorphs are crystalline solids that have the same chemical composition, yet adopt different molecular arrangements in the crystal lattice. The fundamental importance of pharmaceutical solid polymorphism is based on the fact that the distinct solid-state forms of the same chemical compound may have different chemical and physical properties. Thus, it can impact the drug product's manufacturability, bioavailability and stability and therefore has influence on drug product safety and efficacy.1 In the pharmaceutical industry, polymorphism is of critical importance since it has major regulatory and intellectual property implications as well.Crystalline solids may incorporate solvent(s) into the lattice during crystallization to form a solvate. This phenomenon is commonly referred to as pseudopolymorphism, a term introduced by Walter C. McCrone,2 or solvatomorphism. The terminology of multicomponent crystalline solids containing solvent molecules is not uniform and is under constant debate. Some advocate the use of the term pseudopolymorph,3–8 while others argue against it.9,10
In a strict sense, polymorphism denotes systems with the same chemical composition,11,12 and therefore, since a solvate and an unsolvated crystalline form are constitutionally distinct, they cannot be considered as polymorphs by definition. Solid forms crystallized with different amounts or types of solvent molecules are therefore often denoted as pseudopolymorphs.
If the incorporated solvent is water, the form is referred to as hydrate. Hydrates are more acceptable in the pharmaceutical industry, since the presence of most solvents in a therapeutically indicated drug substance might raise toxicity concerns.13 Owing to the ubiquity of water vapour, hydrates are often the thermodynamically stable form under ambient conditions. If a certain hydrate is stable within a large humidity range, it may be much easier to formulate the hydrate in a controlled way and to subsequently store and package it.14
A comprehensive characterization of pharmaceutical hydrates should include a collective knowledge about structure identification, thermodynamic properties and phase diagrams. The investigation of this area requires systematic work and the interplay of various analytical techniques.
Hydrates can be described in terms of their stoichiometry relative to the host molecule, i.e. they are either stoichiometric (where a definite but not necessary integer ratio of solvent to molecule exists) or non-stoichiometric (where the ratio of solvent to molecule may but not necessarily vary continuously over a given range).12
While the solvent in stoichiometric hydrates is usually an integral part of the crystal structure and is essential for the maintenance of the molecular network in the case of non-stoichiometric hydrates, it might be located in certain structural voids and can act mostly as a space filler.6
The desolvation of stoichiometric solvates always leads to a different crystal structure or results in a disordered or amorphous state, while in non-stoichiometric hydrates the structure of the parent hydrate can be retained.
The way how the water sorbs and the location of water on a molecular level are specific for a system and are useful information to have during drug development. The following three categories of crystalline hydrates are recognized:
• In isolated site hydrates, the water molecules are isolated from direct contact with other water molecules by intervening drug molecules.
• In channel hydrates, the water molecules form channels through the crystal, where they can interact through weak interactions.
• In metal ion-associated hydrates, the water molecules are bound directly to a metal ion.15
The characterization of channel hydrates can be challenging as their behavior is not inevitably unambiguous. Depending on the extent of hydrogen bonding and the size of the channels, these hydrates can be either stoichiometric or non-stoichiometric.16 Their water content can vary as a function of relative humidity in the surrounding atmosphere but not necessarily have to. They can also crystallize with well-defined water/compound ratios like caffeine 0.8 hydrate.17
In this paper, the term solvate is used as a general term for crystalline solvent adducts including the subclass of hydrates.
Sitagliptin (7-[(3R)-3-amino-1-oxo-4-(2,4,5-trifluorophenyl)butyl]-5,6,7,8-tetrahydro-3-(trifluoromethyl)-1,2,4-triazolo[4,3-a]pyrazine) is an oral antihyperglycemic of the dipeptidyl peptidase-4 (DPP-4) inhibitor class. It is one of the most popular type 2 diabetes drugs on the market. The original product is marketed by Merck & Co. under the trade name Januvia® as sitagliptin phosphate monohydrate. In this paper, L-tartaric acid was used as a salt-forming agent and the obtained hydrogen tartrate salts (Fig. 1) were studied. The L-tartaric salt of sitagliptin showed comparable pharmaceutical properties and therefore could be used as a potential candidate for a new drug product.
 | ||
| Fig. 1 Chemical structure of sitagliptin L-tartrate. | ||
Due to the propensity of water to interact and coordinate with ions, salts often form hydrates.18
Since hydrogen tartrate anions are prone to link into infinite chains, these salts frequently exhibit a channel hydrate structure like L-histidinium-L-tartrate hemihydrate,19 saxagliptin hydrogen tartrate hemihydrate,20 LY297802 tartrate,21 varenicline L-tartrate22 and silver hydrogen L(+)-tartrate.23
Sitagliptin L-tartrate can crystallize in five different hydrate forms. The thermodynamically preferred crystal form is designated as phase 1.24 This modification and two other crystal forms, phase 2 (ref. 25) and phase 3 (ref. 26), are claimed by Zentiva. The fourth modification is described in a patent filed by Merck27 and referred to as hemihydrate. Therefore, this phase is named in this paper as phase M. We succeeded in growing single crystals of phase 1 and phase 2 that are suitable for X-ray structure determination. Unexpectedly, a few crystals of the tetrahydrate, phase 4, were obtained and its structure was solved by single crystal X-ray diffraction. This phase will be discussed only in the crystal structure section because its synthesis could not be repeated.
Our aim was to provide a molecular level understanding of the arrangement of water molecules in the lattice and the way how water uptake can influence the physical and chemical stability of a pharmaceutical compound. Such knowledge is invaluable, particularly early in drug development when the solid state form is selected. The work presented here aims to demonstrate how the combination of the techniques applied can provide a useful means for defining the hydrate category and for deriving conclusions about the arrangement of water molecules.
Therefore, all modifications – except for the tetrahydrate, phase 4 – were characterized by differential scanning calorimetry (DSC), thermogravimetric analysis (TG), variable humidity X-ray powder diffraction (VH-XRPD), and dynamic vapour sorption (DVS) studies.
Since the stability of a hydrate is governed by the role played by water molecules in the crystal structures, understanding the dehydration mechanism is essential to rationalize form selection in drug development. Are non-stoichiometric hydrates necessarily undesirable?
2. Experimental section
Materials and methods
Phase 1 was obtained when SLT was crystallized from a 2-propanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (8:2, v/v) mixture. Crystallization of SLT from a methanol:water (6:4, v/v) mixture resulted in phase 2 crystals. Crystallization of SLT from an acetone:water (9:1, v/v) mixture led to phase 3. SLT phase M was obtained from pure methanol. Phase 4 crystallized from an ethanol–water (3:1, v/v) mixture at −20 °C in the freezer.
