
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion-templated 2D frameworks from hexahydroxytriphenylene†
Mahbod
Morshedi
,
Anthony C.
Willis
and
Nicholas G.
White
*
Research School of Chemistry, The Australian National University, 137 Sullivan's Creek Road, Acton, 2601, ACT, Australia. E-mail: nicholas.white@anu.edu.au; Web: www.nwhitegroup.com
First published on 6th April 2016
Abstract
Hexahydroxytriphenylene (HHTP) forms 2D frameworks through O–H⋯anion hydrogen bonds with a range of anions. In all cases, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 co-crystals of HHTP and tetraalkylammonium·anion salts are obtained, which have a layered 2D structure. When HSO4− was used as anion, an unprecedented methylation reaction was observed giving crystals containing the methylsulfate anion (MeOSO3−).
:1 co-crystals of HHTP and tetraalkylammonium·anion salts are obtained, which have a layered 2D structure. When HSO4− was used as anion, an unprecedented methylation reaction was observed giving crystals containing the methylsulfate anion (MeOSO3−).
The vast majority of research into the supramolecular chemistry of anions has focused on their solution phase behaviour spurred by the key role negatively-charged species play in a range of biological and environmental processes.1 However recently, a handful of reports have demonstrated that anions can also be used to template the formation of self-assembled structures.2
While the interaction of anions with O–H hydrogen bond donors has received comparatively little attention, the groups of Smith,3 and Kass and Wang,4 have demonstrated that receptors containing these groups can be potent anion hosts. Very recently, White and MacLachlan have demonstrated that O–H⋯anion hydrogen bonds can also be used to form extended structures including 1D polymers and nanotubes.5
Hexahydroxytriphenylene (HHTP, Scheme 1) is an important precursor for a range of supramolecular framework structures. When reacted with transition metals in the presence of base, HHTP gives two or three-dimensional metal organic frameworks (MOFs) containing the sextuply-deprotonated form of the ligand [i.e. triphenylene-hexakis(olate)].6 Some of these MOFs show very high electrical or proton conductivity. Additionally, HHTP has been used to form highly stable covalent organic frameworks (COFs) with boronate derivatives, which show impressive gas storage properties.7
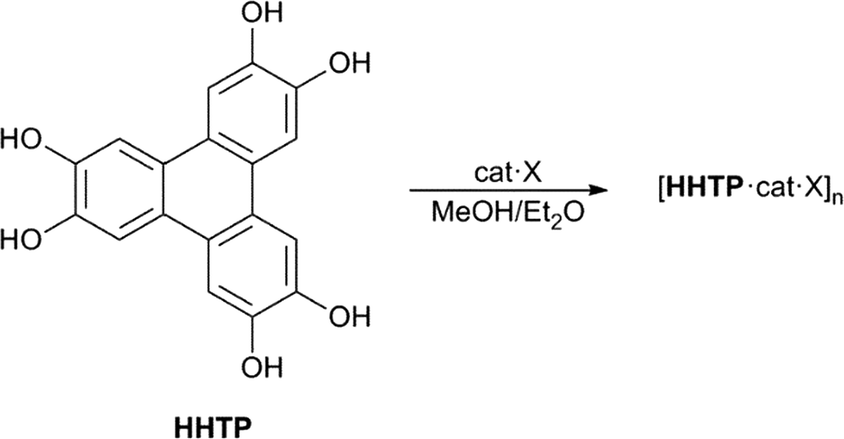 | ||
| Scheme 1 Structure of HHTP and its assembly into anion-templated 2D polymeric structures (cat = tetraalkylammonium cation, X = anion). | ||
Herein, we show that O–H⋯anion hydrogen bonding from the six hydroxyl groups of HHTP can be used to prepare 2D layered structures. The complexes form sheets with one sheet comprised of anions and HHTP ligands, and the second made up of cations and solvent molecules.
We initially tried to prepare HHTP directly from catechol using ammonium persulfate as oxidant,8 but recovered only unreacted catechol. Instead we found that high purity HHTP could be readily prepared by FeCl3-mediated oxidation of 1,2-dimethoxybenzene (veratrole),9 followed by demethylation using BBr3 (see ESI†).