:water (8:2, v/v) mixture. Crystallization of SLT from a methanol:water (6:4, v/v) mixture resulted in phase 2 crystals. Crystallization of SLT from an acetone:water (9:1, v/v) mixture led to phase 3. SLT phase M was obtained from pure methanol. Phase 4 crystallized from an ethanol–water (3:1, v/v) mixture at −20 °C in the freezer.
Differential scanning calorimetry. DSC measurements were performed using a Mettler-Toledo 822e DSC. Samples were placed into standard aluminum pans (40 μL) sealed with a pierced lid. The sample cell was heated at a rate of 10 °C min−1, from 25 °C up to a final temperature of 300 °C with 50 mL min−1 nitrogen purge.
Thermogravimetric analysis. TGA analyses were performed using a NETZSCH TG 209 thermogravimetric analyser (NETZSCH-Gerätebau GmbH, Germany). Each sample was placed in an aluminum sample pan and inserted into the TG furnace. The furnace was heated under nitrogen purge at a rate of 10 °C min−1, from 25 °C up to a final temperature of 300 °C.
Dynamic vapour sorption. Gravimetric moisture sorption analysis was carried out using a humidity and temperature-controlled microbalance DVS apparatus, DVS Advantage 1 (Surface Measurement Systems, U.K.) with a Cahn D200 recording ultra-microbalance with a mass resolution of ±0.1 μg. Samples of approximately 20 mg were dried at 0% RH under a nitrogen stream at 25 °C. Moisture uptake (reported relative to the dry weight) was monitored over a sorption/desorption range of 0–90% RH in increments of 10%.
Variable humidity X-ray powder diffraction. X-Ray powder diffraction (XRPD) patterns were recorded at room temperature in Bragg–Brentano geometry using a PANalytical X'Pert Pro diffractometer (X'Pert Pro PANalytical, Netherlands). A continuous 2θ scan was performed in the range of 2–40° with a step size of 0.01 2θ using an incident beam of CuKα radiation at 40 mA and 45 kV. An Anton Paar temperature–humidity chamber was used to collect in situ XRPD patterns as a function of relative humidity. The humidity was generated by an RH200 relative humidity generator (VTI Inc., USA) and carried by a flow of nitrogen gas. Samples were held at 25 °C, and the RH was ramped in steps of 10% RH. To assess the influence of humidity, the measurements were carried out in the same fashion as the DVS analyses and samples were monitored over a sorption/desorption range of 5–90% RH in increments of 10% (5, 10, 20, 30, 40, 50, 60, 70, 80, 90% RH).
Karl Fischer titration. Karl Fisher titrations for water content determination were performed using a Metrohm 835 Titrando titrator (Metrohm Ltd., Herisau, Switzerland). HYDRANAL®-Methanol dry (Sigma-Aldrich Laborchemikalien GmbH) was used as working medium, and HYDRANAL®-Composite (Sigma-Aldrich Laborchemikalien GmbH) was used as a titration agent.
Headspace gas chromatography. A PerkinElmer AutoSystem XL gas chromatographic system (PerkinElmer, Waltham, MA, USA) equipped with a flame ionization detector and a PerkinElmer TurboMatrix 40 headspace was used for residual organic solvent content determination. Chromatographic data were collected and processed using TotalChrom Navigator v. 6.3.2 software. A CP-Select 624 (30 m, 0.32 mm ID, 1.8 μm df) column (Agilent, Santa Clara, CA, USA) was used for chromatographic determination of the organic solvents. Helium was used as a carrier gas at a flow rate of 2.0 mL min−1. The injector and detector temperatures were set at 165 and 300 °C, respectively. The headspace oven temperature was set at 110 °C with a thermostating time of 30 min. The needle temperature was 115 °C. The pressurization time was 1.0 min, and the vial pressure was set at 21 psi (1.45 bar).
Data collection – single crystal. The single crystal X-ray diffraction analysis was carried out at a temperature of 120 K using a Gemini diffractometer with a Cu-Ultra mirror collimator and an Atlas S1 CCD detector, with Cu Kα radiation from a sealed tube with a wavelength of 1.5418 Å. The collection and data reduction program used was CrysAlisPro, Agilent Technologies, version 1.171.36.28. An empirical correction for absorption was carried out by using the scaling algorithm SCALE3 ABSPACK. The structures were solved by direct methods (program SIR92) and refined in the program CRYSTALS 14.40b. All non-hydrogen atoms were refined anisotropically. In case of more complicated structure features, structure determination was continued using the program Jana2006.20 The simulated XRPD patterns were calculated using the MERCURY software (version 3.3; Cambridge Crystallographic Data Center, Cambridge, UK), and structures were visualized using Diamond (versions 3 and 4; Crystal Impact).
Hydrogen atoms of carbon were attached geometrically. Hydrogen atoms of nitrogen or oxygen were found from difference Fourier maps and their positions were refined with a restraint on distances. Hydrogen atoms bonded to carbon were kept in their theoretical positions. For all hydrogen atoms, the isotropic ADP was calculated as 1.2 multiple of Ueq of the parent atom.
The structures of SLT phase 1 and phase 4 (tetrahydrate) are disordered. In the case of phase 1, the fluoro substituted phenyl rings are disordered by a 180 degree rotation around the C–C bond connecting the ring to the aliphatic moiety. The disorder was described using the rotation of the rigid body. In the case of phase 4, the CF3 groups exhibit strong disorder. See the ESI† for details.
Structure refinement from powder data. The sample was ground and placed into a 0.3 mm borosilicate glass capillary and was measured at room temperature in transmission mode using a PANanalytical Empyrean powder diffractometer from 4° to 80° 2θ with CuKα radiation from a sealed tube (λ = 1.5418 Å, focusing mirror, the step size was 0.013° 2θ).
The details of the refinement can be found in the ESI.†
3. Results and discussion
The water content of the samples was determined by Karl Fischer titrimetry under ambient humidity and temperature. In general, the samples have 1.7% w/w water content (Table 1), but in a few cases slightly different values were observed. Headspace gas chromatography was applied for identification and quantification of residual solvents, but organic solvent contents were all below the detection limit.| Sample | Phase 1 | Phase 2 | Phase 3 | Phase M |
|---|---|---|---|---|
| Water content | 1.7–1.9% | 1.6–1.7% | 1.7–1.9% | 1.7–1.9% |
| TG weight loss | 1.25% | 1.27% | 1.5% | 1.2% |
| DSC solvent loss range | 29–115 °C | 29–112 °C | 28–102 °C | 28–109 °C |
| Melting point | T peak = 202.4 °C | T peak = 197.9 °C | T peak = 196.4 °C | T peak = 202.1 °C |
The water content was used to calculate the hydrate stoichiometry of the phases as given by eqn (1):
Hydrate stoichiometry
 | (1) |
The stoichiometric water content indicates site-specific solvent incorporation.