We attempted to determine if HHTP binds anions in solution using 1H NMR spectroscopy, but were unable to obtain quantitative data. We have found HHTP only shows solubility in acetone or polar solvents (alcohols, DMSO), however in polar solvents, solution binding is too weak to be observed, while addition of anions to HHTP in acetone causes precipitation. Previous studies have shown that the interactions between catechol3 or catechol-containing5b molecules and anions are modest in acetonitrile, so we suggest that more competitive solvents such as DMSO or alcohols disrupt the interactions between HHTP and anions allowing them to remain soluble. Less competitive acetone presumably does not do this, and therefore some kind of oligomeric or polymeric HHTP·anion complex forms and precipitates from solution.
We then investigated the crystallisation of HHTP with anions: dissolving 1:1 stoichiometric mixtures of HHTP and TBA salts or TEA·Cl in methanol gave clear solutions, and vapour diffusion of diethyl ether into these solutions gave single crystals when X = Cl−, I−, OAc− or H2PO4− and twinned crystals when X = HSO4−. When X = Br−, crystals were obtained but diffraction studies show that these appear to be modulated (see ESI†).
Single crystal X-ray diffraction‡ (SCXRD) studies revealed that all of the crystals had the formula [HHTP·X·alkylammonium salt]n and contained very similar supramolecular structures (when we started with TBA·HSO4, we obtained crystals containing the methylated anion MeOSO3−, see later). In all crystals, HHTP and the anion form a 2D sheet architecture, which is assembled through O–H⋯anion hydrogen bonding between HHTP's O–H groups and the anion, as well as intermolecular O–H⋯O hydrogen bonds between adjacent HHTP molecules. This 2D sheet alternates with layers comprised of the alkylammonium cations (these various levels of supramolecular assembly are shown in Fig. 1 for the structure of [HHTP·TBA·Cl]n; full diagrams of all of the complexes are provided in the ESI†).
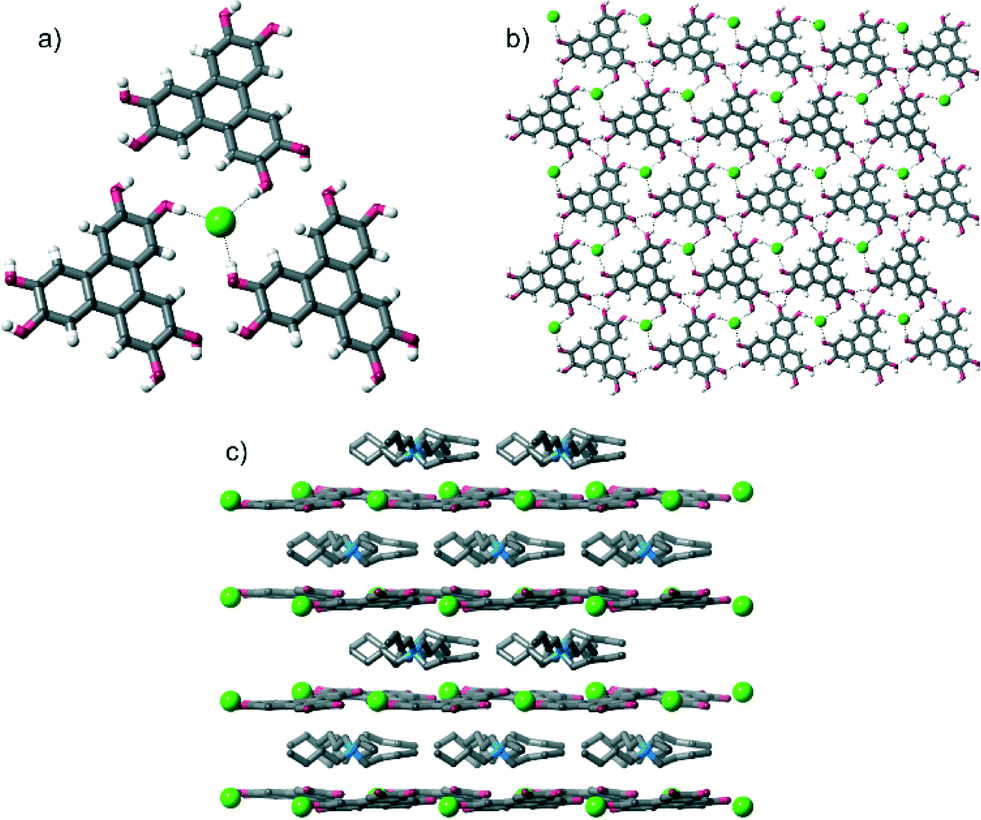 | ||
| Fig. 1 Views of the solid state structure of [HHTP·TBA·Cl]n: a) environment around Cl− anion; b) 2D sheet formed from HHTP molecules and anions; c) packing viewed along c axis (with hydrogen atoms omitted for clarity). TBA cation disorder is omitted for clarity. | ||
These O–H⋯anion hydrogen bonds are typically short: with H⋯X distances as little as 1.74 Å, 64%vdW (where %vdW refers to the distance as a percentage of the sum of the van der Waals radii10 of hydrogen and the interacting atom on the anion§). Generally the H⋯X distances expressed as %vdW increase slightly with decreasing anion basicity, so that the mean H⋯X bond length is 67%vdW for OAc−, 68%vdW for H2PO4−, 71%vdW for MeOSO3−, 77%vdW for Cl− and 82%vdW for I−. The intermolecular O–H⋯O hydrogen bonds are comparable in length in all structures, and are relatively short, H⋯O distances: 1.91–2.13 Å, 70–79%vdWH,X.
More specifically, in the three complexes containing halide anions, [HHTP·TBA·Cl]n, [HHTP·TBA·I]n and [HHTP·TEA·Cl]n, each anion receives three O–H hydrogen bonds from three different HHTP molecules (Fig. 2a). In [HHTP·TBA·H2PO4]n (Fig. 2b) and [HHTP·TBA·MeOSO3]n (Fig. 2c), a similar structure is observed with the larger oxoanions again sitting between three HHTP molecules. In the crystals containing MeOSO3−, the anion receives one hydrogen bond from each of the three HHTP moieties, while in the H2PO4− structure, two HHTP molecules and a methanol solvate act as O–H donors to the anion, which in turn donates two hydrogen bonds to HHTP. The structure of [HHTP·TBA·OAc]n (Fig. 2d) is slightly different with the acetate anion and a methanol solvent molecule located in a pocket between four HHTP molecules. In this pocket, the acetate anion receives hydrogen bonds from two of these molecules, as well as the methanol solvent, while the other two HHTP groups in this pocket donate H-bonds to the methanol molecule.
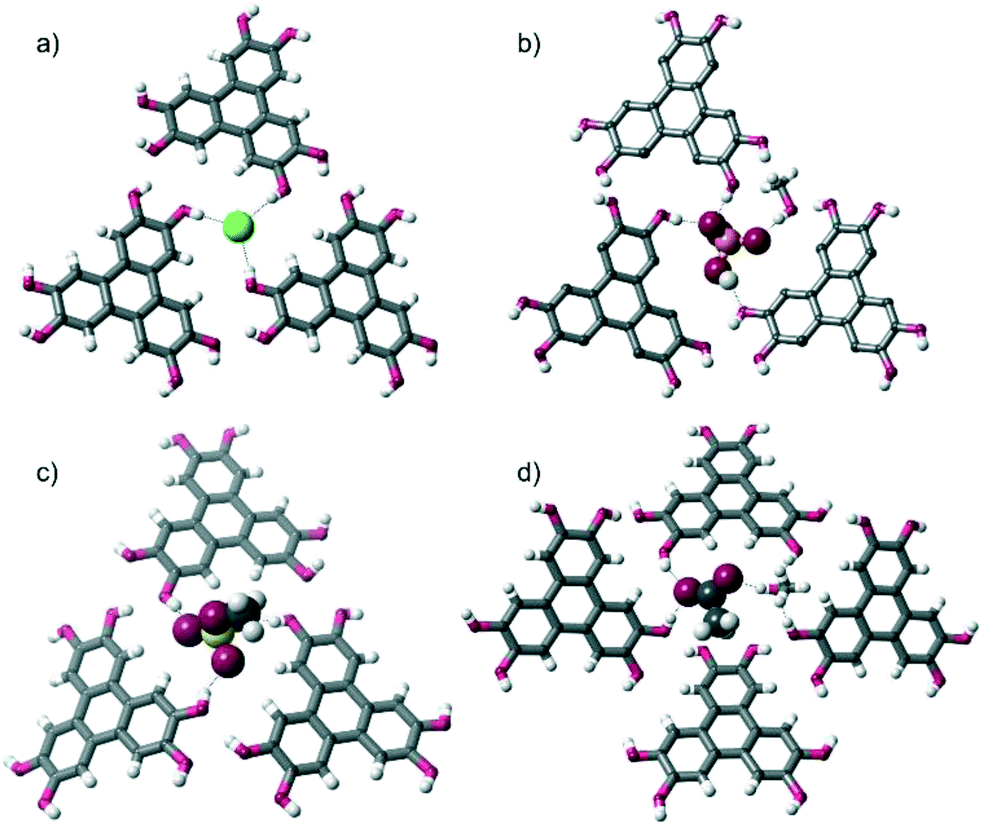 | ||
| Fig. 2 Comparison of the anion environments in solid state structures of [HHTP·TBA·X]n: a) [HHTP·TBA·Cl]n (the anion environments in [HHTP·TBA·I]nand [HHTP·TEA·Cl]nare very similar); b) [HHTP·TBA·H2PO4]n; c) [HHTP·TBA·MeOSO3]n; d) [HHTP·TBA·OAc]n. | ||