The thermoanalytical parameters of the SLT hydrates are summarized in Table 1. Their DSC curves demonstrate some of the general characteristics of channel hydrate dehydration28 as shown in Fig. 2. The water located in the channels begins to evolve at relatively low temperature and is being released continuously at temperatures up to 115 °C in correlation with the one-step, gradual weight loss process in the TG curve (Fig. 3). The dehydrated materials are thermally stable up to the melting onset, and above this temperature, the materials lose their weight sharply while the compounds decompose. The observed endothermic event following the melting peak indicates that decomposition of the compound took place. Infrared detection of the evolved gases revealed the presence of ammonia and carbon dioxide.
 | ||
| Fig. 2 Comparison of the DSC scans of SLT phase 1, phase 2, phase 3 and phase M measured at 10 °C min−1. | ||
 | ||
| Fig. 3 Overlay of the TG curves of the SLT hydrates measured at 10 °C min−1. | ||
The thermal deamination of sitagliptin was investigated by Sonune et al.,29 and the thermal decomposition of a pharmaceutical L-tartrate salt is reported by Murphy et al. and Brahadeeswaran et al.22,30 In both cases, thermal decomposition took place above the melting onset. In Sonune's study, the mass of the thermal degradation products indicated the loss of mass (17; NH3+) from the parent drug molecule. In their study, the 1H NMR spectra of the degradation products also confirmed the deamination of the sitagliptin molecule.
Dynamic vapour sorption
Vapour sorption analysis can provide information on the order of the water molecules incorporated in the crystal lattice and on the hydration/dehydration behaviour of the phases. Sorption isotherms need to be tied to the mechanisms by which water is taken up to understand the way of water inclusion and its impact on the solid form.For ease of interpretation, isotherms are often identified by Brunauer's classifications.31 All SLT hydrates fall under the type I category, which is typical of very hygroscopic materials. This kind of curve indicates localized water in the lattice.32
Examples of type I adsorption have been frequently reported in the literature: paroxetine HCl,33 cromolyn disodium salt,34 azithromycin,35 celiprolol HCl, cefaclor, spirapril HCl,32 erythromycin A,36 and donepezil HCl monohydrate.37
The isotherms of phase 1 (Fig. 4), phase 2 (Fig. 5) and phase M (Fig. 6) show significant uptake at low partial pressures. Approximately 80% of the total water uptake takes place between 0 and 20% RH indicating strong lattice binding. The initial part of the curve is very steep implying that the interactions with the firstly adsorbed molecules (∼1.2% w/w) are stronger; therefore, these molecules should be more localized. These water molecules do not play only a space-filling role and the water–solid interactions are expected to be relatively strong.38
 | ||
| Fig. 4 DVS isotherm of SLT phase 1. | ||
 | ||
| Fig. 5 DVS isotherm of SLT phase 2. | ||
 | ||
| Fig. 6 DVS isotherm of SLT phase M. | ||
This substantial mass gain is followed by slight but progressive sorption at intermediate and high vapour concentrations. The subsequently absorbed molecules are more mobile and less firmly held in the crystal lattice. On the basis of the weight change of ∼1.7% in the moisture sorption isotherm, the phases contain 0.5 molar equivalent of water when they are fully hydrated at 90% RH. The isotherms present only one plateau, corresponding to one stable hydration state.
The sorption and desorption isotherms coincide, and hysteresis is not observed in the case of phase 1, phase 2 and phase M. This confirms that these hydrates do not undergo phase transformation during the analysis, and the water uptake and loss are completely reversible. It was found that these phases remain stable across a wide range of humidity levels: 10–90% RH. Dehydration takes place at a very low RH level (<10%) in a reversible reaction as indicated by the superimposable profiles. Examination by optical microscopy showed that the hydrate crystals exhibit no morphological change upon dehydration.
The DVS isotherm of phase 3 (Fig. 7) differs from those of the above described phases. The step shaped curve indicates that until 20% RH the hemihydrate structure is created. At low relative humidity levels, the sorption branch does not agree with the desorption branch; a hysteresis is observed. It is followed by slight but progressive adsorption at intermediate vapour concentrations, and above 70% RH, another loop is observed. A total of 2.3% weight gain was observed during the adsorption process, which is consistent with approximately 0.7 molar equivalent of water (135% of the hemihydrate equivalent).
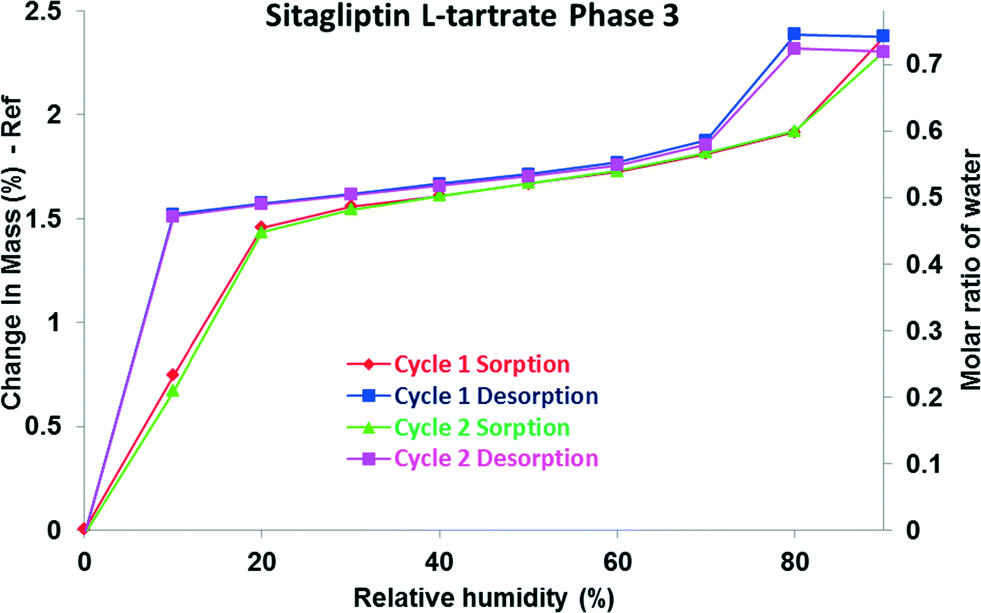 | ||
| Fig. 7 DVS isotherm of SLT phase 3. | ||
There can be more reasons for the occurrence of this hysteresis. First of all, it is possible to obtain isotherm data which appear to show hysteresis by failing to allow a sample to equilibrate. Hysteresis can also occur due to swelling effects.39 The presence of hysteresis between the sorption and desorption isotherms indicates that the diffusion of water molecules out from the structure is slower as the lattice swells upon hydration.