Despite these differing HHTP⋯anion bonding arrangements, 2D sheets are formed in each case. In the structure of [HHTP·TBA·Cl]n (Fig. 1), these sheets are close to planar (the angle between the mean plane of the HHTP moieties is 9°), while in the other TBA-containing structures they show varying degrees of corrugation (angles between mean planes range from 27–40°, see Fig. S13–S18 of the ESI†). In the structure containing the TEA cation, the corrugation is even more pronounced (angle between mean planes = 50°). Presumably this buckling, or lack of it, occurs to maintain favourable hydrogen bonding interactions between HHTP and the anion, and between adjacent HHTP molecules, while also accommodating the differently-sized anions and cations as well as the solvent or absence of it.
It is interesting that the same gross structure is observed when TEA·Cl or TBA·Cl is used; this is in contrast to related systems prepared from tetrahydroxytriptycene and anions, in which markedly different structures were obtained when the cation was changed from TBA to TEA.5a In the structure of [HHTP·TEA·Cl]n, substantial solvent is present, located within the TEA cation layer, so it would appear that the crystal compensates for the smaller cation by filling the empty space with solvent molecules. To investigate this further, we also attempted to crystallise HHTP in the presence of TEA·Br and TEA·I, but did not obtain any singly crystalline material containing HHTP.11 While we cannot be certain, we hypothesise that a range of factors determines whether framework crystallisation is favourable, and that the comparatively strong O–H⋯Cl− interactions are sufficiently strong to overcome the otherwise unfavourable crystallisation with TEA cations and associated solvents. Conversely, the O–H⋯anion interactions are weaker with the less basic anions, Br− and I− and so the framework structures do not form.
In all cases, we were able to prepare the framework materials in bulk, generally in good yields (67–83%); in the case of the framework prepared from HSO4−, the yield was lower (41%). These materials were investigated by thermogravimetric analysis (TGA), and IR and NMR spectroscopies, which revealed that all organic solvent had been removed from the structures on drying (see ESI†).
PXRD patterns for the bulk materials containing TBA cations were generally consistent with those simulated from the single crystal structures, albeit with some loss of crystallinity upon drying (Fig. 3a and S19–S24, see ESI† for further details regarding PXRD experiments). In the TEA-containing structure [HHTP·TEA·Cl]n there is a significant loss of crystallinity upon drying (Fig. 3b), which is perhaps not surprising given the large amount of weakly-interacting solvent present in the lattice (this structure contains diethyl ether, whereas the TBA containing structures contain H-bonded methanol or water molecules).12
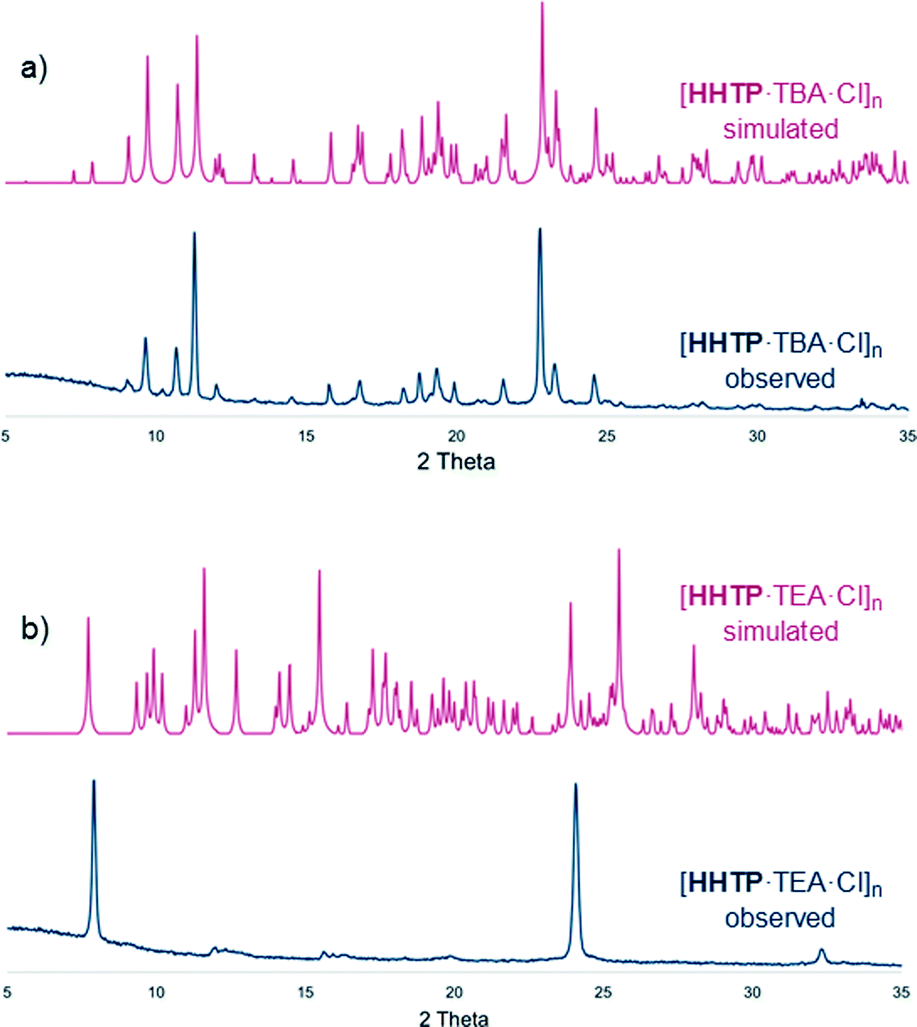 | ||
| Fig. 3 PXRD traces of dried, bulk samples of a) [HHTP·TBA·Cl]n and b) [HHTP·TEA·Cl]n as well as traces simulated from SCXRD data (see ESI† for full details). | ||
As mentioned above, crystallisation of HHTP and TBA·HSO4 gives a crystalline product containing the MeOSO3− anion. A search of the CSD13 reveals 60 structures14 containing this anion; of which the vast majority result from methylation of a nitrogen-containing species with dimethyl sulfate giving a cationic nitrogen centre as the MeOSO3− salt. In a handful of cases, this anion was formed from the dehydration of methanol by sulfuric acid (i.e. H2SO4 + MeOH → MeOSO3H + H2O, followed by deprotonation of MeOSO3H by a basic group within the molecule),15 but we could not find any examples where the HSO4− anion has been methylated to give MeOSO3−. NMR analysis of bulk material obtained from crystallisation of HHTP with TBA·HSO4 (Fig. S9 and S10†) reveals that approximately half of the anion is present as MeOSO3−, with the remainder presumably unreacted HSO4−. Interestingly, when HHTP and TBA·HSO4 are dissolved in CD3OD, no methylsulfate formation is observed by 13C NMR spectroscopy, even after seven days in solution,16 suggesting that crystallisation may be important in the methylation process.