The additional uptake and the hysteresis could be explained by the porous structure of phase 3. A probable explanation is that it may be based on capillary condensation in the mesoporous structure. Materials, whose structures contain mesopores, show a rapid uptake of water at higher humidity. The absorbed water facilitates capillary condensation, and the subsequent water release on decreasing humidity occurs more slowly resulting in hysteresis.40
An empirical classification of hysteresis loops given by IUPAC correlates the hysteresis loops (types H1–H4) with the texture of the adsorbent. Materials that give rise to this kind of hysteresis observed in the case of phase 3 (type H2) contain complex – not uniform – pore networks consisting of pores with ill-defined shape and wide pore size distribution, also referred as ink-bottle pores.41 This type of hysteresis is observed if along with delayed condensation, pore blocking/percolation effects are present.42 In the desorption branch, the water molecules do not tend to leave the pores, and a higher driving force is required to pull the adsorbed molecules out of the structure. The loop between the branches is characteristic of the distributions of the pore size and of the neck size, respectively.43 Thus, the hysteresis observed might reflect the hindered release of the water molecules from the pores. The water uptake and loss occur in a completely reversible process evidenced by the superimposable cycles.
Variable humidity X-ray powder diffraction
Variable humidity X-ray powder diffraction is becoming a generally applied technique for relative stability studies.44 The VH-XRPD results of the phases are given in Fig. 8–11. The data obtained by VH-XRPD measurements are in accordance with the data obtained by DVS. The water uptake and loss are completely reversible whether one proceeds from high to low relative humidity or vice versa. | ||
| Fig. 8 VH-XRPD patterns of phase 1 at various relative humidity values. | ||
 | ||
| Fig. 9 VH-XRPD patterns of phase 2 at various relative humidity values. | ||
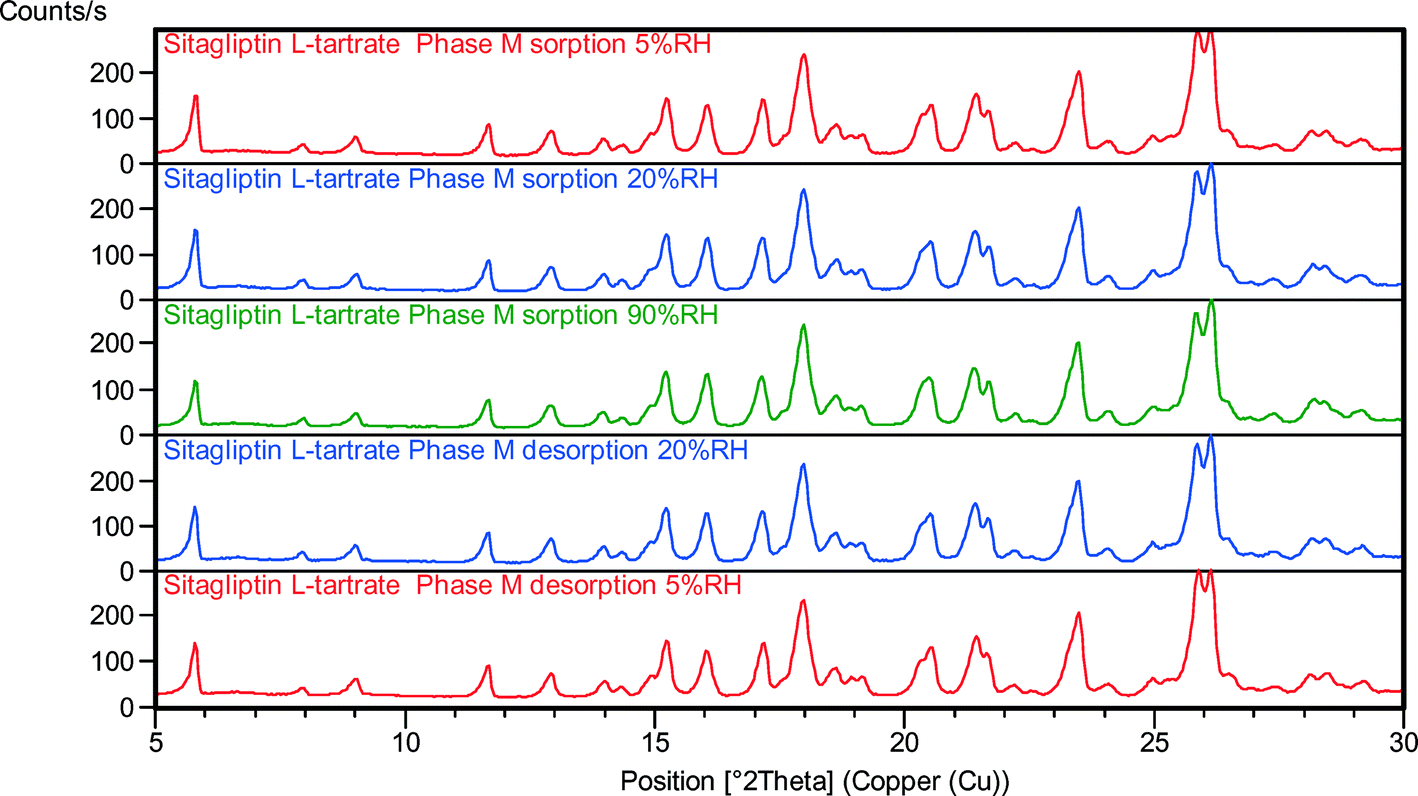 | ||
| Fig. 10 VH-XRPD patterns of phase M at various relative humidity values. | ||
 | ||
| Fig. 11 VH-XRPD patterns of phase 3 at various relative humidity values. | ||
During the desorption in the humidity chamber, the structures of SLT phase 1 (Fig. 8), phase 2 (Fig. 9), phase M (Fig. 10) and phase 3 (Fig. 11) have not changed, and a so-called isomorphic dehydrate formed without significantly altering the structure. The existence of an isostructural dehydrate is a typical characteristic of non-stoichiometric hydrates. The fact that water uptake and loss are unhindered proves that water is located along channels in the lattice. It is apparent that RH has no significant impact on the crystal structure, and no irreversible effect is involved which is in correlation with the lack of hysteresis in the sorption isotherms.
The structures of the parent hydrates are retained, and the crystalline lattices do not collapse. The crystal forms are stable regardless of the RH level. In the case of phase 1 and phase 2, the expansion of the unit cell caused the reflections to shift toward lower angles in the sorption curve, while during the desorption cycle only the phase 2 peaks were shifted toward higher angles (Fig. 9) indicating smaller interplanar distances. In the case of phase 1, the expanded lattice seems to be maintained even in the desorption branch. The crystals swell in the humidity chamber during water uptake, but the shrinkage of the material is less efficient which could result in a pseudo-sustained expanded state (Fig. 8). The major difference can be found in the expansion of the channel diameter, and a significant shift in the 2θ values was observed in the case of phase 1. In the case of phase 2, it is less significant but detectable, and in the case of phase M and phase 3, the expansion and contraction of the unit cell are not notable.
The ease with which the water molecules can enter and exit the crystal lattice is also evidenced by the superimposable sorption–desorption cycles in the DVS isotherm and explains why hydration/dehydration occurs without a major change in the crystal structure.
Their VH-XRPD behavior in accordance with crystal data and solid-state characterization demonstrates that SLT hydrates are channel hydrates containing water in tunnel-like arrangement in the crystal lattice. Water can reversibly get in and out of the crystal lattice as a function of temperature and RH. Therefore, their crystallographic order is retained even under very anhydrous conditions.
The results indicated that SLT phase 1 and phase 2 can be classified as a class C water type where desorption and adsorption cause only lattice contraction and/or expansion, according to the classification advocated by Mimura et al.45 Phase M and phase 3 can be classified as a class D water type where desorption and/or adsorption take place without significant changes in the crystal lattice.
When the water is removed, the crystal lattices retain their three-dimensional order, and the structures created are in a high energy state relative to the original hydrated structure. This dehydrated lattice can reduce its internal energy by resorbing moisture from the surrounding atmosphere. The isostructural dehydrated forms of the SLT phases are extremely hygroscopic as rehydration occurs on exposure to ambient humidity. It is also manifested in the very steep, nearly vertical part of the sorption isotherms in the range of 0–20% RH.
Our results show that SLT phases have a fixed API framework, and the inclusion and the removal of the water molecules do not affect the crystal lattice significantly. This was confirmed by removing the samples from the DVS instrument after the experiments and obtaining powder X-ray diffraction patterns, which matched the reference, hydrated patterns.
Considering non-stoichiometric hydrates, the lack of a stable anhydrous form is anticipated according to the Gibbs phase rule. Since there is a continuous change in the solid phase composition with partial vapour pressure, the number of solid phases is necessarily one.32,46 Hence, the hydration and dehydration of non-stoichiometric hydrates are not accompanied by a phase transition, and all hydration states represent the same crystal form.
The terminology of crystalline solids that contain a solvent as a second component is not uniform and very confusing.6 Sometimes the behaviour of the API does not allow a clear classification. The classification's main advantage is that it can imply practical consequences. Non-stoichiometric hydration is generally undesirable, but the term non-stoichiometric hydrate does not necessarily reflect continuously changing water content. In exceptional cases, like this, however the hydrates are classified as non-stoichiometric hydrates, and still they can act as a suitable candidate for further development.
Crystal structure
Crystal structure determinations were performed to confirm the assumptions derived from the previous examinations. Single crystals of phase 1 and phase 2 were successfully prepared by slow evaporation. Phase 1 was obtained from pure water, while the slow evaporation of the solvent mixture of methanol and water with a volumetric ratio of 84:16 resulted in phase 2 crystals. The identity of the phases was verified by the comparison of the calculated powder pattern to the experimental powder pattern (see the ESI†). In the case of phase M, despite the extensive efforts to determine its structure (see the ESI†) only the crystallographic parameters of the unit cell were resolved by X-ray powder diffraction indexation by the Le Bail refinement. The comparison of the powder patterns of the hydrates is illustrated in Fig. 12, and the summary of the crystal structure data of the four SLT crystal forms is given in Table 2.
 | ||
| Fig. 12 XRPD patterns of the SLT hydrates. | ||
| Name | Sitagliptin L-tartrate phase 1 | Sitagliptin L-tartrate phase 2 | Sitagliptin L-tartrate phase M | Sitagliptin L-tartrate phase 4 |
|---|---|---|---|---|
| Formula | C40H42F12N10O14·1H2O | C20H21F6N5O7·0.5H2O | C20H21F6N5O7·0.5H2O | C20H21F6N5O7·4H2O |
| Temperature (K) | 120 | 293 | 293 | 120 |
| Cell parameters (Å) | a = 7.2124 | a = 7.21076(11) | a = 7.19853(17) | a = 45.2368(4) |
| b = 29.0912 | b = 11.66321(14) | b = 11.6983(3) | b = 8.2403(4) | |
| c = 11.686 | c = 15.64522(12) | c = 15.4294(3) | c = 7.1817(18) | |
| Angles (°) | α = 90 | α = 99.3299(18) | α = 97.697(3) | α = 90 |
| β = 106.181 | β = 98.8646(13) | β = 95.548(6) | β = 95.326(4) | |
| γ = 90 | γ = 105.9897(12) | γ = 106.831(5) | γ = 90 | |
| Volume (unit cell) (Å3) | 2354.8 | 1220.43 | 1219.04 | 2665.53 |
| Volume (asymmetric unit) (Å3) | 588.7 | 610.215 | 609.52 | 666.38 |
| Density (g cm−3) | 1.598 | 1.517 | — | 1.555 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Monoclinic |
| Space group | P21 | P1 | P1 | C2 |
| R-factor (%) | 4.38 | 5.83 | — | 6.63 |
| Z | 2 | 1 | — | 4 |
The structures of phase 1 and phase 2 are analogous. Even though the unit cell of phase 1 has a double volume, the unit cell parameters are similar (it is more obvious after applying the transformation matrix [1 0 0; 0 0 1; 0–0.5 0] to the direct space unit cell vectors (columns) of phase 2, as well as from the conformation of the API which is almost identical (Fig. 15)). Since the packing of the API molecules is different, they represent an example of packing polymorphism (Fig. 13).
 | ||
| Fig. 13 Packing motif of SLT phase 1 (left) and phase 2 (right). Water–O atoms are represented in space-fill style. | ||
Although the structure of phase M was not elucidated, the similarity between phase 2 and phase M is obvious based on their same space group and comparable unit cell size. All these three forms, phase 1, phase 2 and phase M, demonstrate a unique crystal structure, and the volumes of the asymmetric units are comparable. It seems a reasonable assumption that the conformation of the sitagliptin molecule in phase M is similar to those in phase 1 and phase 2, as well.