In conclusion, we have demonstrated that O–H⋯anion hydrogen bonds can be used to assemble the triphenylene-containing ligand HHTP into framework materials. The materials contain 2D sheets formed from HHTP and anions, which adopt different levels of corrugation dependent on the anion and cation size. Despite the fact that O–H⋯anion interactions are weak in solution (in polar solvents), they reproducibly give crystalline frameworks in good yields. These results demonstrate the importance of weak interactions in crystal engineering and we suggest that if anion-templated materials can be prepared using stronger anion⋯receptor interactions, these systems may prove useful for a range of materials applications.
Acknowledgements
We thank Dr Lasse Noren (Australian National University) for assistance with PXRD studies, Dr Leesa Smith and Prof. Michelle Coote (Australian National University) for assistance with TGA analysis, and the Australian National University for supporting this research.Notes and references
- (a) P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS; (b) N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754 CrossRef CAS PubMed; (c) N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
- (a) Y. Haketa, S. Sasaki, N. Ohta, H. Masunaga, H. Ogawa, N. Mizuno, F. Araoka, H. Takezoe and H. Maeda, Angew. Chem., Int. Ed., 2010, 49, 10079–10083 CrossRef CAS PubMed; (b) S. Li, C. Jia, B. Wu, Q. Luo, X. Huang, Z. Yang, Q.-S. Li and X.-J. Yang, Angew. Chem., Int. Ed., 2011, 50, 5721–5724 CrossRef CAS PubMed; (c) Y. Haketa and H. Maeda, Chem. – Eur. J., 2011, 17, 1485–1492 CrossRef CAS PubMed; (d) B. Wu, F. Cui, Y. Lei, S. Li, N. d. S. Amadeu, C. Janiak, Y.-J. Lin, L.-H. Weng, Y.-Y. Wang and X.-J. Yang, Angew. Chem., Int. Ed., 2013, 52, 5096–5100 CrossRef CAS PubMed; (e) K. Pandurangan, J. A. Kitchen, S. Blasco, E. M. Boyle, B. Fitzpatrick, M. Feeney, P. E. Kruger and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2015, 54, 4566–4570 CrossRef CAS PubMed; (f) D. Yang, J. Zhao, Y. Zhao, Y. Lei, L. Cao, X.-J. Yang, M. Davi, N. Amadeu de Sousa, C. Janiak, Z. Zhang, Y.-Y. Wang and B. Wu, Angew. Chem., Int. Ed., 2015, 54, 8658–8661 CrossRef CAS PubMed; (g) H. Maeda, K. Chigusa, R. Yamakado, T. Sakurai and S. Seki, Chem. – Eur. J., 2015, 21, 9520–9527 CrossRef CAS PubMed.
- (a) D. K. Smith, Org. Biomol. Chem., 2003, 1, 3874–3877 RSC; (b) K. J. Winstanley, A. M. Sayer and D. K. Smith, Org. Biomol. Chem., 2006, 4, 1760–1767 RSC.
- (a) A. Shokri, J. Schmidt, X.-B. Wang and S. R. Kass, J. Am. Chem. Soc., 2012, 134, 16944–16947 CrossRef CAS PubMed; (b) A. Shokri, X.-B. Wang and S. R. Kass, J. Am. Chem. Soc., 2013, 135, 9525–9530 CrossRef CAS PubMed; (c) A. Shokri, S. H. M. Deng, X.-B. Wang and S. R. Kass, Org. Chem. Front., 2014, 1, 54–61 RSC.
- (a) N. G. White and M. J. MacLachlan, Cryst. Growth Des., 2015, 15, 5629–5636 CrossRef CAS; (b) N. G. White and M. J. MacLachlan, Chem. Sci., 2015, 6, 6245–6249 RSC; (c) N. G. White, V. Carta and M. J. MacLachlan, Cryst. Growth Des., 2015, 15, 1540–1545 CrossRef CAS.