Phase 1 belongs to the monoclinic crystal system, P21 space group, while phase 2 and phase M belong to the triclinic crystal system, P1 space group. The asymmetric units of phase 1 and phase 2 contain two molecules of sitagliptinium cation, two molecules of hydrogen tartrate ion and one molecule of water. The asymmetric unit of phase 4 contains one molecule of sitagliptinium cation, one molecule of hydrogen tartrate ion, one molecule of firmly attached water and some additional water constituting the channel across the structure. This phase exhibits disorder in the trifluoromethyl moiety (Fig. 14).
 | ||
| Fig. 14 Packing motif of SLT phase 4. Channel water–O atoms are represented in space-fill style. | ||
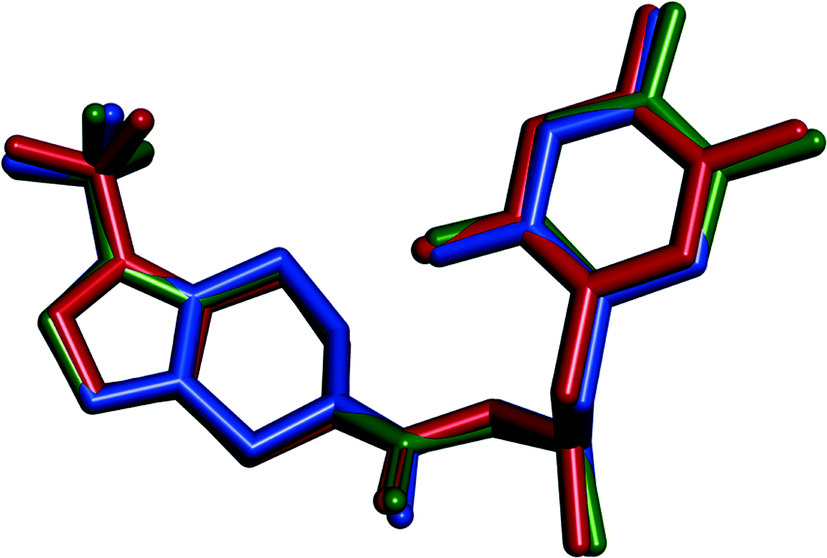 | ||
| Fig. 15 Overlay of sitagliptin molecules in the crystal structures of SLT phase 1 (blue), phase 2 (green) and phase 4 (red). Hydrogen atoms are omitted. | ||
The calculated density of phase 1, 1.598 g cm−3, is significantly higher than that of phase 2, 1.517 g cm−3. Thus, this density difference suggests stronger intermolecular interactions taking place in phase 1, which confirms the higher stability of this form at 0 K. Since the forms differ in the packing of identical molecular units, we can expect that the density rule47 holds. Exceptions to this rule are mainly observed in pairs of polymorphs with conformationally distinct molecules in the asymmetric unit or strong differences in hydrogen bonding.48
The crystal structures show that in all solid forms infinite sheets are formed by hydrogen tartrate anions linked by hydrogen bonds. The anions display a zigzag configuration with a C–C–C–C torsion angle close to 180°. The hydrogen tartrate molecules are bonded into an infinite chain through a head-to-tail arrangement by strong hydrogen bonds between carboxyl and carboxylate groups (see Fig. S6–7 in the ESI†). The neighbouring chains are then crosslinked by hydrogen bonds to generate a two-dimensional framework.
The cation is engaged in intermolecular N–HO hydrogen bonds with the surrounding hydrogen tartrate ions and O–HO interaction with the water molecule to yield a complex, hydrogen-bonded structure. In all structures, the API molecules are bent in a way that the 2,4,5-trifluorophenyl moiety and the trifluoromethyl group of the triazolopiperazine moiety are oriented towards a lipophilic central core region, while the polar groups are oriented towards the hydrogen tartrate and water molecules. In the structure of phase 1, the packing of the molecules allows the phenyl rings to form π–π-stacking, where interacting rings are tilted by an angle of 24°.
Hydrogen bonded water molecules lie between the hydrogen tartrate anionic sheets, but the incorporation of the water molecule O(W) is not identical in phase 1, phase 2 and phase 4.
In the structure of phase 1, the coordinated water molecule O(W) is involved in hydrogen bonding interactions with both carboxylate and hydroxyl oxygens with the hydrogen tartrates and with the carbonyl group of the cation in a tetrahedral arrangement. In the structures of SLT phase 2 and phase 4, the water molecules are held by two hydrogen tartrate ions and by the carbonyl group of the API in a trigonal arrangement.
In all refined structures, the O⋯O distance between adjacent water molecules along the water channel is 7.21 Å, which is clearly too long to represent hydrogen bonding between neighbouring water molecules.
The structures confirm the findings of the solid state analytical techniques about their channel hydrate structures. Referring to Fig. 13 and 14, it is easy to understand that water has an easy exit from the channel type hydrates, and water can simply enter and exit through the tunnels it occupies.
The water molecules stabilize the structures by providing additional hydrogen bonds between the cation and the anion, explaining the observed high affinity to water.
The forms phase 1, phase 2, phase 3 and phase M are all non-stoichiometric channel hydrates exhibiting similar water content close to the hemihydrate equivalent. The term polymorphism is used to describe different crystal forms of exactly the same chemical composition42 and the crystallographic results revealed that phase 1 and phase 2 are true, packing polymorphs.
The structure of phase 4 confirms that the higher the crystallization temperature of the modification, the more closed is the space occupied by guest molecules.49 Since phase 4 crystallized at ∼70 °C lower temperature than the other forms, it exhibits significantly wider channels. One molecule of water is firmly held by strong hydrogen bonds by the carbonyl group of the sitagliptinium cation and by other two hydrogen bonds with the tartrate ions. Three other water molecules are located in a wide channel and are not accurately positioned (Fig. 14).
Conclusion
In this work, we present five hydrated forms of the L-tartrate salt of the pharmaceutical compound sitagliptin. Four of the hydrates contain 0.5 equivalent of water per molecule of API, and their hydration/dehydration behaviour was studied by thermal methods (DSC and TGA), dynamic vapour sorption analysis (DVS), variable humidity X-ray powder diffraction (VH-XRPD) and X-ray crystallography aiming at understanding the role of water molecules in the structures. Our most interesting finding lies in the contradiction between the stoichiometric and non-stoichiometric characteristics of the studied hydrates.As defined,5,6,25 the stoichiometric hydrates have a fixed API/water ratio and the water molecules are strongly bound in the crystalline lattice (e.g. by H-bonds). Therefore, stoichiometric hydrates have to undergo a phase transformation upon dehydration.
On the other hand, generally, the non-stoichiometric hydrates have a variable water content and exhibit reversible dehydration without any significant change in the crystal structure (as evidenced, for example, by the lack of hysteresis in the sorption isotherm).
In this particular case, the more or less fixed API/water ratio under ambient conditions corresponds to stoichiometric behaviour. This is further supported by the fact that the water molecules are held by H-bonds in the crystal structures and stabilize them by providing additional interactions between the sitagliptinium cations and the hydrogen tartrate anions. This interaction substantiates the high affinity of water molecules to the API framework.
On the contrary, their non-stoichiometric channel-like character is evidenced by the VH-XRPD results and the superimposable sorption/desorption curves.
The final deciding factor, in our opinion, is the existence of isostructural dehydrates and the non-destructive process of dehydration. It is an unambiguous proof of the non-stoichiometric nature of the studied hydrates of sitagliptin L-tartrate.
Abbreviations
| SLT | Sitagliptin L-tartrate |
| API | Active pharmaceutical ingredient |
| DPP-4 | Dipeptidyl peptidase-4 |
| DSC | Differential scanning calorimetry |
| TG | Thermogravimetry |
| VH-XRPD | Variable humidity X-ray powder diffraction |
| DVS | Dynamic vapour sorption |
| RH | Relative humidity |
| ADP | Anisotropic displacement parameters |
| NMR | Nuclear magnetic resonance spectroscopy |
| IUPAC | International Union of Pure and Applied Chemistry |
| CSD | Cambridge Structural Database |
Acknowledgements
We would like to express sincere thanks to the Development Department, Zentiva k. s., Prague for giving us the chance to complete this research. We would also like to express our gratitude to Tomáš Gurgut, Márton Harsányi, Ondřej Dammer and Lukáš Krejčík for their analytical support. The authors thank Lukáš Palatinus for devoting his time and effort in examining the structure of phase M. This work was supported by the Grant Agency of Czech Republic (grant no. 106/14/03636S). The equipment for X-ray diffraction has been supported by Project CZ.2.16/3.1.00/24510 within the EU Operation program Prague Competitiveness.References
-
J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, 2008 Search PubMed
.
-
W. C. McCrone, Polymorphism in Physics and Chemistry of the Organic Solid State, ed. D. Fox, M. M Labes and A. Weissberger, Interscience, New York, 1965, vol. II, pp. 728–731 Search PubMed
- T. Threlfall, Analysis of organic polymorphs. A review, Analyst, 1995, 120, 2435 RSC
- D. Giron, Investigations of Polymorphism and Pseudo-polymorphism in Pharmaceuticals by Combined Thermoanalytical Techniques, J. Therm. Anal. Calorim., 2001, 64(1), 37–60 CrossRef CAS
- G. R. Desiraju, Counterpoint: What's in a name?, Cryst. Growth Des., 2004, 4, 1089–1090 CAS
-
U. Griesser, The Importance of Solvates, in Polymorphism in the Pharmaceutical Industry, ed. R. Hilfiker, Wiley-VCH, Weinheim, 2006, pp. 211–234 Search PubMed
- A. Nangia, Pseudopolymorph: Retain This Widely Accepted Term, Cryst. Growth Des., 2006, 6, 2–4 CAS
-
H. G. Brittain, Polymorphism in Pharmaceutical Solids, Taylor & Francis, 2nd edn, 2009 Search PubMed
- K. R. Seddon, Pseudopolymorphism: A polemic, Cryst. Growth Des., 2004, 4, 1087 CAS
- J. Bernstein, And Another Comment on Pseudopolymorphism, Cryst. Growth Des., 2005, 5, 1661–1662 CAS
- R. K. Khankari and D. J. W. Grant, Pharmaceutical hydrates, Thermochim. Acta, 1995, 248, 61–79 CrossRef CAS
-
H. G. Brittain, Polymorphism in Pharmaceutical Solids, Taylor & Francis, 2nd edn, 2009 Search PubMed
- ICH Q3C Impurities: Guideline for Residual Solvents, International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva, Switzerland, 1997 Search PubMed.
-
H. G. Brittain, Polymorphism in Pharmaceutical Solids first Edition, Taylor & Francis, 1999 Search PubMed
- S. R. Vippagunta, H. G. Brittain and D. J. W. Grant, Crystalline solids, Adv. Drug Delivery Rev., 2001, 48, 3–26 CrossRef CAS PubMed
- K. Fucke and J. W. Steed, X-ray and Neutron Diffraction in the Study of Organic Crystalline Hydrates, Water, 2010, 2, 333–350 CrossRef CAS
-
D. C. Lee and M. L. Webb, Pharmaceutical Analysis, Wiley-Blackwell, 2009 Search PubMed
- D. E. Braun, P. G. Karamertzanis and S. L. Price, Which, if any, hydrates will crystallise? Predicting hydrate formation of two dihydroxybenzoic acids, Chem. Commun., 2011, 47, 5443–5445 RSC
- M. K. Marchewkaa, S. Debrus, A. Pietraszko, A. J. Barnes and H. Ratajczak, Crystal structure, vibrational spectra and nonlinear optical properties of L-histidinium-L-tartrate hemihydrate, J. Mol. Struct., 2003, 656, 265–273 CrossRef
-
J. Z. Gogoutas, M. F. Malley, J. D. Dimarco, S. Xiaotian, C. Wei, J. Yu, T. C. Vu, G. S. Jones and S. A. Savage, Crystal forms of saxagliptin and processes for preparing same EP 2481726 A2, Bristol-Myers Squibb Company, 2008 Search PubMed
- S. M. Reutzel and V. A. Russell, Origins of the Unusual Hygroscopicity Observed in LY297802 Tartrate, J. Pharm. Sci., 1998, 87(12), 1568–1571 CrossRef CAS PubMed
- B. J. Murphy, J. Huang, J. Casteelm, A. Cobani and J. F. Krzyzaniak, Varenicline L-tartrate Crystal Forms: Characterization Through Crystallography, Spectroscopy, and Thermodynamics, J. Pharm. Sci., 2010, 99(6), 2766–2776 CrossRef CAS PubMed
- R. C. Bott, G. Smith, D. S. Sagatys, D. E. Lynch, A. N. Reddy and H. L. Lennard, Structures of the hydrogen (+)-tartrates of sodium, thallium (I) and silver (I), Z. Kristallogr., 1994, 209, 803–807 CAS
- J. Richter, P. Lehnert, K. Jarrach, O. Dammer and L. Krejcík, Stabilní polymorf soli (2R)-4-oxo-4-[3-(trifluorometyl)-5,6-dihydro[1,2,4]triazolo[4,3-α]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorofenyl)butan-2-aminu s kyselinou L-vinnou, National Patent Application number CZ 2013/842, 01.11.2013.