- (a) M. Hmadeh, Z. Lu, Z. Liu, F. Gándara, H. Furukawa, S. Wan, V. Augustyn, R. Chang, L. Liao, F. Zhou, E. Perre, V. Ozolins, K. Suenaga, X. Duan, B. Dunn, Y. Yamamto, O. Terasaki and O. M. Yaghi, Chem. Mater., 2012, 24, 3511–3513 CrossRef CAS; (b) N. T. T. Nguyen, H. Furukawa, F. Gándara, C. A. Trickett, H. M. Jeong, K. E. Cordova and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 15394–15397 CrossRef CAS PubMed.
- (a) A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed; (b) H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed; (c) H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed; (d) J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228–231 CrossRef CAS PubMed; (e) G. H. V. Bertrand, V. K. Michaelis, T.-C. Ong, R. G. Griffin and M. Dincă, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4923–4928 CrossRef CAS PubMed; (f) D. D. Medina, J. M. Rotter, Y. Hu, M. Dogru, V. Werner, F. Auras, J. T. Markiewicz, P. Knochel and T. Bein, J. Am. Chem. Soc., 2015, 137, 1016–1019 CrossRef CAS PubMed.
- D. Hao, J. Zhang, H. Lu, W. Leng, R. Ge, X. Dai and Y. Gao, Chem. Commun., 2014, 50, 1462–1464 RSC.
- E. Voisin and V. E. Williams, Macromolecules, 2008, 41, 2994–2997 CrossRef CAS.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- When TEA·Br was used we obtained a poorly crystalline material, which did not diffract X-rays. When TEA·I was used, we obtained a crystalline material, which gave a unit cell consistent with TEA·I: E. Wait and H. M. Powell, J. Chem. Soc., 1958, 1872–1875 RSC.
- [HHTP·TBA·Cl]n shows the best match between observed and simulated PXRD spectra, presumably because no solvent is present in the single crystalline form of this compound. [HHTP·TBA·I]n, [HHTP·TBA·H2PO4]n and [HHTP·TBA·OAc]n show some loss of crystallinity, presumably due to removal of the water and/or methanol solvents present in the crystal structure upon drying. [HHTP·TEA·Cl]n contains a significant amount of solvent in the crystal structure, and PXRD suggests that removal of this solvent upon drying causes a significant loss of crystallinity.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- CSD version 5.36 with one update Search PubMed.
- (a) S. Blösl, W. Schwarz and A. Schmidt, Z. Anorg. Allg. Chem., 1982, 495, 177–185 CrossRef; (b) D. A. Baldwin, J. C. A. Boeyens, R. G. Copperthwaite, J. H. N. Loubser and A. J. Markwell, J. Crystallogr. Spectrosc. Res., 1984, 14, 157–167 CrossRef CAS; (c) S. G. Bott, A. W. Coleman and J. L. Atwood, J. Am. Chem. Soc., 1988, 110, 610–611 CrossRef CAS; (d) A. J. Blake, P. Hubberstey, U. Suksangpanya and C. L. Wilson, J. Chem. Soc., Dalton Trans., 2000, 3873–3880 RSC; (e) J. R. Deschamps, D. A. Knight, E. R. Goldman, J. B. Delehanty and E. L. Chang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, m916–m918 CAS; (f) L. Zhang, Y. Di and W. Dan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o1051 CAS; (g) J. K. Nath and J. B. Baruah, New J. Chem., 2013, 37, 1509–1519 RSC.
- I.e. significantly longer than the time taken for crystals formation, isolation and analysis.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization, details of instrumentation, details of SCXRD experiments and additional figures of solid state structures, details of PRXD experiments and PXRD patterns, thermogravimetric analysis data. CCDC 1451578–1451583. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce00297h |
| ‡ For further details of SCXRD studies, see the ESI.† CIF files for all structures are also provided as ESI† (CSD no.: 1451578–1451583). |
| § This parameter allows useful comparison between hydrogen bonds to differently-sized anions. It is relatively imprecise, as the position of hydrogen atoms is poorly-determined by SCXRD and so values are given to only two significant figures. For a more detailed discussion, see the ESI,† and N. G. White, C. J. Serpell and P. D. Beer, Cryst. Growth Des., 2014, 14, 3472–3479. |
| This journal is © The Royal Society of Chemistry 2016 |