- J. Richter, K. Jarrach, V. Kiss, E. Tieger, J. Havlícek and O. Dammer, Krystalická modifikace 2 L-vínanu (3R)-3-amino-1-[3-(trifluormethyl)-6,8-dihydro-5H-[1,2,4,]triazolo[4,3-a]pyrazin-7-yl]-4-(2,4,5-trifluorfenyl) butan-1-onu, Registration number UV 27898, 01.13.2015.
- J. Richter, K. Jarrach, V. Kiss, E. Tieger, J. Havlícek and O. Dammer, Krystalická modifikace 3 L-vínanu (3R)-3-amino-1-[3-(trifluormethyl)-6,8-dihydro-5H-[1,2,4]triazolo[4,3-a]pyrazin-7-yl]-4-(2,4,5-trifluorfenyl)butan-1-onu, Registration number UV 27930, 01.13.2015.
- R. Ferlita, K. Hansen, V. Vydra, C. M. Lindemann and Y. Wang, Novel crystalline salts of a dipeptidyl peptidase-iv inhibitor, International Publication Number WO 2005/072530, 11.08.2005.
-
D. Q. M. Craig and M. Reading, Thermal analysis of pharmaceuticals, CRC Press, 2007 Search PubMed
- D. P. Sonune and M. K. Mone, Isolation, characterization of degradation products of sitagliptin and development of alidated stability-indicating HPLC assay method for sitagliptin API and tablets, Int. J. Pharm. Sci. Res., 2013, 4(9), 3494–3503 Search PubMed
- S. Brahadeeswaran, V. Kannan, R. Rakhikrishna and J. Philip, Studies on thermophysical and mechanical properties of hydrazonium l-tartrate, J. Therm. Anal. Calorim., 2014, 116(1), 339–347 CrossRef
-
S. Brunauer, The Adsorption of Gases and Vapours, Princenton Unviversity Press, 1945 Search PubMed
- J. R. Authelin, Thermodynamics of non-stoichiometric pharmaceutical hydrates, Int. J. Pharm., 2005, 303(1–2), 37–63 CrossRef CAS PubMed
- M. F. Pina, J. F. Pinto, J. J. Sousa, L. Fábián, M. Zhao and D. Q. M. Craig, Identification and Characterization of Stoichiometric and Nonstoichiometric Hydrate Forms of Paroxetine HCl: Reversible Changes in Crystal Dimensions as a Function of Water Absorption, Mol. Pharmaceutics, 2012, 9(12), 3515–3525 CrossRef CAS PubMed
- G. A. Stephenson and B. A. Diseroad, Structural relationship and desolvation behavior of cromolyn, cefazolin and fenoprofen sodium hydrates, Int. J. Pharm., 2000, 198(2), 167–177 CrossRef CAS PubMed
- S. Timoumi, D. Mangin, R. Peczalski, F. Zagrouba and J. Andrieu, Stability and thermophysical properties of azithromycin dihydrate, Arabian J. Chem., 2014, 7(2), 189–195 CrossRef CAS
- K. Fujii, H. Uekusa and M. Aoki, Solid-State Hydration/Dehydration of Erythromycin A Investigated by ab Initio Powder X-ray Diffraction Analysis: Stoichiometric and Nonstoichiometric Dehydrated Hydrate, Cryst. Growth Des., 2013, 13(5), 2060–2066 CAS
- T. Liu, Y. Ran, B. Wang, W. Dong, S. Wu and J. Gong, The dehydration behavior and non-isothermal dehydration kinetics of donepezil hydrochloride monohydrate Form I, Front. Chem. Sci. Eng., 2014, 8(1), 55–63 CrossRef CAS
- H. G. Brittain, S. J. Bogdanowich, D. E. Bugay, J. DeVincentis, G. Lewen and A. W. Newman, Physical characterization of pharmaceutical solids, Pharm. Res., 1991, 8(8), 963–973 CrossRef CAS
- C. Hill, A. Norton and G. Newman, The Water Vapour Sorption Behavior of Natural Fibers, J. Appl. Polym. Sci., 2009, 112(3), 1524–1537 CrossRef CAS
-
R. L. Hassel and N. D. Desse, Characterization of Water Adsorption and Absorption in Pharmaceuticals, TA Instruments 2007, http://www.tainstruments.com.tw/library_download.aspx?file=TA338+Charact+of+Water+Adsorption+Desorption+in+Pharm.pdf, (accessed 03 August, 2014) Search PubMed
- K. Sing, D. Everett, R. Haul, L. Moscou, R. A. Piierotti, J. Rouquérol and T. Siemieniewska, Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity, Pure Appl. Chem., 1985, 57(4), 603–619 CrossRef CAS
- Physical Adsorption Characterization of Mesoporous Materials: Comments on Hysteresis Scanning By AZoM.com Staff Writers, http://www.azom.com/article.aspx?ArticleID=9439 (accessed 22, November 2014)
- A. Grosman and C. Ortega, Capillary Condensation in Porous Materials. Hysteresis and Interaction without Pore Blocking/Percolation Process, Langmuir, 2008, 24(8), 3977–3986 CrossRef CAS PubMed
- P. Lang, E. Varkonyi, J. Ulrich, P. Szabó-Révész and Z. Aigner, Analysis of the polymorph changes of a drug candidate, J. Pharm. Biomed. Anal., 2015, 102, 229–235 CrossRef CAS PubMed
- H. Mimura, S. Kitamura, T. Kitagawa and S. Kohda, Characterization of the non-stoichiometric and isomorphic hydration and solvation in FK041 clathrate, Colloids Surf., B, 2002, 26(4), 397–406 CrossRef CAS
-
S. R. Byrn, R. R. Pfeiffer and J. G. Stowell, Solid State Chemistry of Drugs, SSCI Inc., 2nd edn, 1999 Search PubMed
- A. Burger and R. Ramberger, On the polymorphism of pharmaceuticals and other molecular crystals. II, Microchim. Acta, 1979, 72(3–4), 273–316 CrossRef
- D. E. Braun, T. Gelbrich, V. Kahlenberg, G. Laus, J. Weiser and U. J. Griesser, Packing polymorphism of a conformationally flexible molecule (aprepitant), New J. Chem., 2008, 32(10), 1677–1685 RSC
- B. Ibragimov, A Simple Correlation between the Structures of Different Crystal Modifications of a Given Host–Guest Complex and their Crystallization Temperatures, J. Inclusion Phenom. Macrocyclic Chem., 1999, 34(3), 345–353 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1439802–1439804. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce00322b |
| This journal is © The Royal Society of Chemistry 2016 |